

Abstract
Mutations in the human ether-a-go-go-related gene (hERG) cause type 2 long QT syndrome. In this study, we investigated the mechanism of the hERG splice site mutation 2398+1G>C and the genotype-phenotype relationship of mutation carriers in three unrelated kindreds with long QT syndrome. The effect of 2398+1G>C on mRNA splicing was studied by analysis of RNA isolated from lymphocytes of index patients and using minigenes expressed in HEK293 cells and neonatal rat ventricular myocytes. RT-PCR analysis revealed that the 2398+1G>C mutation disrupted the normal splicing and activated a cryptic splice donor site in intron 9, leading to the inclusion of 54 nt of the intron 9 sequence in hERG mRNA. The cryptic splicing resulted in an in-frame insertion of 18 amino acids in the middle of the cyclic nucleotide binding domain. In patch clamp experiments the splice mutant did not generate hERG current. Western blot and immunostaining studies showed that the mutant expressed an immature form of hERG protein that failed to reach the plasma membrane. Coexpression of the mutant and wild-type channels led to a dominant negative suppression of wild-type channel function by intracellular retention of heteromeric channels. Our results demonstrate that 2398+1G>C activates a cryptic site and generates a full-length hERG protein with an insertion of 18 amino acids, which leads to a trafficking defect of the mutant channel.
Keywords: long QT syndrome, splicing mutation, arrhythmia, sudden death, myocytes
1. Introduction
Long QT syndrome type 2 (LQT2) is caused by mutations in the human ether-a-go-gorelated gene (hERG) [1]. hERG encodes the pore-forming subunit of the rapidly activating delayed rectifier K+ channel (IKr), which is one of the major ion channel currents contributing to the repolarization of the cardiac action potential [2–4]. A wide spectrum of hERG mutations has been identified including missense, nonsense, in frame insertion or deletion, frameshift and splice site mutations [5–9]. The effects of missense, nonsense, and frameshift mutations on hERG channel function have been studied experimentally [10–14]. However, less is known about the pathogenic mechanisms of splice site mutations in LQT2. To date, more than 10 splice site mutations have been identified in hERG from LQT2 patients [5–9]. For most splice site mutations in hERG, the alterations in the mRNA transcripts were not reported. Although splice site mutations are expected to cause abnormal splicing, it is not possible to predict with certainty how the splice site mutations affect pre-mRNA splicing by genomic sequence analysis alone. Studies of splicing defects in a variety of diseases have shown that splice site mutations can result in different abnormal splicing patterns including exon skipping, use of nearby cryptic splice sites, and whole intron retention [15,16]. Cryptic splice sites are silent splice sites that are activated when the authentic splice site is destroyed by mutations. In order to understand the causative role of splice site mutations in LQT2, it is crucial to perform functional analysis of these mutations at the mRNA level. To date, only the 1945+6T>C splice site mutation in hERG has been experimentally characterized at the mRNA level [8]. This splice site mutation has been shown to cause intron 7 retention and exon 7 skipping. Intron 7 retention resulted in the truncation of the hERG channel protein and nonfunctional channels [8].
In this study we investigated the pathogenic mechanism of the hERG splice site mutation 2398+1G>C and genotype-phenotype relationship of the mutation carriers. The 2398+1G>C mutation has been identified in three unrelated LQT2 kindreds [1,5,7]. This mutation affects the consensus sequence of the donor splice site of intron 9. The donor splice site is defined by a consensus sequence of 9 bp at the exon-intron boundary. The 2398+1G>C mutation involves the G at +1 position, which is 100 % conserved. Although this mutation is predicted to disrupt normal splicing of intron 9 [1], how it alters splicing is unknown. Our results demonstrate that the 2398+1G>C mutation disrupts the normal splicing and leads to the activation of a downstream cryptic splice site. The cryptic splicing results in a full-length hERG protein with an insertion of 18 amino acids, leading to a trafficking defect of the mutant channel.
2. Materials and Methods
2.1. Subjects
The study was approved by the institutional review board and carried out on receipt of informed consent from all study participants. The participants were from three kindreds previously identified as having the 2398+1G>C mutation. Phenotyping was performed based on the history of LQTS-related cardiac events, clinical assessment according to the enhanced ECG criteria in regard to QT intervals and T-wave morphology, and pedigree analysis [8]. Normal unrelated individuals served as controls.
2.2. hERG minigenes and cDNA constructs
Human genomic DNA was used as a template for PCR amplification of a fragment spanning from hERG exons 8 to 11. The PCR products were cloned into pCRII vector using TA cloning kit (Invitrogen, Carlsbad, CA), and verified by DNA sequencing. The minigene was then subcloned into a mammalian expression vector pcDNA5/FRT (Invitrogen), containing a cytomegalovirus (CMV) promoter. The N-terminus of the minigene was tagged by the Myc epitope preceded by a Kozak sequence with a translation start codon, which is in-frame with the hERG translation sequence. The 2398+1G>C mutation in the minigene was generated using pAlter in vitro site-directed mutagenesis system (Promega, Madison, WI). hERG cDNA with the in-frame insertion of 18 amino acids was made by subcloning the RT-PCR fragment containing the 54 bp insertion into the hERG cDNA backbone by SphI and StuI sites. The Flag- and Myc-tagged hERG cDNA constructs were previously described [12,17].
2.3. Transfection of HEK293 cells
The minigene constructs in pcDNA5/FRT vector were stably transfected into HEK293-Flp-In cells as previously described [18]. The hERG cDNA constructs in pcDNA3 vector were stably or transiently transfected into HEK293 cells as previously described [4,11].
2.4. Construction and use of recombinant adenovirus
The AdEasy vector kit was used to generate WT and 2398+1G>C minigene recombinant adenoviruses (Stratagene, La Jolla, CA). First, the WT and 2398+1G>C minigenes were subcloned into pShuttle-CMV vector and recombined with the pAdEasy plasmid in Escherichia coli strain BJ5183. The pAdEasy/minigene plasmids were transfected into HEK293 cells. After 2 days, the transfected cells were cultured in growth medium containing 1.25% Seaplaque-agarose to promote the formation of recombinant viral plaques. Approximately two to three weeks later, individual plaques were picked, amplified in HEK293 cells, and purified over a discontinuous CsCl gradient.
2.5. Primary culture of neonatal rat ventricular myocytes
Neonatal rat ventricular myocytes were prepared as described [18]. Briefly, hearts were removed from 1 to 3-day-old Sprague-Dawley rat pups. The ventricles were trimmed free of atria, fat and connective tissues. Myocytes were dissociated by several 20-minute cycles of collagenase/pancreatin treatment. Myocytes were cultured in DMEM with 17% Media 199, 1% penicillin/streptomycin, 10% horse serum and 5% fetal bovine serum. After one day in culture, myocytes were infected with the recombinant adenoviruses.
2.6. RT-PCR analysis of RNA splicing
For endogenous hERG RNA splicing assays, total RNA was isolated from lymphocytes of normal subjects and patients carrying the 2398+1G>C mutation using the RiboPure Blood kit (Ambion, Austin, TX). Contaminating genomic DNA was removed by DNase I treatment according to the protocol provided by the supplier. For minigene splicing assays, cytoplasmic RNA was isolated from transfected HEK293 cells or neonatal rat ventricular myocytes using the Qiagen RNeasy kit. After reverse transcription (RT) using the SuperScript III First-Strand DNA Synthesis kit (Invitrogen), PCR was performed with primers in exon 8 (forward 5'-CTCGAGGA GTACTTCCAGCACG-3') and exon 10 (reverse 5'-TTGCCAGGCCTTGCATACAG-3') for RNA isolated from lymphocytes and HEK293 cells. For RT-PCR analysis of RNA isolated from neonatal rat ventricular myocytes, a reverse primer complementary to the sequence in the recombinant adenovirus (reverse 5'-GATCCGGTGGATCGGATATCT-3') was used to distinguish between the mRNA transcripts of the infected minigene and endogenous rat ERG. The PCR products were analyzed by electrophoresis on agarose gels, and cloned into pCRII vector for sequence analysis.
2.7. Patch clamp recordings
Membrane currents were recorded in whole cell configuration using suction pipettes as previously described [4,11]. All patch clamp experiments were performed at 22–23°C. The patch clamp data are presented as mean±SEM and analyzed by Student’s t-test. P<0.05 is considered statistically significant.
2.8. Western blot and immunoprecipitation
Western blot and immunoprecipitation were performed as previously described [12,17]. The cell lysates were immunoprecipitated with anti-Myc antibody followed by Western blot with anti-Flag antibody. The membranes were also probed with anti-Myc antibody after stripping out the anti-Flag antibody.
2.9. Immunofluorescence microscopy
The immunofluorescence microscopy was performed as previously described [17]. Cells were fixed with 4% paraformaldehyde and incubated with anti-hERG antibody (1:3000) at 4°C overnight followed by Alexa 488 conjugated goat anti-rabbit IgG secondary antibody (Molecular Probes, Eugene, OR). Immunofluorescence staining was viewed with a Nikon fluorescence microscope.
3. Results
3.1. Phenotyping
Phenotyping was characterized in 7 patients carrying the 2398+1G>C mutation from three kindreds based on medical history, ECG assessment, and pedigree analysis. The genotyping of these families has been reported previously [1,5,7]. QTc of initial ECG was 477±34 ms (n=7, one patient from family 1, five patients from family 2, and one patient from family 3). Four patients had syncope episodes and one of them developed cardiac arrest due to tosade de points with subsequent ventricular fibrillation. Five patients were on beta-blockers, and two of them also had ICD therapy.
3.2. Analysis of RNA splicing in lymphocytes of patients carrying the 2398+1G>C mutation
When the 2398+1G>C mutation was first identified in 1995, it was predicted to disrupt normal splicing of intron 9 [1]. However, it is not possible to predict how this splice site mutation affects pre-mRNA splicing by genomic sequence analysis alone. It is important to analyze the 2398+1G>C mutation at the mRNA level. Because the affected heart tissue from the mutation carriers was not available for this study, we analyzed hERG mRNA transcripts isolated from lymphocytes of the patients carrying the 2398+1G>C mutation. Expression of hERG transcripts in lymphocytes has been reported previously [18,19]. Figure 1A shows the RT-PCR analysis using the primers in exon 8 and exon 10 as described in methods. RT-PCR analysis of RNA isolated from lymphocytes of a normal subject yielded a single PCR product of 362 bp, corresponding to the normal splicing. In contrast, RT-PCR analysis of RNA isolated from lymphocytes of the proband of family 2 showed two bands at 362 bp and 416 bp. To better define the identity and the origin of the spliced transcripts, we sequenced the PCR products from the lymphocytes. Sequence analysis of the 362 bp PCR products confirmed that the normal 5' donor splice site of intron 9 was used in the both normal subject and the proband. Sequence analysis of the 416 bp PCR product from the proband revealed that a cryptic 5' donor site located 54 nt downstream of the normal site was used (Fig. 1B). The same result was observed in another mutation carrier of this family. These findings strongly suggest that the 2398+1G>C mutation activates a cryptic site in intron 9. The cryptic splicing results in the inclusion of 54 nt of the intron 9 sequence in hERG mRNA (Fig. 1C) and leads to an in-frame insertion of 18 amino acids (AMGWGAGTGLEMPS AASR) in the middle of the cyclic nucleotide binding domain (between residue 799 and 800). Thus, the 2398+1G>C mutation produces a full-length hERG channel with a large in-frame insertion.
Figure 1.
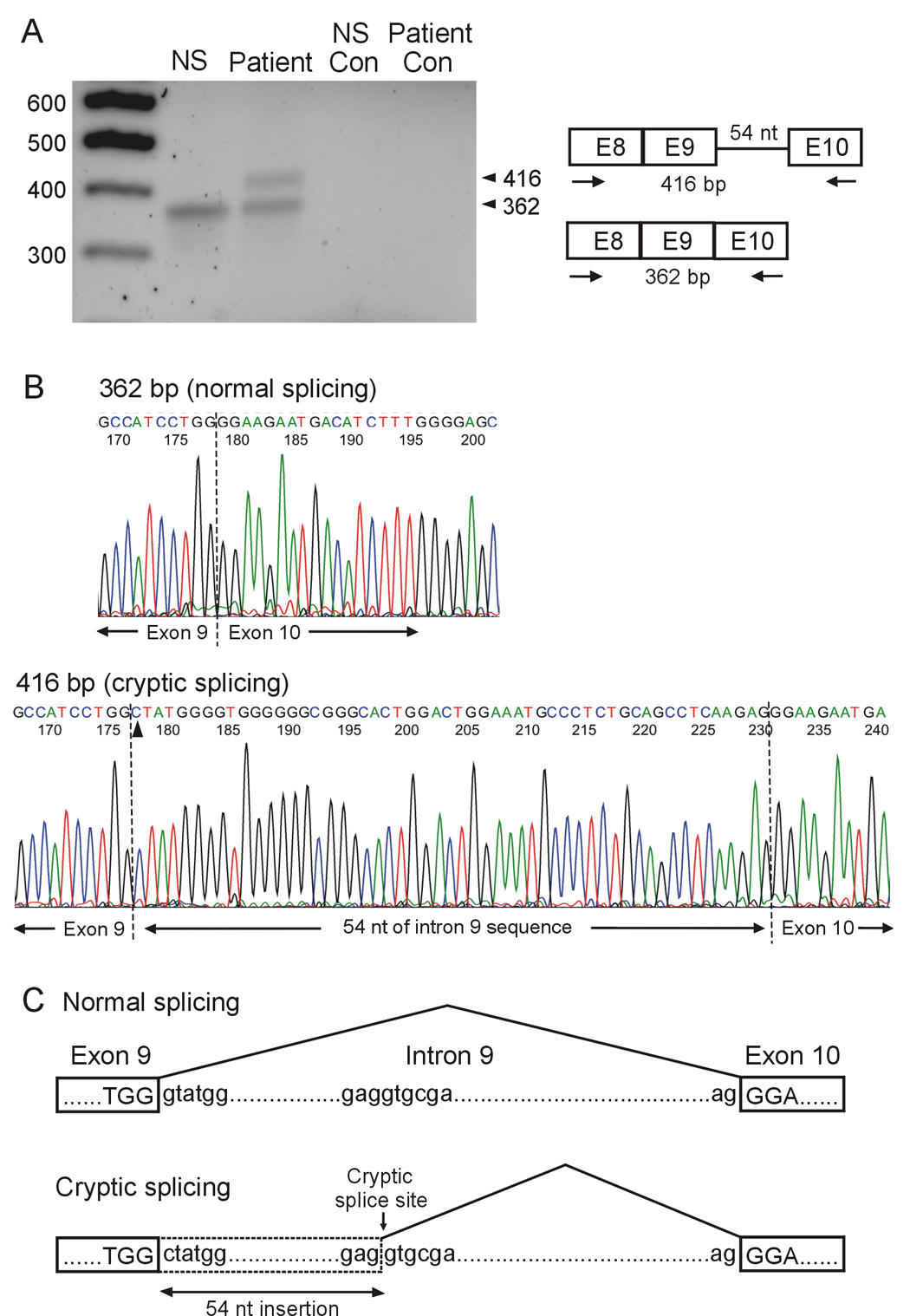
RT-PCR analysis of hERG mRNA isolated from lymphocytes. A: mRNA was isolated from a normal subject (NS) and the proband of family 2. RT- PCR was performed using primers in exon 8 and exon 10 as indicated by arrows. Control lanes: no reverse transcriptase in RT reaction. Similar results were obtained from another mutation carrier of this family and two independent experiments were performed for each patient. B: Sequence analysis of the 362 bp product from a normal subject (top panel) and the 416 bp product from the proband (bottom panel). Arrow head indicates the 2398+1G>C mutation. C: Schematic presentation of normal and cryptic splicing.
3.3. Minigene splicing in HEK293 cells and rat neonatal ventricular myocytes
The analysis of RNA from patients indicates that the 2398+1G>C mutation leads to the activation of a cryptic site. However, it is unclear from these experiments whether the 2398+1G>C mutation completely disrupts the normal splicing, because the normally spliced band (362 bp) in the lymphocytes of mutation carriers could come from either the normal or mutant allele. To further characterize the 2398+1G>C mutation, we performed minigene splicing assays in HEK293 cells and neonatal rat ventricular myocytes. The minigene contains the hERG genomic sequence spanning from exon 8 to exon 11 (Fig. 2A). HEK293 cells were transfected with wild-type (WT) and 2398+1G>C mutant minigenes, and the expressed RNA was analyzed by RT-PCR with the same primers as used in Fig 1. RT-PCR analysis of RNA isolated from cells transfected with the WT minigene yielded a PCR product of 362 bp, while in the 2398+1G>C mutant minigene a 416 bp fragment was observed (Fig. 2B). Sequence analysis of the PCR products revealed that the same cryptic 5' donor site as that observed in patient's lymphocyte RNA was used. These results indicate that the 2398+1G>C mutation completely disrupts normal splicing and leads to cryptic splicing.
Figure 2.
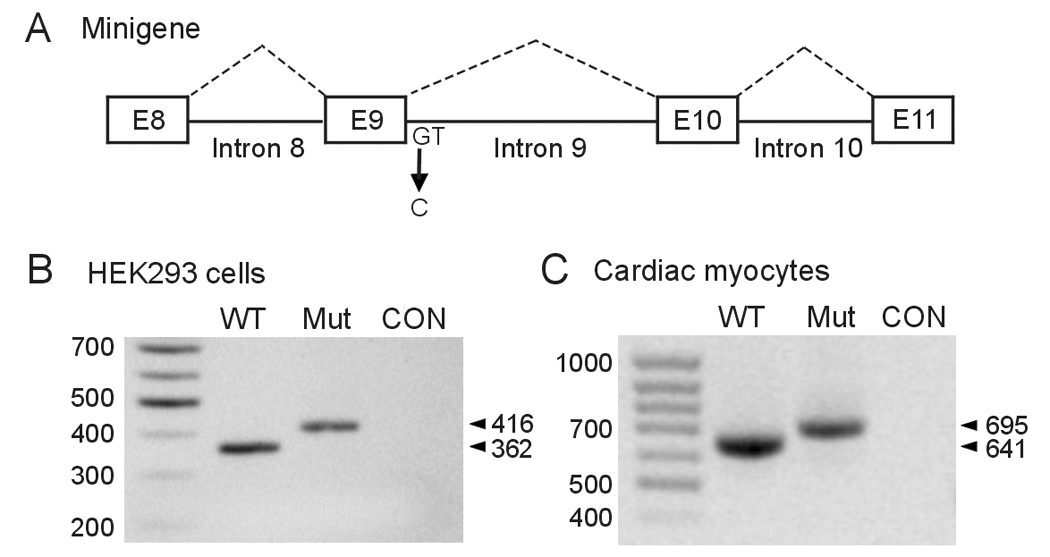
Analysis of the 2398+1G>C mutation using minigenes expressed in HEK293 cells and neonatal rat ventricular myocytes. A: Diagram of the hERG minigene used in transfection experiments. B: The WT and 2398+1G>C minigenes were transfected into HEK293 cells and isolated RNAs were amplified by RT-PCR with the same primers as used in figure 1. CON: untransfected HEK293 cells. C: The WT and 2398+1G>C minigenes were infected into neonatal rat ventricular myocytes using adenovirus constructs. RT-PCR was performed using the same forward primer as above and a reverse primer complementary to the sequence in the recombinant adenovirus. CON: uninfected myocytes. Results shown are representative of three independent experiments.
In order to determine whether the cryptic splicing observed in the lymphocytes and HEK293 cells is also present in the heart, we expressed the WT and 2398+1G>C minigenes in neonatal rat ventricular myocytes using adenovirus constructs. RT-PCR analysis using primers in exon 8 and adenovirus described in methods showed that the WT minigene produced a 641 bp fragment and the mutant minigene yielded a 695 bp fragment (Fig. 2C). Sequence analysis confirmed that the same cryptic 5' donor site as that observed in lymphocytes and HEK293 cells was used by neonatal rat ventricular myocytes. Thus, our results suggest that the cryptic splicing is likely to occur in the heart.
3.4. Functional analysis of the splice mutant
The above experiments at the mRNA level indicate that the cryptic splicing caused by the 2398+1G>C mutation leads to a full-length hERG protein with a large in-frame insertion of 18 amino acids. To study functional consequences of the insertion, we introduced these 18 amino acid residues into hERG cDNA and expressed the splice mutant channel in HEK293 cells. Patch clamp experiments showed that the mutant channel did not produce hERG current (Fig. 3A). Panel B shows the I–V plot of hERG current amplitude present at the end of the depolarizing steps. The maximal outward current in WT hERG was observed at voltage step to 0 mV, and at more positive voltages there was a steep negative slope conductance. The peak current density of WT hERG was 36.7±6.8 pA/pF (n=8). For the 2398+1G>C mutation, the I–V plot of the current present at the end of the depolarizing steps showed a linear relation consistent with a small amplitude leak current as previously reported in untransfected HEK293 cells [4].
Figure 3.
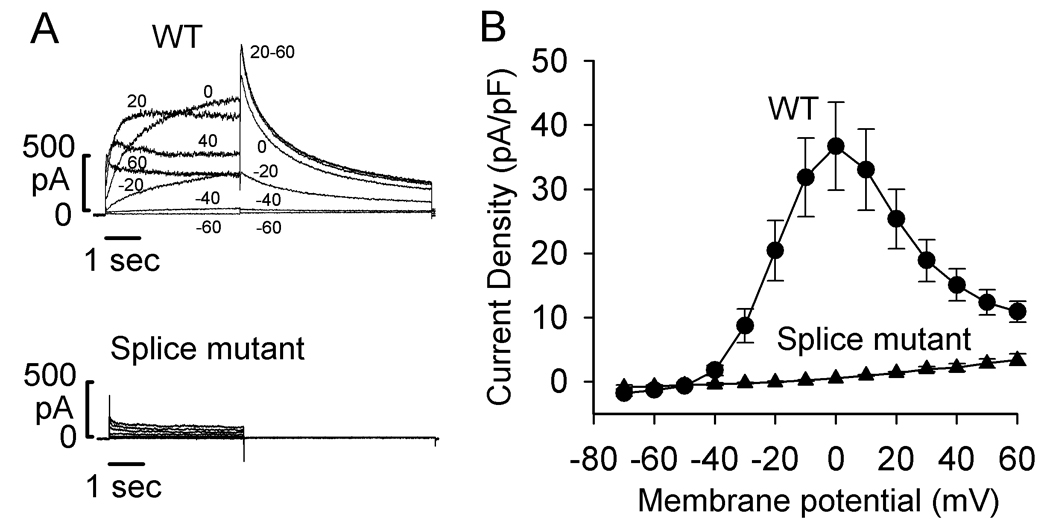
Voltage clamp recordings of WT and the splice mutant expressed in HEK293 cells. A: Representative currents recorded from cells transfected with WT hERG or the splice mutant. Cells were held at −80 mV and depolarized to voltages between −70 and 60 mV for 4 seconds. Cells were then clamped to −50 mV for 6 seconds to record a tail current. For clarity, only selected current traces (as labeled) are shown. B: I–V plot of hERG current recorded at the end of depolarizing steps from WT hERG (circles, n=8) and the splice mutant (triangles, n=6).
3.5. Defective trafficking of the splice mutant
To determine the underlying mechanism responsible for the lack of hERG current in the splice mutant, we performed Western blot analysis of hERG channel proteins (Fig. 4). WT hERG expressed two protein bands at 135 kDa and 155 kDa. The 135 kDa band represents the core-glycosylated immature form of the channel protein located in the ER and the 155 kDa band represents the complex-glycosylated mature form of the channel protein located in the plasma membrane [4,11,20]. The splice mutant expressed a single band at about 135 kDa. To study the cell surface expression of hERG channel proteins, the intact cells transfected with WT hERG or the splice mutant were treated with proteinase K. The 155 kDa band of WT hERG was sensitive to proteinase K treatment with the appearance of digested lower molecular weight bands at 60–75 kDa as described previously [11]. For the splice mutant, the single band was resistant to the proteinase K treatment, suggesting that the mutant channel is not expressed on the cell surface. To confirm the biochemical results, we performed immunofluoresence staining of WT and mutant channels. The cells expressing WT hERG showed a diffused staining pattern throughout the cells including the plasma membrane (Fig. 5 A and B). In the cells expressing the splice mutant the staining pattern was more restricted to a perinuclear region (Fig. 5 C and D). The immunostaining findings are consistent with the Western blot results and suggest that the insertion of 18 amino acids leads to defective trafficking of the mutant channel. Our previous studies have shown that many trafficking defective LQT2 mutations can be rescued by low temperature or hERG channel blocking agent E-4031 [21, 22]. However, low temperature and E-4031 had no effect on the trafficking of the splice mutant (data not shown).
Figure 4.
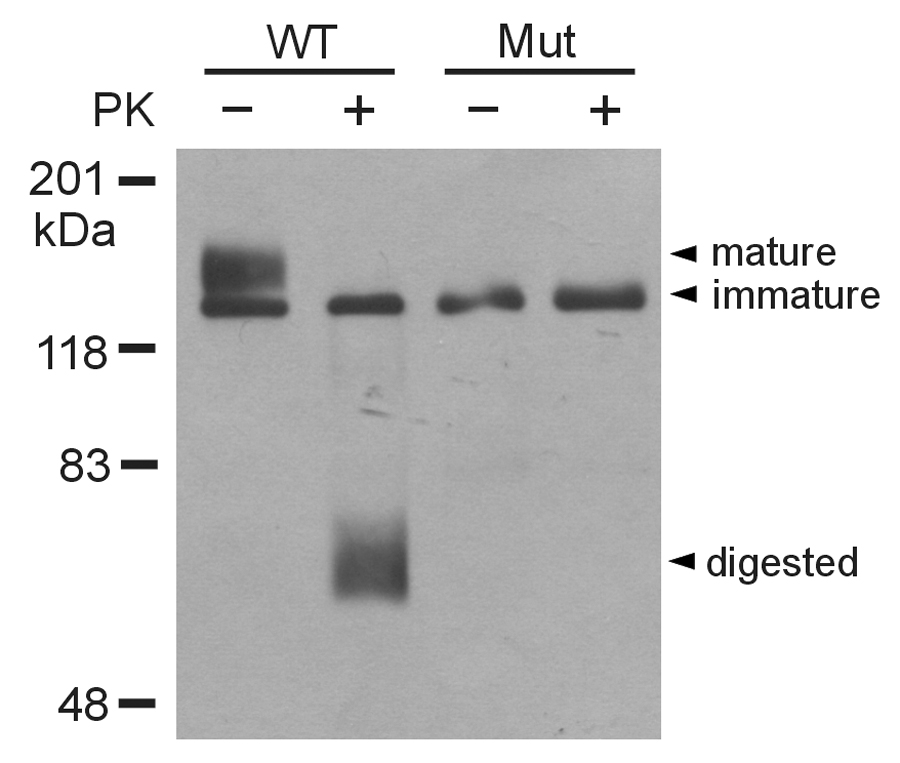
Western blot analysis of WT and splice mutant channels. HEK293 cells were transfected with WT and the splice mutant. The intact cells were treated with (+) or without (−) proteinase K. The isolated membrane proteins were subjected to SDS-PAGE and immunoblotted with anti-hERG antibody. Results shown are representative of three independent experiments.
Figure 5.
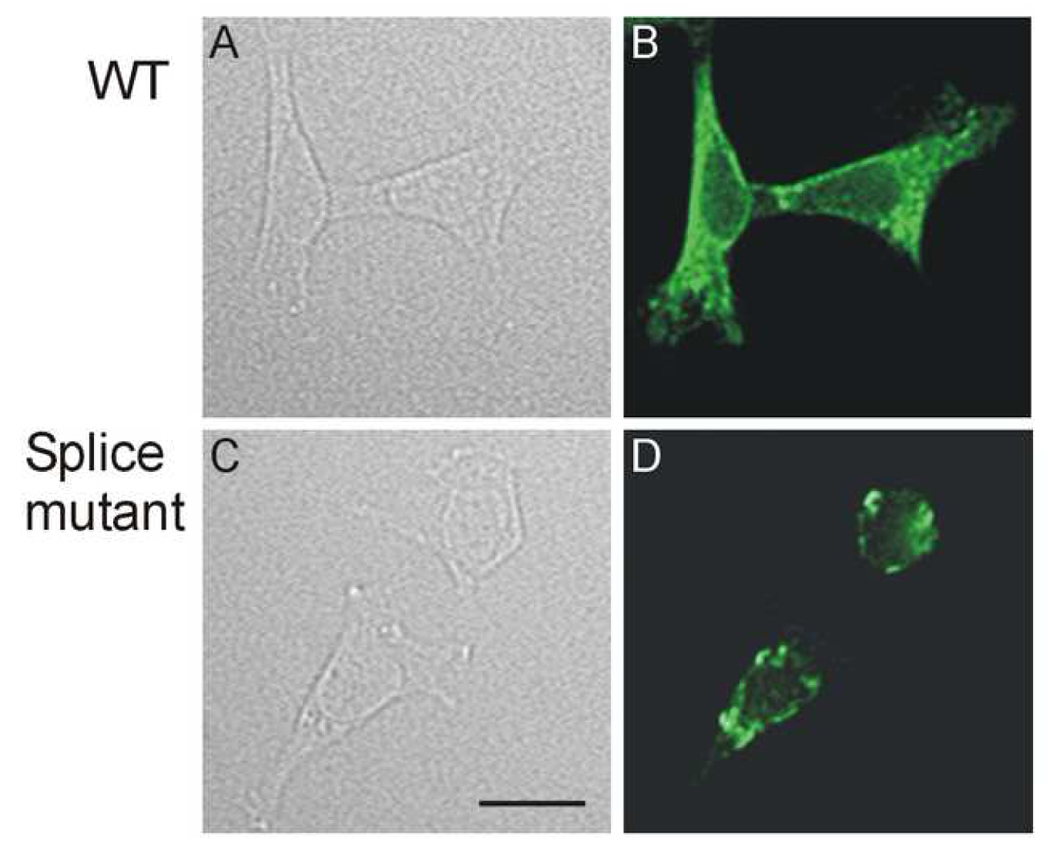
Immunofluorescence staining for localization of WT and splice mutant channels. HEK239 cells were transfected with WT (A and B) and the splice mutant (C and D). For each transfection, a phase contrast photograph (A and C) and anti-hERG antibody staining (B and D) are shown. Calibration bar = 20 µm.
3.6. Dominant negative effect
Because all mutation carriers are heterozygous for the 2398+ 1G >C mutation, both normal and mutant alleles are present in these patients. We next tested whether the mutant channel protein caused by 2398+1G>C exerts a dominant negative effect on WT hERG channel function. We coexpressed the mutant and WT hERG by transiently transfecting the splicing mutant or control plasmid (pcDNA3 vector) into a WT hERG stable cell line [4,12]. When the splice mutant was coexpressed with WT hERG, it caused a significant decrease in hERG current amplitudes (Fig. 6). Panel B shows the I–V plot of peak tail current densities measured at −50 mV against preceding test voltages for WT + vector (n=12) and WT + the splice mutant (n=8, p<0.05 at all voltages more positive than −30 mV). To study the voltage dependence of hERG channel activation, we fit the normalized tail currents with a Boltzmann function. The half-maximal activation voltages (V1/2) and k values for WT + vector (n=12) and WT + the splice mutant (n=8) were not significantly different (−14.4±1.8 mVand −12.6±2.3 mV for V1/2, respectively, p>0.05, and 7.7±0.3 and 8.6±0.7 for k value, respectively, p>0.05).
Figure 6.
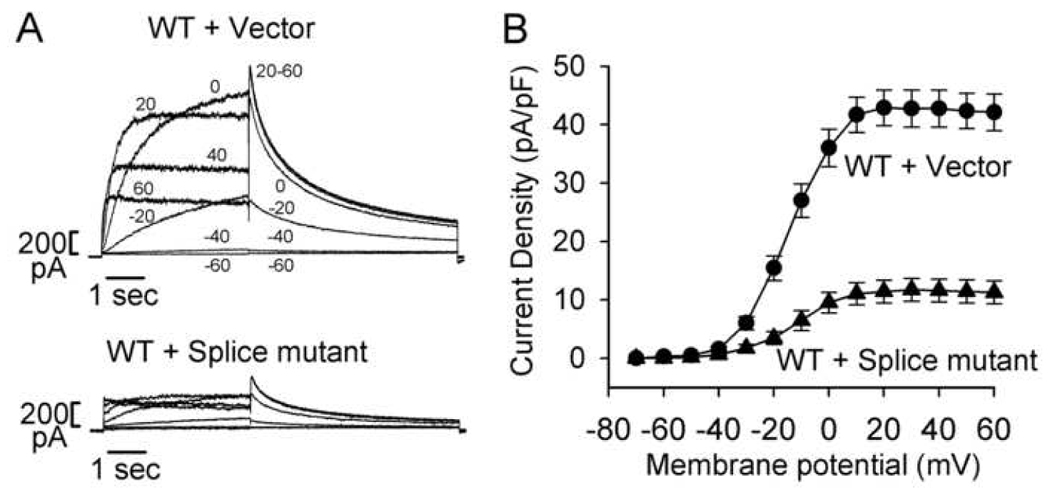
Dominant negative effect of the splice mutant. Control plasmid (pcDNA3 vector) and the splice mutant were transiently transfected into a WT hERG stable cell line. The hERG current was activated with the voltage clamp protocol used in Figure 3. A: Representative currents recorded from the hERG stable cell line following transfection of pcDNA3 vector (WT + vector) and the splice mutant (WT + Splice mutant). For clarity, only selected current traces (as labeled) are shown. B: I–V plot of hERG tail current densities measured at −50 mV following test voltages from −70 to 60 mV for WT + vector (circles, n=12) and WT + Splice mutant (triangles, n=8).
3.7. Coassembly of the splice mutant with WT hERG
The presence of a dominant negative effect for the splice mutant suggests that it can coassemble with WT hERG to form heteromeric channels. To directly demonstrate the coassembly of the splice mutant with WT hERG, we performed coimmunoprecipitation analysis using differentially tagged WT hERG and the splice mutant. We cotransfected Myc-tagged WT hERG (WT-Myc) with Flag-tagged WT hERG (WT-Flag) or Flag-tagged splice mutant (Mut- Flag) in HEK293 cells. Coassembly of Flag-tagged and Myc-tagged hERG channel proteins was determined by immunoprecipitation with anti-Myc antibody followed by Western blot analysis with anti-Flag antibody. As shown in Fig. 7, WT-Flag and Mut-Flag were coimmunoprecipitated with WT-Myc in cotransfected cells. The coimmunoprecipitation was not observed in cells transfected with WT-Myc, WT-Flag or Mut-Flag alone (lanes 1–3). The membranes were also reprobed with anti-Myc antibody to show efficient immunoprecipitation of Myc-tagged hERG proteins. The association of WT-Myc and WT-Flag was observed in both immature and mature forms of hERG channel protein. In contrast, the association of WT-Myc and Mut-Flag was observed primarily in the immature form of hERG channel protein, suggesting that coassembly of the splice mutant and WT hERG occurs prior to the formation of complex glycosylation in the hERG channel biosynthesis pathway. Western blot analysis of the cell lysates without immunoprecipitation showed that coexpression of the splice mutant and WT hERG resulted in a marked decrease in the mature form of Myc-tagged WT hERG. These results suggest that coexpression of the splice mutant with WT hERG causes defective trafficking of WT subunits similar to that of mutant subunits.
Figure 7.
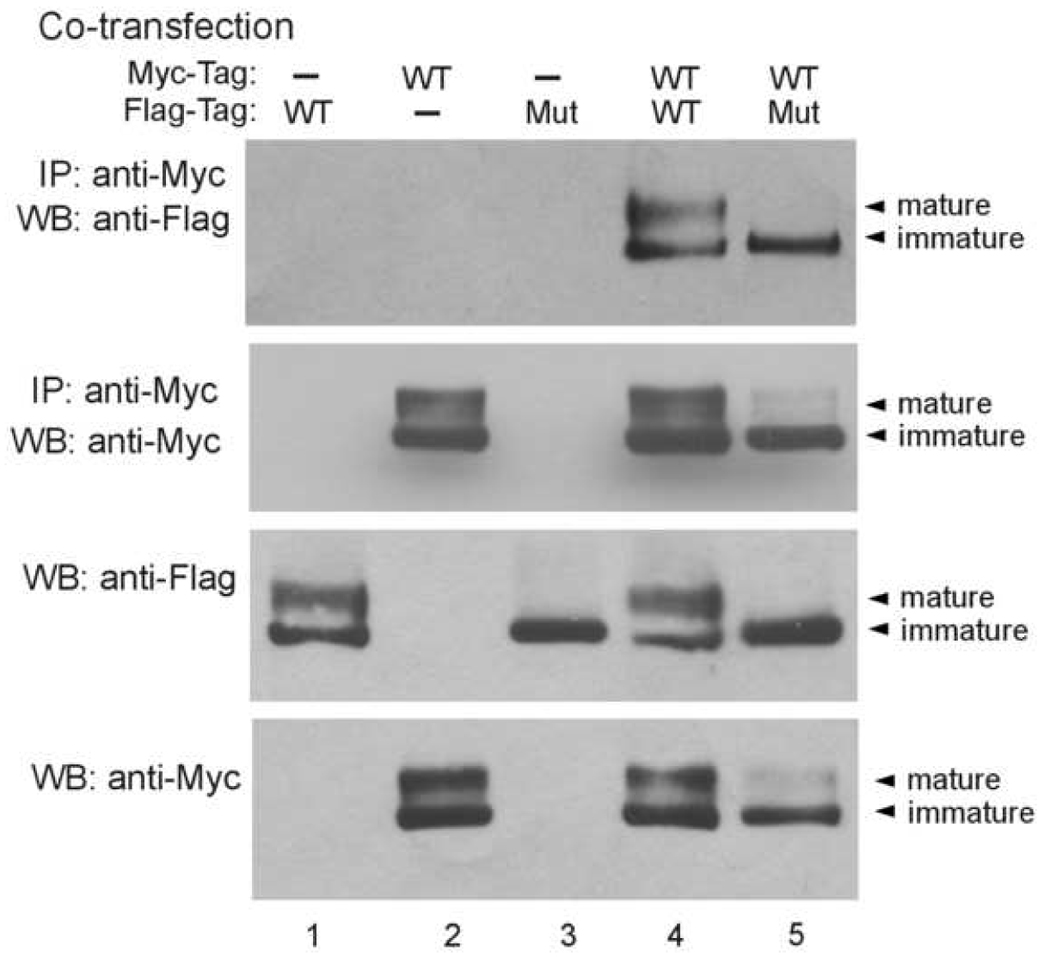
Coassembly of the splice mutant with WT hERG. Flag-tagged WT hERG was cotransfected with Myc-tagged WT hERG or splice mutant in HEK293 cells. Cell lysates were subjected to immunoprecipitation with anti-Myc antibody followed by immunoblotting with anti- Flag or anti-Myc antibody as indicated. Cell lysates were also immunoblotted with anti-Flag or anti-Myc antibody as indicated. Results shown are representative of three independent experiments.
4. Discussion
Accurate pre-mRNA splicing is essential for the conversion of nascent transcripts to mRNA templates for protein synthesis. Pre-mRNA splicing depends upon the recognition of the 5' and 3' splice sites as well as the branch point. These sequences appear to be vulnerable to disease-causing mutations. It has been estimated that up to 15% of all point mutations causing human genetic diseases result in an mRNA splicing defect [15]. The 2398+1G>C mutation in hERG has been identified in three unrelated kindreds with long QT syndrome [1,5,7]. This mutation involves the +1 position of the 5' donor splice site of intron 9. When the 2398+1G>C mutation was first identified, it was predicted to result in protein truncation due to potential abnormal splicing [1]. However, our present analysis at the mRNA level demonstrates that the 2398+1G>C mutation leads to the activation of a cryptic site 54 bp downstream from the normal site of intron 9. Thus, rather than protein truncation this mutation results in the production of a full-length channel with the insertion of 18 amino acids in the middle of the cyclic nucleotide binding domain. This is the first report that a splice site mutation in hERG results in the utilization of a cryptic splice site and leads to a large in-frame insertion in the hERG channel. Our findings underscore the importance of studying LQT2 splice site mutations at the mRNA level.
Using RNA isolated from the lymphocytes of the patients carrying the 2398+1G>C mutation, we were able to show that the endogenous mutant RNA undergoes cryptic splicing. The use of RNA from lymphocytes for studying splice site mutations has been reported in several human heart diseases including KCNQ1 in LQT1 and SCN5A in Brugada syndrome [23–27]. One limitation of using lymphocytes is that the aberrant splicing observed in lymphocytes may not be present in the heart. In the present study we have used a minigene system to demonstrate that the same cryptic site as that observed in lymphocytes was used in neonatal rat ventricular myocytes. This result strongly suggests that the cryptic splicing remains in the mammalian ventricular myocyte and is likely to occur in the human heart of subjects carrying the mutation. Our present findings also show that lymphocytes may be a convenient source of endogenous hERG mRNA for studying other LQT2 splice site mutations when the affected heart tissues are not available.
In general, mutation of a splice site can result in one or a combination of splicing defects. These defects include the failure to recognize the affected exon resulting in exon skipping, activation of a cryptic site, and inability to recognize the affected intron resulting in whole intron retention. For the 2398+1G>C mutation, the activation of a cryptic site in intron 9 might be due to the intrinsic strength of the cryptic site. The sequence of the cryptic 5' splice site in intron 9 is GAG/gtgcga. We calculated the consensus value (CV) scores of the normal, mutant and cryptic splice sites using the method described by Shapiro and Senapathy [28]. The consensus value reflects the similarity of a splice site to the consensus sequence, and most splice donor sites have a score of 70 or higher. The CV scores of WT, 2398+1G>C and the cryptic splice site are 76.3, 58.0 and 79.6, respectively. Thus, the cryptic splice site has an intrinsic strength comparable to that of the normal splice site of intron 9. However, in normal conditions the cryptic site is completely inactive even though its CV score is slightly higher than that of the normal site. The cryptic site is activated when the normal site is disrupted by the mutation with a dramatic decrease in the CV score.
The functional studies indicate that the insertion of 18 amino acids in the cyclic nucleotide binding domain results in defective trafficking of the mutant hERG channel. Defective trafficking of mutant hERG channels has been shown to be the most common mechanism in LQT2 missense mutations [22]. These trafficking deficient hERG mutants are retained in the endoplasmic reticulum due to prolonged association with chaperone proteins calnexin, Hsp70 or Hsp90, and are rapidly degraded by the ubiquitin proteasome pathway [29–31]. It is noted that all LQT2 mutations in the cyclic nucleotide binding domain lead to defective trafficking of mutant channels [22,32]. The large insertion caused by the cryptic splicing involves a highly ordered structure in β5 region of the cyclic nucleotide binding domain, which has been shown to play an important role in hERG channel trafficking [32]. The present results also show that coexpression of mutant and WT channels leads to a dominant negative suppression of WT channel function by intracellular retention of heteromeric channels. The presence of the dominant negative effect is consistent with the observed clinical presentation of the patients carrying the 2398+1G>C mutation. Four of seven mutation carriers had syncope episodes. Previous phenotype-genotype correlation studies have shown that patients with mutations in the pore region of the hERG gene are at markedly increased risk for arrhythmia-related cardiac events compared with patients with nonpore mutations [33]. One of the families (family 2) in the present study was also included in that report. Although this family was assigned to the nonpore mutation group, the mutation carriers from this family appear to have a higher risk for arrhythmia-related cardiac events than other patients with nonpore mutations. Three of five mutation carriers in this family had syncope episodes and one of them developed cardiac arrest. Two patients in this family required ICD therapy. Our present study suggests that the severe clinical phenotype of this family might be explained by the presence of the dominant negative effect observed in the 2398+1G>C mutation.
In conclusion, the 2398+1G>C mutation disrupts the normal splicing and leads to the activation of a downstream cryptic site. The cryptic splicing results in a full-length hERG channel with an insertion of 18 amino acids, leading to a trafficking defect of the mutant channel. These findings indicate that functional analyses at both mRNA and protein levels are important for obtaining a full understanding of putative splice site mutations in hERG. The identification of the pathogenic mechanisms of hERG mutations will facilitate the studies of genotype-phenotype relationship in LQT2.
Acknowledgments
We are grateful to Kristen Tabor, Jennifer L. Robinson and Katherine W. Timothy for recruiting LQT2 families to the study, the LQT2 family members for their cooperation and understanding of research, and Dr. Kent Thornburg for helpful comments on the manuscript. This study was supported in part by NIH grant HL68854 (Dr. Zhou) and the Oregon Clinical and Translational Research Institute (OCTRI), grant number UL1RR024140 01 from the National Center for Research Resources, a component of NIH, and NIH Roadmap for Medical Research. Dr. Zhou is an Established Investigator of American Heart Association.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- 2.Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- 3.Trudeau MC, Warmke JW, Ganetzky B, Robertson GA. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 1995;269:92–95. doi: 10.1126/science.7604285. [DOI] [PubMed] [Google Scholar]
- 4.Zhou Z, Gong Q, Ye B, Fan Z, Makielski JC, Robertson GA. Properties of HERG channels stably expressed in HEK 293 cells studied at physiological temperature. Biophys J. 1998;74:230–241. doi: 10.1016/S0006-3495(98)77782-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, et al. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102:1178–1185. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- 6.Napolitano C, Priori SG, Schwart PJ, Bloise R, Ronchetti E, Nastoli J, et al. Genetic testing in the long QT syndrome: development and validation of an efficient approach to genotyping in clinical practice. JAMA. 2005;294:2975–2980. doi: 10.1001/jama.294.23.2975. [DOI] [PubMed] [Google Scholar]
- 7.Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing testing. Heart Rhythm. 2005;2:507–517. doi: 10.1016/j.hrthm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 8.Zhang L, Vincent GM, Baralle M, Baralle FE, Anson BD, Benson DW. An intronic mutation causes long QT syndrome. J Am Coll Cardiol. 2004;44:1283–1291. doi: 10.1016/j.jacc.2004.06.045. [DOI] [PubMed] [Google Scholar]
- 9.Shang YP, Xie XD, Wang XX, Chen JZ, Zhu JH, Tao QM, et al. A novel splice mutation of HERG in a Chinese family with long QT syndrome. J Zhejiang Univ Sci B. 2005;6:626–630. doi: 10.1631/jzus.2005.B0626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanguinetti MC, Curran ME, Spector PS, Keating MT. Spectrum of HERG K+-channel dysfunction in an inherited cardiac arrhythmia. Proc Natl Acad Sci USA. 1996;93:2208–2212. doi: 10.1073/pnas.93.5.2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou Z, Gong Q, Epstein ML, January CT. HERG channel dysfunction in human long QT syndrome: Intracellular transport and functional defects. J Biol Chem. 1998;273:21061–21066. doi: 10.1074/jbc.273.33.21061. [DOI] [PubMed] [Google Scholar]
- 12.Gong Q, Keeney DR, Robinson JC, Zhou Z. Defective assembly and trafficking of mutant HERG channels with C-terminal truncations in long QT syndrome. J Mol Cell Cardiol. 2004;37:1225–1233. doi: 10.1016/j.yjmcc.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 13.Delisle BP, Anson BD, Rajamani S, January CT. Biology of cardiac arrhythmias: ion channel protein trafficking. Circ Res. 2004;94:1418–1428. doi: 10.1161/01.RES.0000128561.28701.ea. [DOI] [PubMed] [Google Scholar]
- 14.Paulussen AD, Raes A, Jongbloed RJ, Gilissen RA, Wilde AA, Snyders DJ, et al. HERG mutation predicts short QT based on channel kinetics but causes long QT by heterotetrameric trafficking deficiency. Cardiovasc Res. 2005;67:467–475. doi: 10.1016/j.cardiores.2005.05.017. [DOI] [PubMed] [Google Scholar]
- 15.Krawczak M, Reiss J, Cooper DN. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet. 1992;90:41–54. doi: 10.1007/BF00210743. [DOI] [PubMed] [Google Scholar]
- 16.Faustino NA, Cooper TA. Pre-mRNA splicing and human disease. Genes Dev. 2003;17:419–437. doi: 10.1101/gad.1048803. [DOI] [PubMed] [Google Scholar]
- 17.Gong Q, Anderson CL, January CT, Zhou Z. Pharmacological rescue of trafficking defective HERG channels formed by coassembly of wild type and long QT mutant N470D subunits. Am J Physiol Heart Circ Physiol. 2004;287:H652–H658. doi: 10.1152/ajpheart.00052.2004. [DOI] [PubMed] [Google Scholar]
- 18.Gong Q, Zhang L, Vincent GM, Horne BD, Zhou Z. Nonsense mutations in hERG cause a decrease in mutant mRNA transcripts by nonsense-mediated mRNA decay in human long QT syndrome. Circulation. 2007;116:17–24. doi: 10.1161/CIRCULATIONAHA.107.708818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller TE, You L, Myerburg RJ, Benke PJ, Bishopric NH. Whole blood RNA offers a rapid, comprehensive approach to genetic diagnosis of cardiovascular diseases. Genet Med. 2007;9:23–33. doi: 10.1097/gim.0b013e31802d74de. [DOI] [PubMed] [Google Scholar]
- 20.Gong Q, Anderson CL, January CT, Zhou Z. Role of glycosylation in the cell surface expression and stability of HERG potassium channels. Am J Physiol Heart Circ Physiol. 2002;283:H77–H84. doi: 10.1152/ajpheart.00008.2002. [DOI] [PubMed] [Google Scholar]
- 21.Zhou Z, Gong Q, January CT. Correction of defective protein trafficking of a mutant HERG potassium channel in human long QT syndrome: Pharmacological and temperature effects. J Biol Chem. 1999;274:31123–31126. doi: 10.1074/jbc.274.44.31123. [DOI] [PubMed] [Google Scholar]
- 22.Anderson CL, Delisle BP, Anson BD, Kilby JA, Will ML, Tester DJ, et al. Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation. 2006;113:365–373. doi: 10.1161/CIRCULATIONAHA.105.570200. [DOI] [PubMed] [Google Scholar]
- 23.Murray A, Donger C, Fenske C, Spillman I, Richard P, Dong YB, et al. Splicing mutations in KCNQ1: a mutation hot spot at codon 344 that produces in frame transcripts. Circulation. 1999;100:1077–1084. doi: 10.1161/01.cir.100.10.1077. [DOI] [PubMed] [Google Scholar]
- 24.Hong K, Guerchicoff A, Pollevick GD, Oliva A, Dumaine R, de Zutter M, et al. Cryptic 5' splice site activation in SCN5A associated with Brugada syndrome. J Mol Cell Cardiol. 2005;38:555–560. doi: 10.1016/j.yjmcc.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 25.Rossenbacker T, Schollen E, Kuiperi C, de Ravel TJ, Devriendt K, Matthijs G, et al. Unconventional intronic splice site mutation in SCN5A associates with cardiac sodium channelopathy. J Med Genet. 2005;42:e29. doi: 10.1136/jmg.2004.029058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsuji K, Akao M, Ishii TM, Ohno S, Makiyama T, Takenaka K, et al. Mechanistic basis for the pathogenesis of long QT syndrome associated with a common splicing mutation in KCNQ1 gene. J Mol Cell Cardiol. 2007;42:662–669. doi: 10.1016/j.yjmcc.2006.12.015. [DOI] [PubMed] [Google Scholar]
- 27.Zehelein J, Kathoefer S, Khalil M, Alter M, Thomas D, Brockmeier K, et al. Skipping of Exon 1 in the KCNQ1 gene causes Jervell and Lange-Nielsen syndrome. J Biol Chem. 2006;281:35397–35403. doi: 10.1074/jbc.M603433200. [DOI] [PubMed] [Google Scholar]
- 28.Shapiro MB, Senapathy P. RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res. 1987;15:7155–7174. doi: 10.1093/nar/15.17.7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gong Q, Jones MA, Zhou Z. Mechanisms of pharmacological rescue of trafficking-defective hERG mutant channels in human long QT syndrome. J Biol Chem. 2006;280:4069–4074. doi: 10.1074/jbc.M511765200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ficker E, Dennis AT, Wang L, Brown AM. Role of the cytosolic chaperones Hsp70 and Hsp90 in maturation of the cardiac potassium channel HERG. Circ Res. 2003;92:87–100. doi: 10.1161/01.RES.0000079028.31393.15. [DOI] [PubMed] [Google Scholar]
- 31.Gong Q, Keeney DR, Molinari M, Zhou Z. Degradation of trafficking defective long QT syndrome type II mutant channels by the ubiquitin-proteasome pathway. J Biol Chem. 2005;280:19419–19425. doi: 10.1074/jbc.M502327200. [DOI] [PubMed] [Google Scholar]
- 32.Akhavan A, Atanasiu R, Noguchi T, Han W, Holder N, Shrier A. Identification of the cyclic-nucleotide-binding domain as a conserved determinant of ion-channel cell-surface localization. J Cell Sci. 2005;118:2803–2812. doi: 10.1242/jcs.02423. [DOI] [PubMed] [Google Scholar]
- 33.Moss AJ, Zareba W, Kaufman ES, Gartman E, Peterson DR, Benhorin J, et al. Increased risk of arrhythmic events in long-QT syndrome with mutations in the pore region of the human ether-a-go-go-related gene potassium channel. Circulation. 2002;105:794–799. doi: 10.1161/hc0702.105124. [DOI] [PubMed] [Google Scholar]