

Abstract
In Bacillus subtilis, several phenolic acids specifically induce expression of padC, encoding a phenolic acid decarboxylase that converts these antimicrobial compounds into vinyl derivatives. padC forms an operon with a putative coding sequence of unknown function, yveFG, and this coding sequence does not appear to be involved in the phenolic acid stress response (PASR). To identify putative regulators involved in the PASR, random transposon mutagenesis, combined with two different screens, was performed. PadR, a negative transcriptional regulator of padC expression, was identified. padR is not located in the vicinity of padC, and the expression of padR is low and appears constitutive. This is in contrast with what occurs in other gram-positive bacteria, in which padR is autoregulated and induced by phenolic acids. Further screening of the transposon library failed to identify genes other than padR involved in the PASR. Modest inactivation of padR by phenolic acids was obtained in recombinant Escherichia coli expressing padC and padR, and this translates into induction of decarboxylase activity. Gel shift promoter binding assays performed with and without MgCl2, and with and without phenolic acids, demonstrated that phenolic acids were able to abolish the binding of PadR to the yveFG-padC promoter in the absence of MgCl2. Altogether, our results indicate that (i) PadR is inactivated directly by phenolic acids in vitro, (ii) inhibition of PadR in response to phenolic acids may occur without the need for a sensor-like effector in B. subtilis, and (iii) phenolic acids are able to modulate PadR activity in E. coli in the absence of any additional effector.
Phenolic acids, such as ferulic, p-coumaric, and caffeic acids, are lignin-related aromatic acids, which bind the complex lignin polymer to the hemicellulose and cellulose in plant cell walls (9). These acids are toxic for gram-positive bacteria under acidic conditions and are able to specifically and tightly induce expression of padC (designated padA for other gram-positive and lactic acid bacteria), the gene that encodes phenolic acid decarboxylases (PADs) of the gram-positive bacterial species Bacillus pumilus (34), Bacillus subtilis (5), Lactobacillus plantarum (4), and Pediococcus pentosaceus (3). In B. subtilis, salicylic acid, another phenolic compound which induces expression of the bsdBCD operon, encoding phenylacrylic acid decarboxylase B and 4-hydroxybenzoate decarboxylases C and D, is also able to induce the expression of padC. Both BsdD and PadC confer resistance to phenylacrylic acid in B. subtilis (11).
In the phenolic acid stress response (PASR), inducible Pad enzymes decarboxylate phenolic acids into vinyl phenol derivatives that are not toxic for gram-positive bacteria. Expression of padC (or padA) is regulated by a negative transcriptional regulator, PadR, which was identified as the first member of a new class of transcriptional regulators (Pfam PF03551) (3, 14). Although PadRs now form a regulatory family of more than 400 putative PadR-like regulators identified in bacterial genome sequencing programs (Pfam accession number PF03551), only a few of them are currently being studied. AphA is a transcriptional activator of the virulent gene cascade of Vibrio cholerae (18), mediated by quorum sensing (17, 21). However, AphA does not seem to be involved in phenolic acid metabolism in V. cholerae. More recently, the roles of two PadR-like repressors were reported for Listeria monocytogenes: LstR, which is required for effective thermal resistance (35), and LadR, the transcriptional regulator of the mdrL gene, which encodes a multidrug efflux transporter (15).
Despite these recent advances, the inactivation and activation mechanisms of PadR-like regulators have not been elucidated, and the mechanism(s) by which phenolic acids inactivate PadR remains unknown. In P. pentosaceus, padA and padR are organized in an autoregulated bicistronic operon. The operon was heterologously expressed in Escherichia coli, where repression of padA expression by PadR has been confirmed (3). However, phenolic acids remain unable to inactivate PadR in this recombinant strain of E. coli despite the very low expression of the padA-padR operon and the resulting very low level of PadR protein in this strain. To date, genetic studies with P. pentosaceus remain difficult due to the lack of effective transformation methods. In L. plantarum, padA and padR are divergently transcribed. In the absence of phenolic acids, PadR represses the expression of padA by binding to the padA promoter (14). Deletion of the padR gene in L. plantarum leads to the constitutive overexpression of padA where the PadA enzyme is the main protein produced in the cell. As was the case for P. pentosaceus, inactivation of L. plantarum PadR heterologously expressed in E. coli could not be demonstrated. Taken together, these recent results led to the hypothesis that a putative phenolic acid protein sensor could act in conjunction with PadR to regulate phenolic acid metabolism. The goal of the present work is to characterize the genetic and biochemical mechanisms involved in the PASR, one of the most sensitive and specific responses to stress induced by a family of compounds that are ubiquitous in the plant kingdom and that also possess antimicrobial properties.
In this report, we (i) clarify the annotation of the controversial yveE-yveG region located directly upstream of padC and investigate its involvement in the PASR, (ii) integrate our initial results on the identification of PadR with two screening methods designed to identify every gene that could encode an additional transcriptional regulator(s) involved in the PASR, and (iii) demonstrate direct phenolic acid-mediated inactivation of PadR by heterologous expression of padR with a reporter construct including the promoter of the yveFG-padC operon in E. coli and by a DNA mobility shift assay. Our results indicate that inactivation of PadR does not require a phenolic acid protein sensor.
MATERIALS AND METHODS
Plasmids, bacterial strains, and growth conditions.
The plasmids and bacterial strains used in this study are listed in Table 1. E. coli and B. subtilis strains were grown aerobically in Erlenmeyer flasks on a rotary shaker in Luria-Bertani (LB) medium. For selection and growth of transformants, antibiotics were used at the following concentrations: erythromycin, 100 μg/ml for E. coli and 30 μg/ml for B. subtilis; chloramphenicol, 5 μg/ml for B. subtilis; spectinomycin, 100 μg/ml for E. coli and B. subtilis; ampicillin, 100 μg/ml; and kanamycin, 50 μg/ml for E. coli.
TABLE 1.
Plasmids and bacterial strains
| Plasmid or strain | Genotype and/or relevant feature(s) | Source or reference |
|---|---|---|
| Plasmids | ||
| pTZ19R | Ampr ΔlacZ | 28 |
| pTC | pTZ19R with padC under its own promoter | This work (Fig. 4) |
| pTR | pTZ19R with padR under its own promoter | This work (Fig. 4) |
| pTCR | pTRC with padR under its own promoter | This work (Fig. 4) |
| pET28a+ | Kanr; vector for overexpression of His-tagged proteins using the T7 bacteriophage promoter | Novagen |
| pER | pET28a+ containing padR gene beween BspHI and XhoI sites to overproduced PadR His6 tag | This work (Fig. 6) |
| pIC333 | Mini-Tn10 delivering vector used for transposon mutagenesis | 32 |
| pJM103 | Sper; integrative vector used to delete the padR gene | 26 |
| pJMΔR | pJM103 with a truncated padR gene copy | This work (Fig. 4) |
| pJM783 | Ampr Cmr; integrative vector used to construct the lacZ transcriptional fusions F1 to F6 | 26 |
| pJF1 | pJM783 carrying the 148-bp F1 fragment | This work (Fig. 2) |
| pJF2 | pJM783 carrying the 308-bp F2 fragment | This work (Fig. 2) |
| pJF3 | pJM783 carrying the 442-bp F3 fragment | This work (Fig. 2) |
| pJF4 | pJM783 carrying the 568-bp F4 fragment | This work (Fig. 2) |
| pJF5 | pJM783 carrying the 699-bp F5 fragment | This work (Fig. 2) |
| pEFGM | pET28a+ containing yveG gene between NcoI and XhoI sites; the GTG start codon was mutated into ATG | This work |
| pEFGMR | pET28a+ containing yveG gene beween NcoI and XhoI sites; the GTG start codon was mutated into ATG; the stop codon TGA in-frame was mutated into CGA (R) | This work (Fig. 3) |
| pEFGMQ | pET28a+ containing yveG gene beween NcoI and XhoI sites; the GTG start codon changed into ATG; the stop codon TGA in-frame was mutated into CAA (Q) | This work (Fig. 3) |
| Strains | ||
| B. subtilis | ||
| BS168 | trpC2 | Institut Pasteur, France |
| BSΔR | trpC2 Specr ΔpadR mutant | This work (Fig. 4) |
| BS783F1 | trpC2 Cmr Ampr; carrying the F1 padC::lacZ fusion | This work (Fig. 2) |
| BS783F2 | trpC2 Cmr Ampr; carrying the F2 padC::lacZ fusion | This work (Fig. 2) |
| BS783F3 | trpC2 Cmr Ampr; carrying the F3 padC::lacZ fusion | This work (Fig. 2) |
| BS783F4 | trpC2 Cmr Ampr; carrying the F4 padC::lacZ fusion | This work (Fig. 2) |
| BS783F5 | trpC2 Cmr Ampr; carrying the F5 padC::lacZ fusion | This work (Fig. 2) |
| E. coli | ||
| TG1 | SupE hsdΔ5 thiΔ(lac-proAB) F′ traD36 proAB+ lacIqlacZΔM15 | 13 |
| BL21(DE3) star | F−ompT hsdSB (rB− mB−) gal dcm (DE3) | Invitrogen |
| BL21pER | BL21(DE3) star carrying the plasmid pER | This work (Fig. 6) |
| TG1pTC | Ampr; carrying plasmid pTC | This work (Fig. 4) |
| TG1pTR | Ampr; carrying plasmid pTR | This work (Fig. 4) |
| TG1pTCR | Ampr; carring plasmid pTCR | This work (Fig. 4) |
| BL21pEFGM | BL21 with plasmid pEFGM | This work (Fig. 3) |
| BL21pEFGMR | BL21 with plasmid pEFGMR | This work (Fig. 3) |
| BL21pEFGMQ | BL21 with plasmid pEFGMQ | This work (Fig. 3) |
DNA manipulation, PCR amplification, and transformation procedures.
DNA manipulation, purification, ligation, restriction analysis, and gel electrophoresis were carried out as described by Sambrook et al. (30). Genomic DNA was extracted as previously described (24). PCR amplifications were performed with 50-μl reaction mixtures, using 0.1 unit of Taq DNA polymerase (Qbiogen) or PWO (Invitrogen, Roche Molecular Biochemicals) in an automatic thermocycler (Bio-Rad, Richmond, CA), with primers (Table 2) provided by Eurogentec (Belgium). PCR and restriction products were purified using a QIAquick PCR purification kit or a QIAgel agarose gel extraction kit (Qiagen). E. coli was transformed by electroporation as described by Dower et al. (10). B. subtilis was transformed with linear plasmid DNA or chromosomal DNA by using the two-step nutrient downshift procedure described by Msadek et al. (24). DNAs were sequenced by Genome Express (Meylan, France).
TABLE 2.
Primers
| Use and name | Sequence (5′→3′)a | Site(s) created |
|---|---|---|
| Cloning padR in pET28a+ | ||
| BSR1 | GACTCATGAGAGTATTAAAATACGCC | BspHI |
| BSR2 | GCTCTCGAGATCCTTATCTATCATAG | XhoI |
| Cloning in pTZ19R | ||
| BSR3 | TACGTCTAGAGACAGGATTATGTACTGACT | XbaI |
| BSR4 | AAGCTGCAGGATCGACATTGAACCGAAAT | PstI |
| BSD3 | ATCGGATCCTATTGTTTGACAGTTAACTGC | BamHI |
| BSD10 | GACTCCCGGGCGCTGGACTGGCATCATACT | SmaI |
| Determination of position +1 | ||
| BSD8 | GAATCATCTCAGTCCCAGGCTTG | |
| Probe for DNA binding | ||
| BSD1 | CAAAGCTAGCTTCAGACAAGG | |
| BSD8 | GAATCATCTCAGTCCCAGGCTTG | |
| qRT-PCR | ||
| BSDFor (padC) | TTCATAGCGGAATGGTTGCC | |
| BSDRev (padC) | GCTCTGTCCAAGACACTTTAT | |
| BSRFor (padR) | CACAGCCAGATTTACCCTGA | |
| BSRRev (padR) | GCTTTGTGCCCTGAATCGTT | |
| BS16SFor (16S RNA) | AGAACAAAGGGCAGCGAAAC | |
| BS16SRev (16S RNA) | CGATTACTAGCGATTCCAGC | |
| padC::lacZ fusions | ||
| BSDF1 | CCAGAATTCACGGCAAGTCAGCAAGCCGT | EcoRI |
| BSDF2 | CCAGAATTCTCGCGCTAACGGCAGAACAG | EcoRI |
| BSDF3 | CCAGAATTCTCAAGGTTGATCCGCCTATC | EcoRI |
| BSDF4 | CCAGAATTCCTATTGTTTGACAGTTAACTGC | EcoRI |
| BSDF5 | CCAGAATTCATTCAAAGTGAGATTCCGATT | EcoRI |
| BSDFR | TCAGGATCCGATAAAGTTTTCCATCTTACAC | BamHI |
| Sequencing of Tn10 insertions | ||
| TN101 | TGGCCGATTCATTAATGCAGG | |
| TN104 | CGATATTCACGGTTTACCCAC | |
| Deletion of padR | ||
| BSRM1 | ACGTCTAGACCCCCAAAAAATTTTCATACCA | XbaI |
| BSRM2 | CCTGCATGCAAAAGCCCTAATAGGCGTA | SphI |
| BSRM3 | CTAGGATCCATAAGGATTAACCGCAGTTCAG | BamHI |
| BSRM4 | GCAGAATTCATGGTGAGCGGGATGTATTGG | EcoRI |
| TNSPEC1 | TGAGCATGCCTACGGGGTCTGACGCTCAGTGG | SphI |
| TNSPEC2 | GTAGGATCCTGTTATTGCAATAAAATTAGCC | BamHI |
| Codon replacement and cloning of yveFGM | ||
| BSFG1 | ATGCCaTGGTTCTCGTGAAGAAGCb | NcoI, ATG |
| BSFG2 | ATGCTCGAGCTGTACTTCATAAATGTC | XhoI |
| BSFGR | AACGAATCGGAATGAATCgTCTCAGTCCCAGGCTTGTCb | R anticodon |
| BSFGQ | AACGAATCGGAATGAATtgTCTCAGTCCCAGGCTTGTCb | Q anticodon |
Underlined nucleotides correspond to restriction sites given in the right column.
Lowercase, bold, and underlined characters correspond to modified sequences.
RNA extraction, primer extension, and qRT-PCR analysis.
Total RNA extraction and primer extension analysis were performed as described previously (5), except that first-strand cDNA synthesis was carried out using the oligonucleotide BSD8 (Fig. 1 and Table 2). Quantitative reverse transcription-PCR (qRT-PCR) analysis was performed as follows. cDNAs were synthesized using an iScript cDNA synthesis kit (Bio-Rad), following the instructions of the manufacturer. Primers used to quantify relative gene expression were designed using Primer 3 software and are listed in Table 2. Real-time PCR amplifications were carried out with a Bio-Rad I cycler with IQ Sybr green Supermix. A control without reverse transcriptase was used for each reaction to confirm the absence of chromosomal DNA contamination. Three serial dilutions of cDNA were performed for each sample. In each run, a negative control using sterile water was included. An additional cooling step, from 90°C to 60°C (3°C/min), was performed to establish a melting curve in order to verify the homogeneity of the amplicon. Amplification efficiency was determined by running a standard curve with serial dilutions of cDNA. Relative expression levels were calculated using the comparative critical threshold (ΔΔCT) method 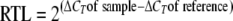 (22). The 16S rRNA was chosen as an internal control to calculate the threshold cycles as previously described for qRT-PCR experiments with B. subtilis (6). To calculate the relative transcript levels (RTL), the ΔCTs of the genes were compared to the ΔCTs of the same genes in the reference conditions. Total RNAs extracted from three independent cultures were analyzed by qRT-PCR for each condition.
(22). The 16S rRNA was chosen as an internal control to calculate the threshold cycles as previously described for qRT-PCR experiments with B. subtilis (6). To calculate the relative transcript levels (RTL), the ΔCTs of the genes were compared to the ΔCTs of the same genes in the reference conditions. Total RNAs extracted from three independent cultures were analyzed by qRT-PCR for each condition.
FIG. 1.

Characterization of the upstream region of padC. The stop codon and transcription terminator (TT; convergent arrows) of racX are indicated. The start of the five transcriptional padC::lacZ fusions F1 to F5 are in bold and underlined (see also Fig. 2A for results of integration in BS168 chromosome). “Put. +1 (1998)” (nucleotide A in bold and underlined) is the putative transcriptional initiation that was obtained with primer BSD2 and previously published (5). “G +1” is the right site of transcription initiation determined with primer BSD8 and padC::lacZ fusion expression results (Fig. 2). “I-R” (ATGT-8 nucleotides-ACAT) in bold corresponds to the inverted-repeat consensus sequence found in promoter sequences of padC or padA genes (3). The putative promoter of yveFG-padC deduced in the present work is indicated upstream of this consensus sequence. RBS, putative ribosome binding sites. TGA in line 400 is the putative stop codon of the previous putative yveF gene or the undetermined X codon indicated in GenBank. The underlined ATG codon located in the middle of line 500 corresponds to the start codon of the previous putative yveG gene indicated in the SubtiList website.
Construction of padC::lacZ transcriptional fusions.
Five padC::lacZ transcriptional fusions were constructed using the pJM783 vector (26), kindly provided by M. Perego (The Scripps Research Institute, La Jolla, CA). Five DNA fragments, 148, 308, 442, 568, and 699 bp upstream from the padC ATG start codon (Fig. 1 and 2A), were PCR amplified using the five sense primers BSDF1, BSDF2, BSDF3, BSDF4, and BSDF5, paired with the antisense primer BSDFR (Table 2). The amplicons were digested with EcoRI and BamHI and cloned into pJM783 at the EcoRI and BamHI sites to produce plasmids pJF1, pJF2, pJF3, pJF4, and pJF5, respectively. These plasmids were then transformed into BS168. Integration of the padC::lacZ fusions into the chromosome by a simple crossover event at the padC locus of BS168 was expected to generate strains BS783F1 to BS783F5, respectively (Fig. 2A and B).
FIG. 2.

Schematic representation of the five chromosomal padC::lacZ transcriptional fusions (A) with analysis of lacZ and padC expression in these strains either induced or not induced with 1 mM ferulic acid (B) and determination of the padA initiation transcription site by reverse transcription with primer BSD8 (C) with RNA from cells either induced (I) or not induced (NI) with 1 mM ferulic acid. T, G, C, and A represent sequencing reaction of this DNA region with primer BSD8. The location of this initiation transcription site is also indicated in panel A. In yveFG, the black vertical bar indicates the stop codon.
Deletion of padR.
Two DNA fragments, encompassing (i) the 5′ end of lipB upstream of padR and the 5′ end of padR (fragment A) and (ii) the 3′ end of padR and the 3′ end of the yfiN open reading frame (fragment B), were generated by PCR amplification using the primer pairs BSRM1-BSRM2 and BSRM3-BSRM4, respectively (Table 2). Fragments A and B were digested with the enzyme couples XbaI-SphI and BamHI-EcoRI, respectively. A third, 1.3-kbp DNA fragment (fragment C), containing the adenyltransferase gene from Tn554 (25), conferring spectinomycin resistance, was PCR amplified from plasmid pIC333 (32) with primers TNSPEC1 and TNSPEC2 and double digested by SphI and BamHI. Taking advantage of the respective double-digested ends of each fragment and of the vector, the ligation was expected to produce the insertion of fragments A, B, and C into the pJM103 vector (26) to generate plasmid pJMΔR. The ligation product was transformed into E. coli TG1, and recombinant E. coli colonies were selected onto LB supplemented with spectinomycin. PCR amplification on Spcr clones allowed the detection of colonies harboring plasmids with the A-B-C insertion. Plasmid pJMΔR was transformed into BS168 to generate, after a double recombination event at the padR chromosomal locus, the replacement of padR with the spectinomycin resistance gene cassette (see Fig. 4A1). The genotype and phenotype of the mutant strain were verified by PCR amplification with appropriate primers and by comparison of PAD activity in bacteria in noninduced and ferulic acid-induced cultures, respectively.
FIG. 4.

Expression of padC and padR in B. subtilis and recombinant E. coli strains. (A1 and A2) Maps of padC DNA of the wild-type strain BS168 and the BSΔR mutant, with their corresponding PAD activities and SDS-PAGE protein profiles. (B1 and B2) Plasmid constructions achieved in the vector pTZ19R that were expressed in E. coli with their corresponding PAD activities and SDS-PAGE protein profiles. In these plasmids, padC and padR were expressed under their own promoters. The PAD activities in crude protein extracts from noninduced cells (NI) and cells induced with 1 mM (∼0.2 g/liter) ferulic acid (I) for 20 min at OD600s of 0.3 or 0.6 and 1.6 are indicated on the right side. Under the noninduced condition (NI), the same results were obtained at the three stages of growth. pTZ, extract from control E. coli strain with the pTZ19R vector without an insert. M, molecular mass markers. Equal amounts of proteins were loaded in each lane of any given gel.
Site-directed mutagenesis of the putative gene yveFG.
Briefly, the yveFG expression plasmid pEFGM was constructed by amplifying the putative yveFG coding sequence, from nucleotide +34 to +496 relative to the transcriptional start site, with the primer pair BSFG1/BSFG2 (Table 2) and ligating the resulting product into the pET28a+ vector. BSFG1 was designed to replace the GTG start codon with ATG and to include an NcoI restriction site. BSFG2 contains the XhoI restriction site for cloning purposes. In order to convert the TGA stop codon in-frame with the yveFG sequence into the sense codons CGA and CAA, corresponding to Arg (R) and Glu (Q), respectively, a two-step PCR-based strategy was used. First, two independent PCRs were performed with primer pairs BSFG1/BSFGR and BSFG1/BSFGQ. The mutagenic BSFGR and BSFGQ primers contain codons CGA and CAA, respectively, instead of the TGA stop codon (Table 2 and Fig. 1). Second, the 120-bp corresponding amplicons were purified (Qiagen kit) and combined with the BSFG2 primer to amplify the 462-bp final products. The final products were then digested with NcoI and XhoI and cloned into the pET28a+ vector to yield plasmids pEFGMR and pEFGMQ, respectively (Table 1). The mutations were verified by sequencing. The three plasmids pEFGM, pEFGMR, and pEFGMQ were transformed into E. coli BL21(DE3) cells and induced with isopropyl β-d-thiogalactopyranoside (IPTG) to monitor protein expression by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
Transposon mutagenesis.
For transposon mutagenesis, the mini-Tn10 delivering vector pIC333 (32), kindly given by S. Aymerich (INRA, Grignon, France), was used according the procedure described by Dartois et al. (8). The culture that showed the best transposition efficiency was diluted and plated onto X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside)-containing LB agar with or without ferulic acid to screen for target mutants. To rescue inserted transposons and adjacent regions, chromosomal DNA was extracted from B. subtilis mutants and digested with EcoRI, which does not cut within the mini-Tn10 insertion sequence. The resulting DNA fragments were self-ligated and used to transform the E. coli TG1 strain. Transformants were selected onto LB agar containing spectinomycin, and the target of insertion was sequenced using primers TN101 and TN102 (Table 2), located at each end of the mini-Tn10 sequence.
Overexpression and purification of PadR.
Expression and purification of the PadR recombinant protein were performed as previously described (14). The coding region of PadR was amplified by PCR with primers BSR1 and BSR2, thus replacing the TAA stop codon by the XhoI restriction site. The amplified DNA fragment was cloned between the NcoI and XhoI restriction sites of the pET28a+ vector, generating plasmid pER. Expression of pER in E. coli BL21(DE3) cells induced with isopropyl IPTG produced a PadR fusion protein containing a His6 tag at the C terminus. The PadR recombinant protein was purified from E. coli BL21(DE3) cell extract by using a Ni-nitrilotriacetic acid column (Novagen).
EMSA.
The 235-bp padC promoter DNA probe was PCR amplified with primers BSD1 and BSD8 (Table 2 and Fig. 1), previously labeled with T4 polynucleotide kinase (Invitrogen) in the presence of [γ-32P]ATP (PerkinElmer). BS168 chromosomal DNA was used as the template. PCR products were purified using a Qiagen kit. Standard electrophoretic gel mobility shift assays (EMSA) were performed as follows. Purified PadR was incubated for 20 min at 28°C in 15 μl of binding buffer containing 10 mM Tris-HCl (pH 7.8), 5% (vol/vol) glycerol, 0.2 mM EDTA, 2.5 mM MgCl2, 50 mM KCl, 2 mM dithiothreitol, 2.5 μg/ml bovine serum albumin as the unspecific protein competitor, and 2.5 μg/ml salmon sperm DNA as the unspecific DNA competitor. To verify the specificity, a 100-fold excess of an unlabeled yveFG-padC promoter fragment or a 1,000-fold excess of competing poly(dI-dC) was tested. The samples were resolved onto 5% (wt/vol) polyacrylamide gels, which were dried and analyzed by autoradiography. To test the effect of phenolic acids on PadR DNA binding, p-coumaric, ferulic, or o-coumaric acid was preincubated with 1 nM of purified PadR in 15 μl of binding buffer for 5 min at room temperature. To test the effect of phenolic acids on the binding, 1 nM of purified PadR was preincubated with p-coumaric, ferulic, or o-coumaric acid in 15 μl of binding buffer for 5 min at room temperature. o-Coumaric acid, a position isomer of p-coumaric acid unable to induce the expression of padC (5), was used as the most appropriate negative control. The probe was then added to the above-mentioned mixture and incubated for 20 min at 28°C before loading in a 5% (wt/vol) polyacrylamide gel.
Cloning of the yveFG-padC operon and padR in E. coli.
The yveFG-padC DNA fragment encompassing the yveFG-padC coding sequence, the promoter sequence comprising up to 576 bp upstream of the ATG start codon of padC and the 116 bp downstream of the TAA stop codon, including the transcriptional terminator, was PCR amplified with primers BSD3 and BSD10. The resulting fragment was digested with BamHI and SmaI and ligated into pTZ19R to generate plasmid pTC. The padR DNA fragment was amplified with primers BSR3 and BSR4, digested with XbaI and PstI, and then inserted between the XbaI and PstI restriction sites of plasmids pTZ19R and pTC to create plasmids pTR and pTCR, respectively (see Fig. 4B1).
Preparation of cell extracts and assays for PAD activity.
Cells of wild-type and mutant strains of B. subtilis and recombinant E. coli strains grown in LB medium were harvested as described previously (2) and then disrupted using glass bead beating. PAD activity in cell extracts was measured by the procedure of Barthelmebs et al. (2). This procedure consists of using UV spectrophotometry to monitor the disappearance of absorption peaks of the substrates (phenolic acids) and the simultaneous appearance of new peaks corresponding to vinyl derivatives. Protein concentrations in cell extracts were determined using a protein assay kit (Bio-Rad, Richmond, CA), with bovine serum albumin as the standard.
RESULTS
Nucleotide sequence analysis and characterization of the upstream region of padC.
The padC gene of BS168 was previously characterized by our group, and a putative transcriptional start site was identified at nucleotide −159 upstream from the ATG start codon, using primer BSD2 located in the 5′ region of padC (Fig. 1) (5). This transcription start site is located within the putative gene yveG, which is in turn preceded by another putative gene, yveF. Recently (11), we demonstrated by Northern blot experiments that the yveF-yveG DNA region was cotranscribed with padC: the stop codon of yveG was found to be located 6 nucleotides upstream of the padC ribosome binding site. Annotation of the yveF-yveG region has led to the publication of conflicting information in the SubtiList World Wide Web server (http://genolist.pasteur.fr/SubtiList/) and in GenBank with regard to the presence or absence of a stop codon between the putative yveF and yveG coding sequences (Fig. 1). In an attempt to better understand the protein coding potential of the yveF-yveG region, we set out to perform three site-directed mutageneses of the yveF-yveG genes. In each mutant, the initial GTG start codon was replaced with a methionine (M) ATG codon. The first construct, named yveFGM, retained the putative stop codon at position 365. In the second (yveFGMR) and third (yveFGMQ) mutants, the TGA stop codon was replaced with CGA (R [arginine]) and CAA (Q [glutamine]), respectively. These substitutions were selected based on residues found at the corresponding positions in the BLR7842 and CAC3321 homologs of Bradyrhizobium japonicum USDA 110 and Clostridium acetobutylicum ATCC 824, respectively. Each mutant gene was cloned into pET28a+ and transformed into E. coli BL21. Protein extracts of IPTG-induced cultures were analyzed by SDS-PAGE (Fig. 3). In recombinant E. coli carrying yveFGM, in which the start codon (GTG) was changed into ATG, no protein could be detected in the molecular mass range of 18 kDa or 3 kDa, which would correspond to either a YveFG fusion peptide (assuming the absence of a stop codon and the presence of an X codon at position 365 [Fig. 1]) or a putative YveF peptide (assuming a TGA stop codon at position 365). In recombinant E. coli harboring yveFGMR and yveFGMQ, an intense protein band of about 18 kDa was produced, suggesting that the absence of a detectable peptide produced by the yveFGM clone was probably due to the stop codon, which was replaced in yveFGMR and yveFGMQ. Taken together, these results indirectly suggest that a stop codon, rather than the undetermined X codon suggested in GenBank, is indeed present in the coding sequence of yveF-yveG. Our experiment also failed to detect the production of stable YveF or YveG peptides from the yveF-yveG coding sequence, recombinantly expressed in E. coli.
FIG. 3.

SDS-PAGE analysis of crude extracts from IPTG-induced cells from recombinant E. coli strains expressing the site-directed mutagenized yveFG gene in the vector pET28a+ under the T7 promoter. M, molecular mass markers. The arrow indicates YveFGMR and YveFGMQ, which were overproduced, while no YveFGM protein was produced in this size range or in the 3-kDa range. Three kilodaltons is the molecular mass corresponding to the putative truncated YveF protein resulting from the TGA stop codon inside yveFG. pET28a+ corresponds to the IPTG-induced strain with the pET28a+ vector without an insert.
Screening of genes potentially involved in the regulation of padC expression by random transposon mutagenesis.
Previous unpublished work by our group (3) led to the identification of yfiO as the negative transcriptional regulator of padC gene expression. The yfiO gene was consequently renamed padR (TrEMBL accession no. P94443; SubtiList). In an attempt to identify additional genes that could be involved in the control of padC expression by ferulic acid, such as effectors involved in signaling to PadR, we set out to perform random transposon mutagenesis by using a padC::lacZ reporter strain. As a prerequisite, we characterized the padC promoter region and determined the position of the initiation site of padC transcription. Five padC::lacZ fusion strains (Fig. 1 and 2) that harbored promoter regions of increasing lengths were constructed. The objective was to select a reporter construct that included all cis-acting regulatory elements to identify genes involved in the regulation of padC expression by transposon mutagenesis. The five strains gave white colonies (LacZ negative) on LB medium containing X-Gal without ferulic acid and deep-blue colonies (LacZ positive) when ferulic acid (1 mM) was present in the medium (Fig. 2B). In liquid cultures lacking ferulic acid (noninduced), PAD activity could not be detected in any of the five strains. In the presence of ferulic acid in the culture medium, the reporter strains BS783F1, BS783F2, and BS783F3 appeared PAD negative, while activities of about 0.90 μmol·min−1·mg−1 were detected in the cultures of strains BS783F4 and BS783F5 (Fig. 2B). Taken together, these results indicate that constructs F1 to F3 do not include the padC promoter (Fig. 2A) and that the promoter is located upstream from the F3 fusion point. To confirm these results, reverse transcription analysis was carried out with primer BSD8 (Fig. 1 and 2C), with mRNA extracted from cultures of the wild-type BS168 strain either noninduced or induced by 1 mM ferulic acid. No signal was detected in the noninduced sample, while a transcription start site was identified at a G residue located 33 bp upstream of the yveFG start codon in the induced sample (Fig. 2C). The corresponding −10 box (TAAAGT) has two mismatches compared to the consensus sequence (TATAAT), and the −35 box is a perfect TTGACA sequence (Fig. 1). The promoter also appears to be of the extended −10 TG variety. This transcription initiation site differs from that previously identified by our group, due to the fact that the primer extension reported in reference 5 was performed using a template that did not include the actual +1 position and a primer located more than 560 nucleotides downstream from the true +1 position.
To identify the gene(s) encoding a putative sensor or otherwise positive regulator of padC expression, transposon mutagenesis for screening LacZ-negative mutants was carried out with the BS783F1 strain, which lacks PAD activity (Fig. 2), on LB medium containing X-Gal and ferulic acid. Among the five constructs (BS168F1 to BS168F5) described above, only BS168F1, BS168F2, and BS168F3, which lacked PAD activity, were suitable because ferulic acid was not consumed, leading to the continuous induction of the padC promoter. With any of the five padC::lacZ fusions used in the absence of ferulic acid, interruption of a gene encoding a hypothetical negative transcriptional regulator of padC expression was expected to produce LacZ-positive mutants among white clones on LB medium with X-Gal. Therefore, the reporter strain BS168F1 was chosen for the double screen by random mutagenesis. Following mutagenesis, cultures were diluted and plated onto LB medium containing spectinomycin and X-Gal, either in the presence of ferulic acid (to identify mutations in a gene encoding a hypothetical sensor or transcription activator) or in the absence of ferulic acid (to isolate mutations in a hypothetical negative regulator gene).
From the screen performed in the absence of ferulic acid, 16 light-blue colonies and 14 deep-blue colonies were isolated among 20,000 LacZ-negative clones. Rescue of the adjacent Tn10 sequences in the 16 light-blue clones revealed that insertion had taken place within the lacR gene, encoding the negative transcriptional regulator of the endogenous β-galactosidase of BS168, as described by Daniel et al. (7). In the 14 deep-blue Tn10 mutants, insertion had occurred in yfiO, subsequently designated padR (for phenolic acid decarboxylase Regulator). As previously determined by our group (14), B. subtilis PadR displays 50% amino acid identity with its homolog in L. plantarum and shares the same predicted coiled-coil domains.
From the screen performed in the presence of ferulic acid, 15 white colonies were isolated among 20,000 LacZ-positive clones. Rescue and sequencing of the adjacent mini-Tn10 regions revealed that all 15 insertions had occurred in the lacZ reporter gene or in the region between the yveFG promoter and padC. For all three families of mutants that were characterized (within the endogenous lacR, padR, or lacZ reporter construct), several positions of the mini-Tn10 insertion were observed, indicating saturation of the chromosome by highly effective random insertion mutagenesis. The mini-Tn10 insertions within lacR and the lacZ reporter genes validate the double-screen methodology in the presence and absence of ferulic acid. Competent wild-type BS168 cells were then transformed with chromosomal DNA from four of the padR insertion mutants to verify the insertion phenotype in a functional padC background. The four mutants obtained in this backcross experiment produced constitutive expression of padC, with a PAD activity of about 1.5 mmol·min−1·mg−1. These results demonstrate that padR is a negative transcriptional regulator of padC. Later in this report, by constructing and characterizing a ΔpadR mutant strain, we confirm that PadR represses expression of padC.
The screening performed with LB medium in the presence of ferulic acid indicated that white mutant colonies resulted from Tn10 insertions in the lacZ reporter gene or in the padC promoter region fused with lacZ (Fig. 2A). Because we have previously demonstrated that the putative yveFG gene, located upstream from padC, is interrupted by a stop codon and does not encode any detectable peptide, the white phenotype of Tn10 insertions in the yveFG sequence is probably due to the absence of LacZ production following interruption of the padC::lacZ mRNA.
Characterization of PadR-mediated expression of padC.
To confirm the negative effect of PadR on padC transcription as observed by transposon mutagenesis, we constructed a padR deletion mutant and studied its effect on PAD activity. The mutant strain, designated BSΔR, displayed constitutive PAD activity that was twice as high as that observed in the wild-type BS168 strain induced by 1 mM ferulic acid (Fig. 4A1). Protein extracts of the wild-type and mutant strains grown under induced and uninduced conditions are presented in Fig. 4A2. A protein band of about 19 kDa, corresponding to the size of the PadC enzyme, is visible in the wild-type extract induced by ferulic acid, while it is not detectable in the corresponding uninduced extract. In the BSΔR mutant, this PadC band is constitutively produced and significantly more intense than in the induced wild-type extract, in agreement with the twofold increase in PAD activity in the BSΔR strain.
In B. subtilis, padR is located between the two oppositely transcribed genes lipB (metabolism of lipids) and yfiN (unknown function, similar to ATP binding proteins). Attempts to detect padR mRNA in B. subtilis by Northern blotting failed in our hands, as did primer extension experiments for identifying its transcriptional start site (data not shown). Therefore, qRT-PCR experiments were performed with mRNA extracts from uninduced and induced cultures, using 1 mM ferulic, p-coumaric, and o-coumaric acids to measure the RTL of the yveFG-padC operon (Fig. 5A) and padR (Fig. 5B) at three different growth stages. For each locus, the lowest value of RTL observed in one set of experiments was used as the baseline for subsequent calculation of induction factors. As expected, the RTL of yveFG-padC were very low in uninduced cultures. In ferulic and p-coumaric acid-induced cells, they increased by factors of 7,500 to 8,200 (±400), while o-coumaric acid, an isomer of p-coumaric acid, was unable to significantly induce expression of yveFG-padC. The RTL of padR under induced conditions was low and not significantly different from that observed in noninduced cultures. The very low expression of PadR in B. subtilis was confirmed by Western blotting experiments using antibodies raised against PadR and purified from immunized rabbits (see the supplemental material), which indicated the absence of a detectable PadR band in an extract from B. subtilis, while a PadR band was well detected by Western blotting with cell extracts from recombinant E. coli expressing PadR.
FIG. 5.

RTL of padC (A) and padR (B) as determined by qRT-PCR from noninduced cultures and cultures induced with ferulic, p-coumaric, or o-coumaric acid for 5 min at three stages of growth. For each gene, the noninduced cultures of each stage were chosen as the reference condition with a value of 1. Vertical bars indicate the standard deviation, with the average value corresponding to three independent determinations for each condition.
Coexpression of the yveFG-padC operon and padR in E. coli.
As previously demonstrated (3), E. coli does not possess a PASR system, but phenolic acids are able to penetrate rapidly into growing and resting cells and eventually to interact with PadR. Therefore, E. coli is a relevant host for determining whether the system comprising yveFG-padC and padR could be functional in the absence of any other factor from B. subtilis and for testing the hypothesis of direct PadR inactivation by phenolic acids. Three plasmids were constructed in the pTZ19R vector and transformed into E. coli TG1 (Fig. 4B1). In each of the resulting strains, PAD activity was measured in uninduced and ferulic acid-induced cultures, and the corresponding protein extracts were resolved by SDS-PAGE (Fig. 4B2). With plasmid pTC, in which padC is used as a reporter of its own promoter in the absence of PadR, E. coli displayed constitutive and high PAD activity (about 5.5 ± 0.5 μmol·min−1·mg−1). A PadC protein band was visible early during growth (at an optical density at 600 nm [OD600] of 0.3) and was also present in the late log phases of growth (OD600 of 1.6). This result indicated that the promoter of the yveFG-padC operon was well recognized by E. coli. With plasmid pTR containing PadR only, SDS-PAGE separation of the protein extract indicated that PadR was not significantly produced during the early growth phase but accumulated during the late log phase of growth. The identity of the PadR band was confirmed using PadR antibodies (see the supplemental material). This result indicated that expression levels of PadR were low and similar throughout all growth phases in B. subtilis and in the early log phase in recombinant pTR and pTCR E. coli strains. With plasmid pTCR containing both padC and padR, a low PAD activity of 0.001 μmol·min−1·mg−1 was detected during the early phase of growth under uninduced conditions, compared to the level (5.5 μmol·min−1·mg−1) observed in the absence of the padR gene. This indicated that PadR was functional in E. coli and that the very low level of PadR produced at this stage of growth (not detectable in the protein profile) was sufficient to repress expression of padC. Significant repression of PadC expression was also observed in the extract harvested at an OD600 of 1.6, where PadR had accumulated in the cells. With the same plasmid (pTCR) and under ferulic acid-induced conditions at an OD600 of 0.3, PAD activity was 30 times higher than in uninduced cultures (0.030 μmol·min−1·mg−1) though still relatively low compared to the value observed in the absence of PadR (5.5 μmol·min−1· mg−1). No further increase in PAD activity was observed during the later growth phases, when the PadR levels were high.
In other words, phenolic acids are only partially able to relieve the PadR-mediated repression of padC in E. coli. This contrasts with the situation observed in B. subtilis, where expression of PadC is relatively similar with PadR and phenolic acids present and in the absence of PadR. Although inactivation of PadR by ferulic acid is not complete in E. coli, this is the first report of phenolic acid-mediated inactivation of PAD activity by a Pad repressor recombinantly expressed in E. coli in the absence of additional effectors from the native organism.
In vitro analysis of the influence of phenolic acids on the interaction of PadR with the padC promoter.
Because inactivation of PadR in recombinant E. coli could be seen following the addition of ferulic acid and since the transposon mutagenesis described above did not produce evidence of a phenolic acid sensor, we hypothesized that phenolic acids could interact directly with PadR without involving an additional effector. To verify this hypothesis, DNA mobility shift assays were performed with the yveFG-padC promoter DNA probe and PadR produced and purified from recombinant E. coli BL21pER (Fig. 6A). The optimal padC DNA probe concentration was found to be around 1 nM, with detectable binding also observed at lower concentrations. At 1 nM, two probe-PadR complexes, named C1 and C2, were observed while a fraction of the probe remained unbound (Pr). At a PadR concentration of 10 nM, a binding complex with a higher molecular weight (C3) was observed with apparent complete binding of the probe (Fig. 6B). A 1,000-fold excess of competing poly(dI-dC) did not affect the observed mobility shift (Fig. 6C), indicating that the interaction between PadR and the yveFG-padC promoter fragment was specific. In addition, when a 100-fold excess of unlabeled promoter fragment was included in the mixture, no shifted radioactive band was detected, indicating that the unlabeled probe had captured all available PadR protein.
FIG. 6.

EMSA of PadR with the yveFG-padC promoter. (A) SDS-PAGE of protein extracts containing PadR and purified PadR used in EMSA. M, molecular mass standards. Lanes: 1, extract from E. coli BL21(DE3) carrying the control vector (pET28a+); 2, extract from E. coli BL21 carrying the vector with padR; 3, purified PadR. (B) EMSA of PadR with the yveFG-padC promoter. P, yveFG-padC promoter DNA probe without PadR; R1 to R8, different concentrations of PadR (R1, 0.02 nM; R2, 0.05 nM; R3, 0.10 nM; R4, 0.25 nM; R5, 0.5 nM; R6, 1 nM; R7, 2.5 nM; R8, 10 nM of purified PadR). C1, C2, and C3 indicate putative complexes formed between the DNA probe and PadR. (C) EMSA with probe and PadR concentrations identical to those used for lane R6 of panel B, with addition of a 1,000-fold excess of competing poly(dI-dC) or -(dIdC R6) or a 100-fold excess of an unlabeled promoter fragment (UP R6). (D) EMSA with probe and PadR concentrations identical to those used for lane R6 of panel B, with 1 nM of PadR preincubated or not preincubated with phenolic acids. P2, P4, O2, O4, F2, and F4 are p-coumaric (P), o-coumaric (O), and ferulic (F) acids at 2 and 4 mM. (E) Same as panel D, but without MgCl2 in the binding buffer. Binding reactions for panels B, C, and D were carried out under standard conditions (with 2.5 mM MgCl2), as described in Materials and Methods.
A concentration of 1 nM of PadR, which produced specific binding with the DNA probe, was used to investigate in vitro inhibition of PadR by phenolic acids in the presence of MgCl2. In this experiment, p-coumaric and ferulic acids were tested because they are known to induce expression of the yveFG-padC gene product, while o-coumaric acid, a position isomer of p-coumaric acid unable to induce PAD activity, was used as a negative control. Under standard binding conditions with 2.5 mM MgCl2 (Fig. 6D), no detectable inhibition of binding was observed even at 4 mM phenolic acid, a concentration that is four times higher than the concentration required to provide complete inactivation of PadR in B. subtilis in vivo. MgCl2 is the most abundant divalent cation in living organisms. It is essential for a wide variety of cellular functions, particularly the structuring of nucleic acids, regulatory proteins, and the complexes that they form together (29). Thus, it is very often used in mobility shift assay buffers to facilitate binding. However, MgCl2 can induce misleading gel shift in EMSA because of its ability to enhance end-to-end DNA interactions and stable base pairing between overhangs as short as 3 bp (33). Taking into account this potential effect of MgCl2 in the regulation of intramolecular interactions, we hypothesized that MgCl2 could counteract the effect of phenolic acids by making the structure of PadR less accessible to phenolic acids and/or by enhancing DNA-protein binding at a level not encountered in living BS168 cells. Binding assays were repeated with identical concentrations of the three phenolic acids but without addition of MgCl2 (Fig. 6E). This time, p-coumaric and ferulic acids were able to abolish binding between the yveFG-padC DNA probe and PadR at concentrations of 2 mM and higher. As observed in vivo, o-coumaric acid remained unable to abolish DNA binding of PadR.
DISCUSSION
The results presented in this report contribute to elucidating the mechanisms involved in the regulation of expression of padC, which encodes the tightly inducible PAD of B. subtilis. The first part of the work focuses on the characterization of the controversial 600-bp yveFG DNA region upstream of padC (11). Gene annotation published in the SubtiList World Wide Web Server (http://genolist.pasteur.fr/SubtiList/) proposes two separate yveF and yveG coding sequences with a stop codon at position 365 (Fig. 1). GenBank, on the other hand, suggests replacing the stop codon with an undetermined X codon. The resulting coding sequence would encode a putative YveFG protein of 156 amino acid residues and 17.4 kDa. The rationale for suggesting a fusion YveFG peptide is based on the following observations. First, there is no clear ribosome binding site sequence upstream of the start codon of the second putative gene, yveG, indicating that effective translational initiation is unlikely. Second, a PSI-BLAST search in GenBank reveals homology between the hypothetical full-length YveFG peptide and two proteins of unknown function, BLR7842 of Bradyrhizobium japonicum USDA 110 and CAC3321 of Clostridium acetobutylicum ATCC 824, with identities of 44% and 46%, respectively. It should be noted that neither of these two species possesses a padC homolog in the vicinity of its yveFG locus.
Site-directed mutagenesis of the yveFG gene region, followed by recombinant expression in E. coli, supports the presence of a stop codon interrupting the yveFG coding sequence in wild-type B. subtilis 168. Failure to detect any translation product from the native yveFGM gene expressed in E. coli—except for the GTG-to-ATG replacement of the initial Met residue—makes it unlikely that a yveFG-encoded peptide(s) is functional and involved in the PASR described in the present work. Construction of a B. subtilis 168 mutant strain with an uninterrupted yveFG coding sequence is in progress to investigate whether homologous expression of a YveFG fusion protein would modulate PASR and/or padC expression in B. subtilis and whether this might have had a role in the PASR during the course of evolution.
Random transposon mutagenesis designed to identify negative regulators of the PASR led to the isolation of 14 independent mutants, each harboring a disrupted padR gene, previously identified as the transcriptional repressor of padC (3). The deletion of padR resulted in constitutive expression of padC where PAD activity, not detectable in noninduced wild-type cultures, was approximately double the PAD activity measured in ferulic acid-induced wild-type cells. The RTL of padR under induced conditions was low and not significantly different from that observed in noninduced cultures, suggesting that padR expression is low and constitutive. This is in contrast with the situation found in L. plantarum, where the PAD activity of a padR mutant is about 50 times higher than that in phenolic acid-induced wild-type cells (14), and the RTL of padR increased 30-fold following addition of p-coumaric acid, suggesting transcriptional autoregulation of padR (20). This differential regulation mechanism may be explained in part by the genetic organization of padC and padR in the different species. In P. pentosaceus (3) and L. plantarum (14), the padA (padC homolog) and padR genes form an operon and a divergon, respectively, where PadR represses its own transcription, while the expression of padR in B. subtilis is very low during all phases of growth and does not appear to be regulated by phenolic acids. This negative-feedback loop of PadR expression also affects padA transcription directly in L. plantarum, since their diverging promoters overlap almost completely. In P. pentosaceus, PadR represses its own transcription as well, along with that of padA, since they form a bicistronic operon (3). In other words, P. pentosaceus, L. plantarum, and Bacillus pumilus (3, 14) seem to have evolved a regulation mechanism that contains an additional “checkpoint” in the form of PadR-negative autoregulation. This implies that exposure to phenolic acids initially causes increases in padR transcription and protein levels, which may cause a delay and/or require higher phenolic acid levels before PadR-mediated repression is relieved and transcription of padC is allowed to proceed. In B. subtilis, on the other hand, low and constitutive levels of PadR are present at all times, suggesting that B. subtilis would be able to trigger phenolic acid decarboxylation in response to phenolic acid stress more rapidly. Why would the two groups of bacteria have evolved different regulation mechanisms in response to phenolic acid exposure? One could speculate that B. subtilis, living in plant/soil ecosystems, (i) thrives in nutrient-poor environments where competition for food is intense and (ii) is more exposed to phenolic acids, which are ubiquitous in the plant kingdom. Some of these phenolic acids have toxic properties, while others may provide a potential carbon source (16). Therefore, B. subtilis has evolved a mechanism that allows it to respond quickly to phenolic acid fluctuations, while this is less critical for bacteria such as B. pumilus or lactic acid bacteria, which are commensal or colonize nutrient-rich and “bacterium-friendly” habitats (12). The search for a positive effector of the PASR by random mutagenesis under ferulic acid-induced conditions did not lead to the identification of insertions other than those located inside lacZ or in the yveFG-padC promoter region to which it was fused. Given the high number of mutants that we obtained at different positions in each target gene, we are confident that the transposition experiment was saturating. However, we cannot exclude the possibility that such a positive effector was missed either due to its essentiality or due to a polar effect of the transposon insertion on an essential downstream cotranscribed gene.
The search for a negative effector led to the identification of PadR only. The essential character of such a negative effector is unlikely for the following reason. We have shown with the BS783F1 construct (Table 1 and Fig. 2) that the absence of PAD activity was not detrimental to the growth of B. subtilis in the presence of 1 mM of ferulic acid at neutral pH on solid LB medium, which was used for screening the transposon library. This indicates that interruption of a putative sensor-like effector responsible for the inactivation of PadR by phenolic acids would not be lethal in B. subtilis, unless it plays an essential role in addition to being involved in the PASR. In other gram-positive species with a PASR, no such sensor has been identified to date. Heterologous expression of the divergently oriented L. plantarum padA-padR couple in E. coli suggested that an additional mediator was required to effect inhibition of PadR by phenolic acids. The present work, however, indicates that intracellular MgCl2 levels may critically influence DNA-protein interactions between PadR and the padA or padC promoter regions. Further experiments are under way in our laboratory to revisit phenolic acid-mediated modulation of PadR in L. plantarum.
To test the hypothesis of direct PadR inhibition by phenolic acids, we expressed the system comprising padR and padC of B. subtilis in E. coli, where phenolic acid metabolism is absent. Heterologous expression of padR in E. coli, along with the yveFG-padC operon, demonstrated that PadR was produced and active in E. coli and was able to effectively repress transcription of yveFG-padC, even when PadR was produced at very low levels during the early log phase of growth (Fig. 4B1). At this stage, only partial inactivation of PadR was obtained by the addition of high concentrations of ferulic acid to the culture medium. No further inactivation of PadR was observed later in the growth cycle when its cellular levels were higher. Altogether, the ability of phenolic acid to abolish PadR-mediated repression of padC was around 1,000 times lower in recombinant E. coli than in the native host. One hypothesis to explain this poor inactivation of PadR in E. coli is that intracellular conditions that affect the PASR may be different in gram-negative and gram-positive bacteria. Among factors that could affect the structures of transcriptional regulators and their affinity for nucleic acids and that are generally involved in phosphorylation mechanisms, Mg2+ is considered to be one of the most important (23, 27). Mg2+ is the most abundant divalent cation in living organisms and is essential for a wide variety of cellular functions (29). The internal concentration of free Mg2+ in E. coli ranges between 1 and 5 mM (1) and is tightly regulated since it fluctuates by less than a factor of 2 when outside concentrations vary over 1,000-fold (31). Therefore, we were unable to modify intracellular Mg2+ levels in recombinant E. coli strains expressing yveFG-padC and padR. To our knowledge, Mg2+ concentrations in gram-positive bacteria, like B. subtilis and lactic acid bacteria, have not been reported. However, Kumarevel et al. (19) indicated that concentrations of 0.25 to 0.5 mM are physiologically relevant in B. subtilis, which would be around 10 times lower than those measured in E. coli.
To study the interaction between the yveFG-padC promoter and PadR, EMSA were carried out with PadR purified from E. coli in the presence and absence of MgCl2 in the binding buffer and either with or without preincubation with phenolic acids. EMSA confirmed the specific binding of PadR to the promoter of yveFG-padC. Several probe-PadR complexes were observed, depending on the concentration of PadR probe involved in the reaction. At a PadR concentration of 10 nM, a binding complex of high molecular weight (C3) with apparently complete binding of the probe was observed (Fig. 6B). This C3 complex is thought to be the result of PadR multimerization (data not shown). Further experiments are in progress to characterize the stoichiometry of PadR binding to the padC promoter region. The EMSA also demonstrated that phenolic acids, which are able to induce expression of the yveFG-padC operon, do so via inactivation of PadR only in the absence of MgCl2 in vitro. The effect of a range of MgCl2 concentrations between 0 and 2.5 mM was tested to simulate relevant intracellular concentrations expected to be found in B. subtilis. The results revealed that inactivation of PadR by p-coumaric acid was detected when the MgCl2 concentration was lower than 0.25 mM (see the supplemental material). It is possible that Mg2+enhances the affinity of PadR for the padC promoter region and thereby attenuates or suppresses the overall effect of phenolic acids. Internal Mg2+ concentrations in B. subtilis could be lower than 0.25 mM.
In summary, we have demonstrated that phenolic acids are able to directly modulate PadR binding to the promoter of padC in vitro. We have also obtained reasonable indications that no additional effector appears to be involved in the PASR in B. subtilis. Structural analysis of the active and inactive forms of PadR is currently in progress, as is the identification of the site of its binding to the yveFG-padC promoter. Chromosomal replacement of the B. subtilis yveFG locus with the two functional genes yveFGMR and yveFGMQ, in which the stop codon has been replaced, is also in progress to evaluate its potential role in other stress response mechanisms that could be linked or not linked to the PASR.
Supplementary Material
Acknowledgments
P. N. Tran and T. K. C. Nguyen were supported by Ph.D. grants from Ambassade de France in Vietnam, H. Seraut was supported by a Ph.D. grant from the French Ministère de l'Education Nationale et de la Recherche, and J. Gury was supported by a Ph.D. grant from the MICA and CEPIA departments of INRA and the Conseil Régional de Bourgogne.
We are very grateful to Marta Perego (The Scripps Research Institute, La Jolla, CA) and Stéphane Aymerich (INRA-CNRS, Thiverval-Grignon, France) for the gift of vectors. We thank also Christine Rojas for her laboratory work and Philip Bastable for checking the English translation.
Footnotes
Published ahead of print on 7 March 2008.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Alatossava, T., H. Jutte, A. Kuhn, and E. Kellenberger. 1985. Manipulation of intracellular magnesium content in polymyxin B nonapeptide-sensitized Escherichia coli by ionophore A23187. J. Bacteriol. 162413-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barthelmebs, L., C. Diviès, and J.-F. Cavin. 2000. Knockout of the p-coumarate decarboxylase gene from Lactobacillus plantarum reveals the existence of two other inducible enzymatic activities involved in phenolic acid metabolism. Appl. Environ. Microbiol. 663368-3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barthelmebs, L., B. Lecomte, C. Diviès, and J.-F. Cavin. 2000. Inducible metabolism of phenolic acids in Pediococcus pentosaceus is encoded by an autoregulated operon which involves a new class of negative transcriptional regulator. J. Bacteriol. 1826724-6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cavin, J.-F., L. Barthelmebs, and C. Diviès. 1997. Molecular characterization of an inducible p-coumaric acid decarboxylase from Lactobacillus plantarum: gene cloning, transcriptional analysis, overexpression in Escherichia coli, purification, and characterization. Appl. Environ. Microbiol. 631939-1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cavin, J.-F., V. Dartois, and C. Diviès. 1998. Gene cloning, transcriptional analysis, purification, and characterization of phenolic acid decarboxylase from Bacillus subtilis. Appl. Environ. Microbiol. 641466-1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan, A. Y., and B. L. Lim. 2003. Interaction of a putative transcriptional regulatory protein and the thermo-inducible cts-52 mutant repressor in the Bacillus subtilis phage φ 105 genome. J. Mol. Biol. 33321-31. [DOI] [PubMed] [Google Scholar]
- 7.Daniel, R. A., J. Haiech, F. Denizot, and J. Errington. 1997. Isolation and characterization of the lacA gene encoding beta-galactosidase in Bacillus subtilis and a regulator gene, lacR. J. Bacteriol. 1795636-5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dartois, V., T. Djavakhishvili, and J. Hoch. 1996. Identification of a membrane protein involved in activation of the KinB pathway to sporulation in Bacillus subtilis. J. Bacteriol. 1781178-1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Vries, R. P., B. Michelsen, C. H. Poulsen, P. A. Kroon, R. H. van den Heuvel, C. B. Faulds, G. Williamson, J. P. van den Hombergh, and J. Visser. 1997. The faeA genes from Aspergillus niger and Aspergillus tubingensis encode ferulic acid esterases involved in degradation of complex cell wall polysaccharides. Appl. Environ. Microbiol. 634638-4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dower, W. J., J. F. Miller, and C. W. Ragsdale. 1988. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res. 166127-6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duy, N. V., U. Mader, N. P. Tran, J.-F. Cavin, L. T. Tam, D. Albrecht, M. Hecker, and H. Antelmann. 2007. The proteome and transcriptome analysis of Bacillus subtilis in response to salicylic acid. Proteomics 7698-710. [DOI] [PubMed] [Google Scholar]
- 12.Farzanfar, A. 2006. The use of probiotics in shrimp aquaculture. FEMS Immunol. Med. Microbiol. 48149-158. [DOI] [PubMed] [Google Scholar]
- 13.Gibson, T. J. 1984. Studies on the Epstein-Barr virus genome. Ph.D. thesis. Cambridge University, Cambridge, United Kingdom.
- 14.Gury, J., L. Barthelmebs, N. P. Tran, C. Diviès, and J.-F. Cavin. 2004. Cloning, deletion, and characterization of PadR, the transcriptional repressor of the phenolic acid decarboxylase-encoding padA gene of Lactobacillus plantarum. Appl. Environ. Microbiol. 702146-2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huillet, E., P. Velge, T. Vallaeys, and P. Pardon. 2006. LadR, a new PadR-related transcriptional regulator from Listeria monocytogenes, negatively regulates the expression of the multidrug efflux pump MdrL. FEMS Microbiol. Lett. 25487-94. [DOI] [PubMed] [Google Scholar]
- 16.Karmakar, B., R. M. Vohra, H. Nandanwar, P. Sharma, K. G. Gupta, and R. C. Sobti. 2000. Rapid degradation of ferulic acid via 4-vinylguaiacol and vanillin by a newly isolated strain of Bacillus coagulans. J. Biotechnol. 80195-202. [DOI] [PubMed] [Google Scholar]
- 17.Kovacikova, G., W. Lin, and K. Skorupski. 2003. The virulence activator AphA links quorum sensing to pathogenesis and physiology in Vibrio cholerae by repressing the expression of a penicillin amidase gene on the small chromosome. J. Bacteriol. 1854825-4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kovacikova, G., and K. Skorupski. 2001. Overlapping binding sites for the virulence gene regulators AphA, AphB and cAMP-CRP at the Vibrio cholerae tcpPH promoter. Mol. Microbiol. 41393-407. [DOI] [PubMed] [Google Scholar]
- 19.Kumarevel, T., H. Mizuno, and P. K. Kumar. 2005. Structural basis of HutP-mediated anti-termination and roles of the Mg2+ ion and L-histidine ligand. Nature 434183-191. [DOI] [PubMed] [Google Scholar]
- 20.Licandro-Seraut, H., J. Gury, N. P. Tran, L. Barthelmebs, and J.-F. Cavin. 2008. Kinetics and intensity of the expression of genes involved in the stress response tightly induced by phenolic acids in Lactobacillus plantarum. J. Mol. Microbiol. Biotechnol. 1441-47. [DOI] [PubMed] [Google Scholar]
- 21.Lin, W., G. Kovacikova, and K. Skorupski. 2007. The quorum sensing regulator HapR downregulates the expression of the virulence gene transcription factor AphA in Vibrio cholerae by antagonizing Lrp- and VpsR-mediated activation. Mol. Microbiol. 64953-967. [DOI] [PubMed] [Google Scholar]
- 22.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2[-ΔΔCt)] Method. Methods 25402-408. [DOI] [PubMed] [Google Scholar]
- 23.McPhee, J. B., M. Bains, G. Winsor, S. Lewenza, A. Kwasnicka, M. D. Brazas, F. S. Brinkman, and R. E. Hancock. 2006. Contribution of the PhoP-PhoQ and PmrA-PmrB two-component regulatory systems to Mg2+-induced gene regulation in Pseudomonas aeruginosa. J. Bacteriol. 1883995-4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Msadek, T., V. Dartois, F. Kunst, M.-L. Herbaud, F. Denizot, and G. Rapoport. 1998. ClpP of Bacillus subtilis is required for competence development, motility, degradative enzyme synthesis, growth at high temperature and sporulation. Mol. Microbiol. 27899-914. [DOI] [PubMed] [Google Scholar]
- 25.Murphy, E. 1985. Nucleotide sequence of a spectinomycin adenyltransferase AAD(9) determinant from Staphylococcus aureus and its relationship to AAD(3′′) (9). Mol. Gen. Genet. 20033-39. [DOI] [PubMed] [Google Scholar]
- 26.Perego, M. 1993. Integrational vectors for genetic manipulations in Bacillus subtilis, p. 615-624. In A. L. Sonensheim, J. A. Hoch, and R. Losick (ed.), Bacillus subtilis and other gram-positive bacteria: biochemistry, physiology, and molecular genetics. American Society for Microbiology, Washington, DC.
- 27.Ramström, H., S. Sanglier, E. Leize-Wagner, C. Philippe, A. Van Dorsselaer, and J. Haiech. 2003. Properties and regulation of the bifunctional enzyme HPr kinase/phosphatase in Bacillus subtilis. J. Biol. Chem. 2781174-1185. [DOI] [PubMed] [Google Scholar]
- 28.Rokeach, L. A., J. A. Haselby, and S. O. Hoch. 1988. Molecular cloning of the cDNA encoding the human Sm-D autoantigen. Proc. Natl. Acad. Sci. USA 854832-4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Romani, A. M., and A. Scarpa. 2000. Regulation of cellular magnesium. Front. Biosci. 5720-734. [DOI] [PubMed] [Google Scholar]
- 30.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring harbor Laboratory, Cold Spring Harbor, NY.
- 31.Silver, S., and D. Clark. 1971. Magnesium transport in Escherichia coli. J. Biol. Chem. 246569-576. [PubMed] [Google Scholar]
- 32.Steinmetz, M., and R. Richter. 1994. Easy cloning of mini-Tn10 insertions from the Bacillus subtilis chromosome. J. Bacteriol. 1761761-1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tagashira, H., M. Morita, and T. Ohyama. 2002. Multimerization of restriction fragments by magnesium-mediated stable base pairing between overhangs: a cause of electrophoretic mobility shift. Biochemistry 4112217-12223. [DOI] [PubMed] [Google Scholar]
- 34.Zago, A., G. Degrassi, and C. V. Bruschi. 1995. Cloning, sequencing, and expression in Escherichia coli of the Bacillus pumilus gene for ferulic acid decarboxylase. Appl. Environ. Microbiol. 614484-4486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang, C., J. Nietfeldt, M. Zhang, and A. K. Benson. 2005. Functional consequences of genome evolution in Listeria monocytogenes: the lmo0423 and lmo0422 genes encode sigmaC and LstR, a lineage II-specific heat shock system. J. Bacteriol. 1877243-7253. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.