

Abstract
Our earlier effort to develop constrained analogues of flexible piperidine analogs for monoamine transporters led to the development of a series of 3,6-disubstituted piperidine derivatives, and a series of 4,8-disubstituted 1,4-diazabicyclo[3.3.1]nonane derivatives. In further structure-activity relationship (SAR) studies on these constrained derivatives, several novel analogues were developed where an exocyclic hydroxyl group was introduced on the N-alkyl-aryl side chain. All synthesized derivatives were tested for their affinities for the dopamine transporter (DAT), serotonin (5-HT) transporter (SERT), and norepinephrine transporter (NET) in the brain by measuring their potency in inhibiting the uptake of [3H]DA, [3H]5-HT, and [3H]NE, respectively. Compounds were also tested for their binding potency at the DAT by their ability to inhibit binding of [3H]WIN 35,428. The results indicated that position of the hydroxyl group on the N-alkyl side chain is important along with the length of the side chain. In general, hydroxyl derivatives derived from more constrained bicyclic diamines exhibited greater selectivity for interaction with DAT compared to the corresponding 3,6-disubstituted diamines. In the current series of molecules, compound 11b with N-propyl side chain with the hydroxyl group attached in the benzylic position was the most potent and selective for DAT (Ki = 8.63 nM; SERT/DAT = 172 and NET/DAT = 48.4).
Introduction
Cocaine binds to several binding sites in the brain including those on monoamine transporter proteins. These proteins transport dopamine (DA), serotonin (5-HT) and norepinephrine (NE) (DAT, SERT, and NET, respectively). 1, 2 However, binding of cocaine to DAT is believed to be responsible for production of its powerful reinforcing effect. As no effective medication is currently available to treat cocaine dependence, the development of an effective pharmacotherapy for this disorder is urgently needed.
The dopamine hypothesis of cocaine addiction received further support from a series of in vivo experiments and also from molecular biological studies involving DAT knockout mice.3, 4 Furthermore, in a recent experiment with knock-in mouse model it was demonstrated that binding to DAT is mainly responsible for its reinforcing effect.5 This recent evidence further validates DAT as a target for drug development for cocaine addiction. DAT has been targeted for the development of pharmacotherapy for cocaine addiction for number of years. However, it is also important to mention that other studies have indicated the additional involvement of the serotonergic system in some of the subjective effects of cocaine.6 The validity of DAT as a target for development of cocaine pharmacotherapy is evident from preclinical results in animal behavior studies which indicated that GBR 12909, a DAT blocker, could attenuate self-administration of cocaine without modulating food reinforcement in monkeys.7 In a human clinical trial GBR 12909 was a non-stimulant.8 However, the clinical trial of GBR 12909 was discontinued due to problems of QTc prolongation. In another ongoing study with a different DAT blocker, the phenyl tropane analogue RTI-336 is being evaluated preclinically as a pharmacotherapy for cocaine abuse.9 Finally, a recent study on the mechanism of interaction of benztropine-like compounds with DAT suggests a link between conformational effects at DAT and their ability to serve in psychostimulant substitution therapy.10, 11
Structurally diverse molecules have been developed for DAT. These molecules are broadly categorized into four main classes depending on their chemical structure, known as the tropane, GBR, methylphenidate and mazindol class of derivatives. Detailed structure-activity relationship (SAR) studies of these different categories of molecules have been described in a recent review paper.12
In our earlier studies for development of novel molecules for DAT, we have developed a large number of flexible piperidine analogs of GBR 12909 exhibiting potent affinity at the DAT.13–15 In order to address poor in vivo activity in these flexible molecules, we modified one of our lead flexible DAT-selective piperidine analogs, compound I in Figure 1, into a series of structurally constrained 3,6-disubstituted piperidine derivatives. The cis isomeric derivative from this novel series exhibited preferential affinity at the DAT over the trans derivative.16 Further SAR exploration based on the novel cis-structure yielded more potent molecules for the DAT (compounds II and III in Figure 1), thus, confirming preferential affinity of this novel cis-template for the DAT.17 In our efforts to further expand our SAR studies in searching for suitable pharmacotherapeutic agents for cocaine addiction, the design of more structurally constrained molecules of 3,6-disubstituted derivatives were undertaken. Thus, a further structural rigidification on this template was carried out by linking the two nitrogen atoms in the molecule by a bis-methylene-chain linker which yielded a novel series of 4,8-disubstituted 1,4-diazabicyclo[3,3,1]nonane derivatives where the piperazine and the piperidine rings were fused into each other. A brief SAR study was conducted which led to the discovery of lead molecules exhibiting high affinity and selectivity for the DAT, paralleling the results obtained from the corresponding lesser constrained 3,6-disubstituted versions.17 However, the more rigid 1,4-diazabicyclo[3,3,1]nonane compounds exhibited greater selectivity for the DAT indicating enhanced effect of rigidity on selectivity (compound IV in Figure 1).18 Furthermore, in vivo locomotor and drug discrimination experiments demonstrated more activity in these constrained derivatives compared to their flexible counterparts, indicating an effect of the constrained structure in efficient penetration of blood brain barrier. In this regard, more constrained 1,4-diazabicyclo[3,3,1]nonane seemed to exhibit higher in vivo potency.
Figure 1.

Molecular structure of dopamine transporter blockers
In the present SAR study we examine the effect of introduction of an exocyclic hydroxyl group in the above constrained derivatives on affinity and selectivity for monoamine transporters. Specifically, we wanted to observe the effect of introduction of an exocyclic hydroxyl functionality in both optically pure (−)-cis-3,6-disubstituted monocyclic diamine and (−)-cis-bicyclic diamine structures to understand the influence of such a hydroxyl group in binding interaction. Introduction of a comparable hydroxyl group in the piperidine ring of our earlier flexible piperidine analogue produced highly potent and selective molecule for DAT.19 In the present study we also determine the influence of chirality of the newly introduced hydroxyl center on interaction with monoamine transporters.
Chemistry
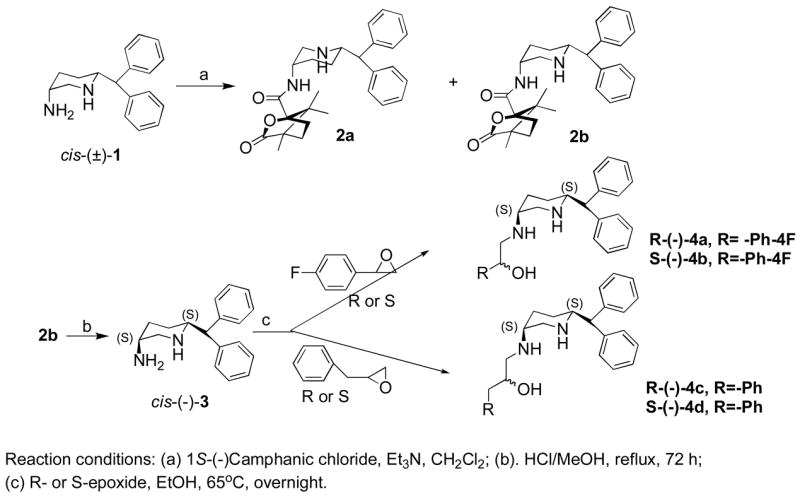
Synthesis of target compounds are shown in Schemes 1, 2 and 3. Scheme 1 describes the synthesis of targets 4a to 4d. As described in our earlier publication,17 optically active cis-amine was synthesized from racemic 1 by converting the amine into diastereoisomeric intermediates followed by separation and hydrolysis of the desired isomers to the corresponding amine cis-(−)-3. Treatment of (−)-3 with optically active epoxides furnished the final hydroxy targets 4a–4d in good yield. The optically active epoxides were synthesized by following the published procedure.20
Scheme 1.
Scheme 2.
Scheme 3.

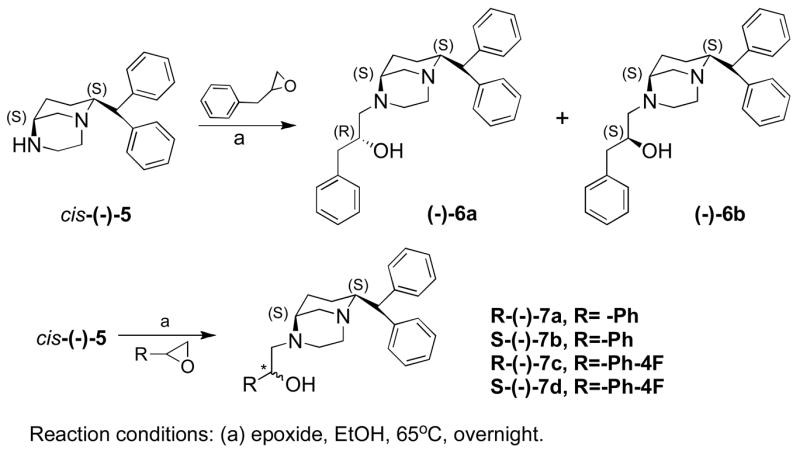
Scheme 2 describes the synthesis of targets 7a–7d. Here optically active amine (−)-5 was synthesized by following our earlier procedure.18 This amine was treated with racemic 2,3-epoxypropyl benzene, which produced two diastereomers 6a and 6b, which were separated by column chromatography. Amine cis-(−)-5 was further treated with enantiomeric (R ans S) 2-phenyloxirane and 2-(4-fluorophenyl)oxirane separately in ethanol to yield the target compounds 7a–7d in reasonably good yield.
Scheme 3 describes the synthesis of targets 11a and 11b. N-alkylation of amine cis-(−)-5 with 3-chloro-4′-fluoro propiophenone under basic condition produced 8 which was reduced by sodium borohydride to produce mixture of both R- and S-alcohols 9. Alcohol 9 was next converted into diastereomeric caphanic esters, which were separated by semi-preparative HPLC process. The final targets, 11a and 11b, were produced after hydrolyzing the esters.
Results and Discussion
Our previous study with 3,6-disubstituted constrained piperidine derivatives produced high-affinity ligands for DAT.17, 18 In the current study, we wanted to explore whether introduction of an exocyclic hydroxyl group can further increase affinity of these compounds for DAT or for other monoamine transporters. We had established earlier that (−)-enantiomeric (S,S) versions of disubstituted diamines exhibited the highest potency. Thus, in our design we selectively wanted to synthesize only the (−)-isomeric versions. Design of compounds 4a–b and 4c–d involved introduction of an exocyclic hydroxyl group to 3,6-disubstituted template. Between compounds 4a and 4b, compound 4b has the hydroxyl group in a S-configuration as it was synthesized from S-epoxide. Compound 4b was more potent in inhibiting uptake of radiolabeled DA and NE by DAT and NET, respectively, compared to 4a (Ki (DAT) = 236 nM Vs. 152 nM for 4a and 4b, respectively and Ki (NET) = 1435 Vs. 306 nM for 4a and 4b, respectively). Thus, the difference in NE uptake inhibitory activity between the two compounds was much greater than the DA uptake inhibitory activity.
In designing the next two compounds, an additional methylene unit was introduced between the phenyl moiety and the hydroxyl center. This transformation made 4c–d more potent DAT uptake inhibitors compared to 4a–b (Table 1). Both 4c and 4d exhibited low nanomolar activity for inhibition of DA uptake activity (Ki (DAT) = 25 nM and 25.3 nM for 4c and 4d, respectively) and also the same relative potency was exhibited in the binding assay with the radiolabeled tropane ligand CFT. It is interesting to note that both compounds exhibited comparable inhibition activity, thus, not exhibiting any preference for chirality of the hydroxyl center. Thus, a minor change in molecular structure resulted in almost ten-fold increase of DAT inhibition potency in 4c–d compared to 4a–b.
Table 1.
Affinity of Drugs at DAT, SERT, and NET in Rat Brain
| Compounds | DAT binding, Ki, nM, [3H]WIN 35, 428a | DATuptake, Ki, nM, [3H]DAa | SERT uptake, Ki, nM, [3H]5-HTa | NET uptake, Ki, nM [3H]NEa |
|---|---|---|---|---|
| GBR 12909b | 10.6 ± 1.9 | 10.6 ± 2.2 | 91.1 ± 12.8 | 102 ± 32 |
| IIIb | 11.3 ± 0.9 | 9.10 ± 1.86 | ||
| IVb | 22.5 ± 2.1 | 18.4 ± 0.9 | ||
| D-228, 4a (S,S,R) | 236 ± 41 | 2,895 ± 755 | 1435 ± 495 | |
| D-227, 4b (S,S,S) | 152 ± 46 | 3,117 ± 757 | 306 ± 89 | |
| D-254, 4c (S,S,R) | 31.8 ± 6.0 | 25.1 ± 2.5 | 1,391 ± 298 | 170 ± 32 |
| D-272, 4d (S,S,S) | 28.9 ± 2.3 | 25.3 ± 6.9 | 2596 ± 718 | 231 ± 50 |
| D-169, 6a (S,S,R) | 148 ± 48 | 82.9 ± 7.2 | 11,216 ± 231 | 730 ± 79 |
| D-170, 6b (S,S,S) | 47.0 ± 5.6 | 16.8 ± 1.3 | 10,336 ± 539 | 259 ± 20 |
| D-250, 7a (S,S,R) | 228 ± 45 | 204 ± 34 | 12,904 ± 1440 | 954 ± 138 |
| D-251, 7b (S,S,S) | 1,039 ±179 | 266 ± 31 | 9,508 ±1,748 | 1,730 ± 387 |
| D-252, 7c (S,S,R) | 142 ± 22 | 66.5 ± 5.7 | 7,414 ± 978 | 319 ± 58 |
| D-253, 7d (S,S,S) | 649 ± 100 | 160 ± 12 | 7344 ± 1,437 | 728 ± 142 |
| D-273 (11a) | 19.9 ± 0.9 | 41.8 ± 6.9 | 11,884 ± 4136 | 388 ± 76 |
| D-274 (11b) | 13.5 ± 2.9 | 8.63 ± 1.36 | 1484 ± 366 | 418 ± 27 |
For binding, the DAT was labeled with [3H]WIN 35, 428. For uptake by DAT, SERT and NET, [3H]DA, [3H]5-HT and [3H]NE accumulation were measured. Results are average ± SEM of three to eight independent experiments assayed in triplicate.
Results from Ref # 12 and 13.
In designing our next analogues, the bicyclic (−)-diamine template was chosen for introduction of exocyclic hydroxyl group. Thus in compounds 6a and 6b, which represent bicyclic versions of 4c and 4d, a hydroxyl functionality was introduced with R- and S-stereocenters. These molecules exhibited differential potencies for inhibition of DA uptake with the S-hydroxyl stereo-center exhibiting greater potency than the R stereo-center (Ki (DAT) = 16.8 nM Vs. 82.9 nM for 6b and 6a, respectively). Thus, this result was somewhat different compared to their 3,6-disubstituted counterparts 4c–d. Compound 6b was 5 times more potent in inhibiting DA uptake than 6a (Table 1). This might indicate an effect of a more constrained bicyclic structure on greater selectivity and affinity for DAT as we observed earlier.18 The next series of compounds 7a–d, which represents more constrained bicyclic versions of 4a–b, yielded results from weak to strong potency for the DAT. However, these molecules, in contrary to previous derivatives, produced preferential interaction with DAT with an exocyclic hydroxyl group in the R-stereo center. The most potent compound identified in the 7-series was fluoro derivative 7c, exhibiting the highest potency for DAT (Ki = 66.5 nM). In general, fluoro derivatives were more potent compared to unsubstituted versions. In general, compounds 7a–d were much less potent than 6a–b, indicating the importance of N-alkyl chain length in transporter interaction.
In the design of next two bicyclic amine analogues, 11a and 11b, the hydroxyl group was introduced in the benzyl position at the terminus of the propyl chain. Thus, in these two compounds the hydroxyl group was located furthest from the N-atom as compared to previous analogues. It is evident from uptake inhibition data that such location of the hydroxyl group produced maximal inhibition of DA uptake in one of the diastereomers, 11b (Ki = 8.63 nM). Thus, compound 11b exhibited maximum potency and selectivity for inhibition of dopamine uptake compared to inhibition of both serotonin and norepinephrine (SERT/DAT and NET/DAT; 172 and 48.4, Table 2). In fact, compound 11b turned out to be the most potent and selective compound in this current series of molecules. The diastereomer 11a, on the other hand, was less potent compared to 11b even though its potency was comparable to the third best compound 4d in the series (Ki (DAT) = 41.8 nM and 25.3 nM, respectively for 11a and 4d). It is evident from this result that the location of the hydroxyl group with respect to the aromatic ring and the N-atom played an important role in uptake inhibition activity.
Table 2.
Selectivity ratio for uptake inhibition
| Compound | SERT uptake/DAT uptakea | NET uptake/DAT uptakea | DAT uptake/DAT Bindinga |
|---|---|---|---|
| GBR 12909 | 8.5 | 9.6 | 1.0 |
| 4a | 12.2 | 6.0 | |
| 4b | 20.5 | 2.0 | |
| 4c | 55.4 | 6.7 | 0.78 |
| 4d | 103 | 9.1 | 0.87 |
| 6a | 135 | 8.8 | 0.56 |
| 6b | 615 | 15.4 | 0.35 |
| 7a | 63.2 | 4.6 | 0.89 |
| 7b | 35.7 | 6.5 | 0.25 |
| 7c | 111 | 4.7 | 0.46 |
| 7d | 45.9 | 4.5 | 0.24 |
| 11a | 284.3 | 9.2 | 2.1 |
| 11b | 171.9 | 48.4 | 0.63 |
Ratio of Ki values
Conclusion
The positional effect of the exo-cyclic hydroxyl group on N-alkyl side chain and the importance of the optimum length of N-alkyl side chain were evaluated for interaction with monoamine transporters. The results indicated that an N-propyl linker length was optimal for interaction with DAT. Position of an exo-cyclic hydroxyl group in the benzylic position of the N-propyl terminus produced the most active and selective DAT compound 11b in the current series. In most cases a stereochemical preference was exhibited for the hydroxyl stereo center. In general, as expected from earlier findings, hydroxyl compound derived from more constrained bicyclic amine produced greater selectivity for DAT than 3,6-disubstituted amine derivatives.
Experimental detail
Analytical silica gel-coated TLC plates (Si 250F) were purchased from Baker, Inc and were visualized with UV light or by treatment with phosphomolybdic acid (PMA). Flash chromatography was carried out on Baker Silica Gel 40 mM. 1H NMR spectra were routinely obtained at Varian 400 MHz FT NMR. The NMR solvent used was CDCl3 as indicated. TMS was used as an internal standard. Elemental analyses were performed by Atlantic Microlab, Inc and were within ± 0.4% of the theoretical value. Optical rotations were measured on Perkin-Elmer 241 polarimeter.
[3H]WIN 35,428 (83.6 Ci/mmol), [3H]dopamine (55.1 Ci/mmol), [3H]serotonin (30.0 Ci/mmol), and [3H]norepinephrine (54.6 Ci/mmol) were obtained from Dupont-New England Nuclear (Boston, MA, U.S.A). WIN 35,428 napthalene sulfonate was purchased from Research Biochemicals, Inc. (Natick, MA, U.S.A.). (−)-Cocaine HCl was obtained from the National Institute on Drug Abuse. GBR 12909 Dihydrochloride (1-[2-[bis(4-Fluorophenyl)methoxy]ethyl]-4-[3-phenylpropyl]piperazine) was purchased from SIGMA-ALDRICH (#D-052; St. Louis, MO).
Synthesis of N-(6- benzhydrylpiperidin-3-yl)-4,7,7-trimethyl-3-oxo-2-oxa-bicyclo [2.2.1]heptane-1-carboxamide (2a and 2b)
To a stirring solution of racemic cis-6-benzhydrylpiperidin-3-ylamine 1 (1.10 g, 4.12 mmol) in anhydrous CH2Cl2 (50 ml), triethylamine (2.08 g, 20.64 mmol) was added drop wise (1S)-(−)-camphanic chloride (1.07 g, 4.95 mmol) dissolved in 10 ml anhydrous CH2Cl2 under nitrogen. The reaction mixture was stirred at 0°C for 30 min and at room temperature for another 3 h under nitrogen atmosphere. The reaction mixture was then diluted with CH2Cl2 (50 ml) and washed with water (20 ml), dried over Na2SO4 and the solvent was evaporated in vacuo to afford a mixture of two diastereomers 2a and 2b. Each diastereoisomer was separated by flash column chromatography over silica gel using hexanes/diethyl ether (12:88) as a mobile phase.
Eluting first was 2a (0.51 g, 55%) 1H NMR (400 MHz, CDCl3): δ 0.89 (3H, s, CH3), 1.09 (3H, s, CH3), 1.11 (3H, s, CH3), 1.37–1.41 (1H, m, H-5), 1.48–1.56 (1H, m, H-5), 1.65–1.72 (2H, m, CCH2C), 1.82–1.97 (3H, m, CCH2C and H-4), 2.48–2.56 (1H, m, H-4), 2.78–2.84 (2H, m, H-2), 3.23 (1H, dt, J = 2.4 Hz, J = 10.4 Hz, H-6), 3.79 (1H, d, J = 10.0 Hz, (Ph)2CH), 4.09–4.12 (1H, m, H-3), 7.13–7.37 (8H, m, ArH), 7.39–7.41 (2H, m, ArH). Eluting second was 2b (0.45g, 49%) 1H NMR (400 MHz, CDCl3): δ 0.82 (3H, s, CH3), 1.02 (3H, s, CH3), 1.05 (3H, s, CH3), 1.32–1.35 (1H, m, H-5), 1.43–1.52 (1H, m, H-5), 1.57–1.64 (2H, m, CCH2C), 1.71–1.90 (3H, m, CCH2C and H-4), 2.41–2.50 (1H, m, H-4), 2.71–2.80 (2H, m, H-2), 3.16 (1H, dt, J = 2.0 Hz, J = 10.4 Hz, H-6), 3.71 (1H, d, J = 10.0 Hz, (Ph)2CH), 4.01–4.07 (1H, m, H-3), 7.07–7.30 (8H, m, ArH), 7.33–7.35 (2H, m, ArH).
Synthesis of (−)-cis-6-Benzhydrylpiperidin-3-ylamine (3)
A solution of 2b (0.55 g, 1.23 mmol) in conc. HCl/MeOH (50 ml, 1:4 ratio v/v) was refluxed for 72 h. Methanol was then evaporated under reduced pressure at 50° C and the remaining aqueous solution was neutralized by saturated NaHCO3 solution. The solution was extracted with CH2Cl2 (3 × 50 ml). All organic layers were combined, washed with brine (50 ml) and dried over Na2SO4, concentrated and purified by flash column chromatography over silica gel using ethyl acetate/MeOH/Et3N (80:15:5) to afford 3 as a white solid (0.26 g, 79%). 1H NMR (400 MHz, CDCl3): δ 1.35–1.43 (2H, m, H-5), 1.59–1.64 (2H, m, H-4), 2.18 (2H, broad singlet, NH), 2.79–2.81 (2H, m, H-2), 2.98–3.04 (1H, m, H-3), 3.25 (1H, dt, J = 4 Hz, J = 10 Hz, H-6ax), 3.80 (1H, d, J = 10.2 Hz, (Ph)2CH), 7.12–7.40 (10H, m, ArH). [α]25D = (−) 41.9° (c 1, MeOH).
Procedure A. Synthesis of (R)-2-[(3S, 6S)-6-benzhydrylpiperidin-3-ylamino)-1-(4-fluorophenyl)ethanol (4a)
To a stirring solution of (−)-cis-6-benzhydrylpiperidin-3-ylamine 3 (0.058 g, 0.217 mmol) in dry ethanol (20 ml), was added R-(−)-4-fluoro styrene oxide (0.045 g, 0.326 mmol). The reaction mixture was refluxed overnight under nitrogen atmosphere. The solvent was evaporated and the product was purified by flash column chromatography over silica gel using diethyl ether/MeOH/Et3N (92:8:0.2) to give 4a (0.023 g, 26%). 1H NMR (400 MHz, CDCl3): δ 1.30–1.38 (2H, m, H-5), 1.48–1.55 (1H, m, H-4ax), 1.75–1.79 (1H, m, H-4eq), 2.44–2.54 (1H, dd, J = 2.0 Hz, J = 10.0 Hz, NHCH2), 2.71–2.78 (2H, m, H-2), 2.86–2.90 (1H, dd, J = 3.2 Hz, J = 12.4 Hz, NHCH2), 2.97–3.00 (1H, m, H-3eq), 3.25 (1H, dt, J = 3.2 Hz, J = 9.6 Hz, H-6ax), 3.75 (1H, d, J = 10 Hz, (Ph)2CH), 4.60–4.64 (1H, dd, J = 3.2 Hz, J = 9.6 Hz, CH-OH), 7.01 (2H, t, J = 8.4 Hz, ArH), 7.13–7.37 (12H, m, ArH). Free base converted into oxalate salt, m.p. 202–204 °C. [α]25D (oxalate salt) = (−) 21.5° (c 0.26, MeOH).
Analysis calculated for (C26H29FN2O. 2(COOH)2, 0.5H2O) C, H, N.
Synthesis of (S)-2-[(3S, 6S)-6-benzhydrylpiperidin-3-ylamino)-1-(4-fluorophenyl) ethanol (4b)
Compound 3 (0.076 g, 0.285 mmol) was refluxed with S-(+)-4-fluoro styrene oxide (0.059 g, 0.427 mmol) (Procedure A) to yield 4b (0.029 g, 25%). 1H NMR (400 MHz, CDCl3): δ 1.25–1.38 (2H, m, H-5), 1.47–1.53 (1H, m, H-4ax), 1.79–1.83 (1H, m, H-4eq), 2.43–2.53 (1H, dd, J = 2.8 Hz, J = 12.0 Hz, NHCH2), 2.71–2.74 (2H, m, H-2), 2.87–2.97 (2H, m, NHCH2 and H-3eq), 3.25 (1H, dt, J = 3.2 Hz, J = 9.6 Hz, H-6ax), 3.74 (1H, d, J = 10 Hz, (Ph)2CH), 4.60–4.63 (1H, dd, J = 2.8 Hz, J = 8.8 Hz, CH-OH), 7.01 (2H, t, J = 8.8 Hz, ArH), 7.16–7.37 (12H, m, ArH). [α]25D (oxalate salt) = (+) 19.2° (c 0.38, MeOH). Free base was converted into oxalate salt 203–205 °C.
Analysis calculated for (C26H29FN2O. 2(COOH)2, 0.9H2O) C, H, N.
Synthesis of (R)-1-[(3S, 6S)-6-benzhydrylpiperidin-3-ylamino)-3-phenylpropan-2-ol (4c)
Compound 3 (0.076 g, 0.285 mmol) was reacted with R-(+)-2,3-epoxypropyl benzene (0.057 g, 0.427 mmol) (Procedure A). The crude product was purified by flash column chromatography using diethyl ether/MeOH/Et3N (90:10:0.2) to give 4c (0.035 g, 30%). 1H NMR (400 MHz, CDCl3): δ 1.25–1.34 (2H, m, H-5), 1.44–1.52 (1H, m, H-4ax), 1.72–1.75 (1H, m, H-4eq), 2.32–2.38 (1H, dd, J = 2.8 Hz, J = 9.6 Hz, NHCH2), 2.64–2.83 (5H, m, H-2, NHCH2, Ph-CH2), 2.90–2.93 (1H, m, H-3eq), 3.23 (1H, dt, J = 3.2 Hz, J = 10.0 Hz, H-6ax), 3.73–3.83 (2H, m, (Ph)2CH and CH-OH), 7.12–7.31 (13H, m, ArH), 7.35–7.36 (2H, m, ArH). Optical rotation of free base, [α]25D = (−) 38.9 ° (c 0.57, MeOH). Free base was converted into oxalate salt 205–207 °C.
Analysis calculated for (C27H32N2O. 2(COOH)2, 0.5H2O) C, H, N.
Synthesis of (S)-1-[(3S, 6S)-6-benzhydrylpiperidin-3-ylamino)-3-phenylpropan-2-ol (4d)
Compound 3 (0.075 g, 0.281 mmol) was reacted with S-(−)-2,3-epoxypropyl benzene (0.056 g, 0.422 mmol) (Procedure A) and was purified by flash column chromatography over silica gel using diethyl ether/MeOH/Et3N (90:10:0.2) to yield 4d (0.037 g, 33%). 1H NMR (400 MHz, CDCl3): δ 1.23–1.35 (2H, m, H-5), 1.41–1.49 (1H, m, H-4ax), 1.75–1.78 (1H, m, H-4eq), 2.36–2.41 (1H, dd, J = 3.2 Hz, J = 8.8 Hz, NHCH2), 2.66–2.84 (5H, m, H-2, NHCH2, Ph-CH2), 2.89–2.92 (1H, m, H-3eq), 3.23 (1H, dt, J = 2.4 Hz, J = 10.0 Hz, H-6ax), 3.75 (1H, d, J = 10.0 Hz, (Ph)2CH) 3.78–3.85 (1H, m, and CH-OH), 7.12–7.31 (13H, m, ArH), 7.35–7.36 (2H, m, ArH). Optical rotation, [α]25D = (−) 49.3 ° (c 0.96, MeOH). Free base was converted into oxalate salt 206–209 °C
Analysis calculated for (C27H32N2O. 2(COOH)2, 0.3H2O) C, H, N.
Synthesis of 1-((5S, 8S)-8-benzhydryl-1,4-diazabicyclo [3.3.1]nonane-2-yl)-3-phenylpropan-2-ol (6a and 6b)
To a stirring solution of cis-(−)-8-benzydryl-1,4-diazabicyclo[3.3.1]nonane 5 (0.100 g, 0.341 mmol) in dry ethanol was added 2,3-epoxypropyl benzene (0.068 g, 0.512 mmol). The reaction mixture was stirred overnight at 65°C (Procedure A). The diastereomers were separated by preparative TLC using acetone/diethyl ether (20:80) as mobile phase to afford 6a and 6b. Upper Fraction gave 6a (0.026 g, 18%). 1H NMR (400 MHz, CDCl3): δ 1.24–1.43 (2H, m, H-7), 1.54–1.66 (1H, m, H-6ax), 2.03–2.06 (1H, m, H-6eq), 2.28 (1H, t, J = 12.4 Hz, NCH2CH), 2.39 (1H, bs, H-5), 2.57–2.87 (6H, m, NCH2CH, NCH2CH2N, Ph-CH2), 2.98–3.01 (1H, m, H-9ax), 3.10 (1H, bd, J = 11.8 Hz, NCH2CH2N), 3.22–3.25 (1H, m, H-9eq) 3.77 (1H, dt, J = 4.8 Hz, J = 11.2 Hz, H-8ax), 3.84–3.91 (2H, m, CH-OH, (Ph)2CH), 7.11–7.15 (2H, m, ArH), 7.19–7.31 (11H, m, ArH), 7.35–7.38 (2H, m, ArH). [α]25D (oxalate salt) = (−) 24.7 ° (c 0.42, MeOH).
Analysis calculated for (C29H34N2O. 2(COOH)2) C, H, N.
Lower fraction gave 6b (0.024g, 16%). 1H NMR (400 MHz, CDCl3): δ 1.22–1.29 (1H, m, H-7ax), 1.34–1.42 (1H, m, H-7eq), 1.47–1.56 (1H, m, H-6ax), 2.02–2.07 (1H, m, H-6eq), 2.20 (1H, dd, J = 10.8 Hz, J = 2.4 Hz, NCH2CH), 2.48 (1H, bm, H-5), 2.67 (1H, dd, J = 8.0 Hz, J = 5.6 Hz, NCH2CH), 2.75–3.10 (7H, m, NCH2CH2N, Ph-CH2, H-9ax), 3.17–3.20 (1H, m, H-9eq) 3.75 (1H, dt, J = 4.8 Hz, J = 11.3 Hz, H-8ax), 3.83–3.90 (2H, m, CH-OH, (Ph)2CH), 7.12–7.15 (2H, m, ArH), 7.19–7.30 (11H, m, ArH), 7.33–7.38 (2H, m, ArH). [α]25D (oxalate salt)= (−) 31.2 ° (c 0.40, MeOH).
Analysis calculated for (C29H34N2O. 2(COOH)2) C, H, N.
Synthesis of (R)-2-((5S, 8S)-8-benzhydryl-1,4-diazabicyclo[3.3.1]nonane-2-yl)-1-phenylethanol (7a)
To a stirring solution of cis-(−)-8-benzydryl-1,4-diazabicyclo[3.3.1] nonane 5 (0.065 g, 0.222 mmol) in dry ethanol was added R-(+)-2-phenyloxirane (0.04 g, 0.333 mmol) (Procedure A). The compound was purified by flash column chromatography by using diethyl ether/MeOH (9:1) to afford 7a (0.051 g, 56%). 1H NMR (400 MHz, CDCl3): δ 1.26–1.33 (1H, m, H-7ax), 1.38–1.47 (1H, m, H-7eq), 1.54–1.63 (1H, m, H-6ax), 2.00–2.03 (1H, m, H-6eq), 2.38–2.47 (2H, m, NCH2CH, H-5), 2.77 (1H, dd, J = 3.6 Hz, J = 12.4 Hz, NCH2CH), 2.82–2.99 (3H, m, NCH2CH2N) 3.03–3.06 (1H, m, H-9ax), 3.17–3.19 (1H, m, NCH2CH2N), 3.29–3.33 (1H, m, H-9eq) 3.80 (1H, dt, J = 4.4 Hz, J = 10.8 Hz, H-8ax), 3.91 (1H, d, J = 11.6 Hz, (Ph)2CH), 4.69 (1H, dd, J = 3.2 Hz, J = 11.2 Hz, CHOH), 7.10–7.38 (15H, m, ArH). [α]25D = (−) 52.3 ° (c 0.52, MeOH). Free base was converted into oxalate salt 194–197 °C
Analysis calculated for (C28H32N2O. 2(COOH)2) C, H, N.
Synthesis of (S)-2-((5S, 8S)-8-benzhydryl-1,4-diazabicyclo[3.3.1]nonane-2-yl)-1-phenylethanol (7b)
Compound 5 (0.060 g, 0.205 mmol) was reacted with S-(−)-2-phenyloxirane (0.036 g, 0.307 mmol) (Procedure A) to yield 7b (0.045 g, 53%). 1H NMR (400 MHz, CDCl3): δ 1.25–1.34 (1H, m, H-7ax), 1.38–1.49 (1H, m, H-7eq), 1.54–1.65 (1H, m, H-6ax), 2.22–2.27 (1H, m, H-6eq), 2.34 (1H, dd, J = 2.8 Hz, J = 10.4 Hz, NCH2CH), 2.46–2.49 (1H, m, NCH2CH2N), 2.64 (1H, bs, H-5), 2.91–3.11 (6H, m, NCH2CH2N, NCH2CH, H-9ax), 3.24–3.27 (1H, m, H-9eq) 3.72–3.83 (1H, m, H-8ax), 3.88 (1H, d, J = 11.6 Hz, (Ph)2CH), 4.67 (1H, dd, J = 3.6 Hz, J = 10.0 Hz, CHOH), 7.10–7.37 (15H, m, ArH). [α]25D = (−) 45.8 ° (c 0.52, MeOH). Free base was converted into oxalate salt 193–195 °C
Analysis calculated for (C28H32N2O. 2(COOH)2, 0.3H2O) C, H, N.
Synthesis of (R)-2-((5S, 8S)-8-benzhydryl-1,4-diazabicyclo[3.3.1]nonane-2-yl)-1-4-fluorophenyl)ethanol (7c)
Compound 5 (0.060 g, 0.205 mmol) was reacted with R-(−)-2-(4-fluorophenyl)oxirane (0.042 g, 0.307 mmol) (Procedure A) to yield 7c (0.034 g, 39%). 1H NMR (400 MHz, CDCl3): δ 1.25–1.32 (1H, m, H-7ax), 1.35–1.47 (1H, m, H-7eq), 1.54–1.64 (1H, m, H-6ax), 2.00–2.03 (2H, m, H-6eq), 2.32–2.38(1H, dd, J = 11.2 Hz, J = 0.8 Hz, NCH2CH), 2.44 (1H, bs, H-5), 2.73 (1H, dd, J = 3.6 Hz, J = 12.8 Hz, NCH2CH), 2.79–2.96 (3H, m, NCH2CH2N), 3.02–3.06 (1H, m, H-9ax), 3.16–3.19 (1H, bm, NCH2CH2N), 3.27–3.31 (1H, m, H-9eq), 3.75–3.86 (1H, m, H-8ax), 3.90 (1H, d, J = 11.2 Hz, (Ph)2CH), 4.66 (1H, dd, J = 3.2 Hz, J = 10.8 Hz, CHOH), 7.03 (2H, t, J = 8.4 Hz, ArH), 7.10–7.38 (12H, m, ArH). [α]25D (free base) = (−) 45.5 ° (c 0.57, MeOH). Free base was converted into oxalate salt 189–191 °C
Analysis calculated for (C28H31FN2O. 2(COOH)2, 0.2H2O) C, H, N.
Synthesis of (S)-2-((5S, 8S)-8-benzhydryl-1,4-diazabicyclo[3.3.1]nonane-2-yl)-1-4-fluorophenyl)ethanol (7d)
Compound 5 (0.070 g, 0.239 mmol) was reacted with S-(+)-2-(4-fluorophenyl)oxirane (0.049 g, 0.358 mmol) (Procedure A) to yield 7d (0.040 g, 39%). 1H NMR (400 MHz, CDCl3): δ 1.27–1.34 (1H, m, H-7ax), 1.37–1.49 (1H, m, H-7eq), 1.59–1.68 (1H, m, H-6ax), 2.20–2.32 (2H, m, H-6eq, NCH2CH), 2.45–2.49 (1H, m, NCH2CH2N), 2.62 (1H, bs, H-5), 2.91–3.11 (5H, m, NCH2CH2N, NCH2CH, H-9ax), 3.23–3.28 (1H, m, H-9eq), 3.75–3.82 (1H, m, H-8ax), 3.88 (1H, d, J = 11.2 Hz, (Ph)2CH), 4.64 (1H, dd, J = 3.2 Hz, J = 10.0 Hz, CHOH), 6.99–7.04 (2H, m, ArH), 7.10–7.37 (12H, m, ArH). [α]25D (free base) = (−) 44.0 ° (c 0.56, MeOH). Free base was converted into oxalate salt 191–193 °C
Analysis calculated for (C28H31FN2O. 2(COOH)2, 0.3H2O) C, H, N.
Synthesis of 3-((1S, 6S)-6-benzhydryl-2,5-diazabicyclo[3.3.1]nonane-2-yl)-1-(4-fluorophenyl)propan-1-one (8)
Into a stirred solution of cis-(−)-8-benzydryl-1,4-diazabicyclo[3.3.1] nonane 5 (0.250 g, 0.854 mmol) in dry acetonitrile was added K2CO3 (0.354 g, 2.56 mmol) followed by 3-chloro-4′-fluoro propiophenone (0.207 g, 1.11 mmol). Catalytic amount of potassium iodide was added and the reaction mixture was refluxed for 3 h under nitrogen atmosphere. After evaporation of solvent, the residue was dissolved in water and extracted with CH2Cl2 (2 × 100 ml), dried over Na2SO4 and evaporated. The compound was purified by flash column chromatography using diethyl ether/MeOH/Et3N (93:7:0.2) to afford 8 (0.260 g, 68%). 1H NMR (400 MHz, CDCl3): δ 1.25–1.34 (1H, m, H-7ax), 1.34–1.45 (1H, m, H-7eq), 1.48–1.58 (1H, m, H-6ax), 2.20–2.23 (1H, m, H-6eq), 2.52 (1H, bs, H-5), 2.62–2.65 (1H, m, NCH2CH2), 2.80–3.16 (8H, m, NCH2CH2N, NCH2CH2, CH2CO, H-9ax), 3.20–3.23 (1H, m, H-9eq) 3.77 (1H, dt, J = 4.4 Hz, J = 11.2 Hz, H-8ax), 3.89 (1H, d, J = 11.8 Hz, (Ph)2CH), 7.08–7.15 (2H, m, ArH), 7.20–7.28 (6H, m, ArH), 7.36 (2H, d, J = 7.2 Hz, ArH), 7.95–7.99 (2H, dt, J = 2.0 Hz, J = 5.2 Hz, ArH).
Synthesis of 3-((1S, 6S)-6-benzhydryl-2,5-diazabicyclo[3.3.1]nonane-2-yl)-1-(4-fluorophenyl)propan-1(R & S)-ol (9)
To a stirred solution of compound 8 (0.250 g, 0.564 mmol) dissolved in 25 ml of THF was added NaBH4 (0.025 g, 0.677 mmol) followed by addition of 0.5 ml of water. The reaction mixture was stirred for 3 h under nitrogen atmosphere at RT. Water (5 ml) was added next into the reaction mixture. The solvent was evaporated and the residue was dissolved in water and extracted with CH2Cl2 (2 × 50 ml), dried over Na2SO4 and evaporated in vacuo. The crude product was purified by flash column chromatography using diethyl ether/MeOH/Et3N (93:8:0.2) to afford 9 (0.195 g, 79%). 1H NMR (400 MHz, CDCl3): δ 1.25–1.41 (2H, m, H-7ax, H-7eq), 1.47–1.95 (3H, m, CH2CHOH, H-6ax), 2.09–2.18 (1H, m, H-6eq), 2.59–2.96 (6H, m, H-5, NCH2CH2, NCH2CH2N), 3.04 (1H, d, J = 13.2 Hz, H-9ax), 3.12–3.25 (2H, m, NCH2CH2N, H-9eq), 3.73–3.82 (1H, m, H-8ax), 3.88 (1H, dd, J = 1.6 Hz, J = 11.6 Hz, (Ph)2CH), 4.84–4.95 (1H, m, CHOH), 7.00 (2H, dt, J = 1.6 Hz, J = 8.4 Hz, ArH), 7.10–7.16 (2H, m, ArH), 7.19–7.37 (10H, m, ArH).
Synthesis of 3-((1S, 6S)-6-benzhydryl-2,5-diazabicyclo[3.3.1]nonane-2-yl)-1-(4-fluorophenyl)propyl-4,7,7-trimethyl-3-oxo-2-oxabicyclo[2.2.1]heptane-1-carboxylate (10a and 10b)
To a stirred solution of 3-((1S, 6S)-6-benzhydryl-2,5-diazabicyclo [3.3.1]nonane-2-yl)-1-(4-fluorophenyl)propan-1 (R & S)-ol 9 (0.195 g, 0.438 mmol), Et3N (0.088 g, 0.876 mmol), and DMAP (10 mg) in 100 ml of dry CH2Cl2 under nitrogen atmosphere at 0°C was added (1S)-(−)-camphanic chloride (0.123 g, 0.569 mmol) dissolved in 10 ml dry CH2Cl2. The reaction mixture was stirred at 0°C for 30 min and at room temperature for 3 h under nitrogen. The reaction mixture was quenched with water (20 ml) and then diluted with CH2Cl2 (50 ml). Organic layer was separated and the aqueous layer was extracted with CH2Cl2 (2 × 50 ml), dried over Na2SO4 and evaporated in vacuo. The crude product was purified by flash column chromatography over silica gel using diethyl ether/MeOH (95:5) to afford mixture of two diastereoisomers 10a and 10b (0.250 g, 91%). The diastereoisomers were separated by semipreparative HPLC using a normal phase column (Nova-pack silica 6μm). Hexanes/2-propanol/Et3N (92:8:0.3) was used as a mobile phase with a flow rate of 12 mL/min. The two fractions were eluted with retention time 2.83 min and 3.30 min for 10a and 10b, respectively.
Eluting first was 10a (0.115 g, 42%). 1H NMR (400 MHz, CDCl3): δ 0.79 (3H, s, CH3), 0.97 (3H, s, CH3), 1.07 (3H, s, CH3), 1.24–1.49 (2H, m, H-7ax, H-7eq), 1.62–1.68 (1H, m, H-6ax), 1.84–2.07 (3H, m, CH2CHO, H-6eq), 2.16–2.49 (5H, m, H-5, CCH2C, NCH2CH2N), 2.59–2.63 (1H, m, NCH2CH2N), 2.68–2.75 (1H, m, NCH2CH2), 2.86–2.98 (2H, m, NCH2CH2, H-9ax), 3.09–3.12 (1H, m, NCH2CH2N), 3.18–3.21 (1H, m, H-9eq), 3.75 (1H, dt, J = 4.4 Hz, J = 11.2 Hz, H-8ax), 3.87 (1H, d, J = 11.2 Hz, (Ph)2CH), 5.94 (1H, t, J = 7.2 Hz, CHOCO), 6.97–7.03 (2H, m, ArH), 7.09–7.14 (2H, m, ArH), 7.19–7.27 (6H, m, ArH), 7.30–7.37 (4H, m, ArH).
Eluting second was 10b (0.105 g, 38%). 1H NMR (400 MHz, CDCl3): δ 0.88 (3H, s, CH3), 0.98 (3H, s, CH3), 1.09 (3H, s, CH3), 1.23–1.52 (2H, m, H-7ax, H-7eq), 1.62–1.68 (1H, m, H-6ax), 1.84–1.99 (2H, m, CH2CHO), 2.04–2.07 (1H, m, H-6eq), 2.12–2.52 (5H, m, H-5, CCH2C, NCH2CH2N), 2.68–2.75 (1H, m, NCH2CH2), 2.84–2.92 (1H, m, NCH2CH2), 2.95–2.99 (1H, m, H-9ax), 3.08–3.11 (1H, m, NCH2CH2N), 3.18–3.22 (1H, m, H-9eq), 3.76 (1H, dt, J = 4.8 Hz, J = 11.2 Hz, H-8ax), 3.87 (1H, d, J = 11.6 Hz, (Ph)2CH), 5.92 (1H, t, J = 6.8 Hz, CHOCO), 6.98–7.03 (2H, m, ArH), 7.09–7.15 (2H, m, ArH), 7.19–7.37 (10H, m, ArH).
Procedure B. Synthesis of (−)11a
The first eluting camphanic ester fraction 10a (0.075 g, 0.120 mmol) was hydrolyzed with K2CO3 (20 mg) in methanol (20 ml) at room temperature for 12 h. Methanol was evaporated, water (20 ml) was added and the product was extracted with ethyl acetate (2 × 50 ml). Organic layer was dried over Na2SO4 and evaporated under reduced pressure. The crude product was purified by flash column chromatography over silica using diethyl ether/MeOH/Et3N (93:8:0.2) to afford 11a (0.048 g, 90%). 1H NMR (400 MHz, CDCl3): δ 1.25–1.44 (2H, m, H-7ax, H-7eq), 1.54–1.69 (2H, m, CH2CHOH, H-6ax), 1.80–1.90 (1H, m, CH2CHOH), 2.15–2.18 (1H, m, H-6eq), 2.63 (1H, bs, H-5), 2.67–2.974 (1H, m, NCH2CH2), 2.79–2.95 (4H, m, NCH2CH2, NCH2CH2N), 3.04 (1H, d, J = 13.2 Hz, H-9ax), 3.13–3.18 (1H, m, NCH2CH2N), 3.22–3.25 (1H, m, H-9eq), 3.79 (1H, dt, J = 4.8 Hz, J = 11.6 Hz, H-8ax), 3.88 (1H, d, J = 11.6 Hz, (Ph)2CH), 4.86 (1H, dd, J = 2.4 Hz, J = 9.6 Hz, CHOH), 7.00 (2H, t, J = 8.8 Hz, ArH), 7.11–7.16 (2H, m, ArH), 7.19–7.37 (10H, m, ArH). [α]25D = (−) 38.7 ° (c 1.08, MeOH). Free base was converted into oxalate salt 191–193 °C
Analysis calculated for (C29H33FN2O. 2(COOH)2, 1.5H2O) C, H, N.
Synthesis of (−)11b
The second eluting fraction 10b (0.105 g, 0.168 mmol) was hydrolyzed with K2CO3 (20 mg) in methanol (20 ml) to afford 11b (0.071 g, 94%) (Procedure B). 1H NMR (400 MHz, CDCl3): δ 1.24–1.43 (2H, m, H-7ax, H-7eq), 1.48–1.54 (1H, m, H–6ax), 1.71–1.79 (1H, m, CH2CHOH), 1.88–1.95 (1H, m, CH2CHOH), 2.09–2.12 (1H, m, H-6eq), 2.60–2.66 (2H, m, H-5, NCH2CH2), 2.76–2.97 (4H, m, NCH2CH2, NCH2CH2N), 3.04 (1H, d, J = 13.2 Hz, H-9ax), 3.12–3.15 (1H, m, NCH2CH2N), 3.21–3.24 (1H, m, H-9eq), 3.78 (1H, dt, J = 4.8 Hz, J = 11.6 Hz, H-8ax), 3.88 (1H, d, J = 11.6 Hz, (Ph)2CH), 4.94 (1H, dd, J = 3.2 Hz, J = 7.6 Hz, CHOH), 7.00 (2H, t, J = 8.4 Hz, ArH), 7.10–7.16 (2H, m, ArH), 7.20–7.37 (10H, m, ArH). [α]25D = (−) 56.8 ° (c 1.0, MeOH). Free base was converted into oxalate salt 193–195 °C
Analysis calculated for (C29H33FN2O. 2(COOH)2, 0.5H2O) C, H, N.
Transporter assays
The affinity of test compounds in binding to rat DAT and in inhibiting monoamine uptake was monitored as described by us previously.21 Briefly, rat striatum was used for measuring binding of [3H]WIN 35,428 by DAT and uptake of [3H]DA by DAT. Rat cerebral cortex was used for assessing uptake of [3H]serotonin by SERT and hippocampus for uptake of [3H]NE by NET. Nonspecific binding at DAT was defined with 100 uM cocaine; nonspecific uptake at DAT, SERT, and NET with 100 uM cocaine, 10 uM citalopram, and 10 uM desipramine, respectively. Test compounds were dissolved in dimethyl sulfoxide (DMSO), diluted out in 10% (v/v) DMSO, and added to assays resulting in a final DMSO concentration of 0.5% which by itself did not interfere with the assays. At lease five triplicate concentrations of each test compound were studied, spaced evenly around the IC50 value. The latter was estimated by nonlinear computer curve-fitting procedures and converted to Ki with the Cheng-Prusoff equation as described previously16.
Elemental Analysis Results of Final Products
| Compounds | Calculated | Found | ||||
|---|---|---|---|---|---|---|
| C | H | N | C | H | N | |
| 4a 2(COOH)2 0.5H2O | 60.70 | 5.77 | 4.72 | 60.78 | 5.72 | 4.69 |
| 4b 2(COOH)2 0.9H2O | 59.97 | 5.84 | 4.66 | 59.98 | 5.71 | 4.57 |
| 4c 2(COOH)2 0.5H2O | 63.15 | 6.32 | 4.75 | 63.15 | 6.31 | 4.74 |
| 4d 2(COOH)2 0.3H2O | 63.53 | 6.30 | 4.78 | 63.45 | 6.42 | 4.79 |
| 6a 2(COOH)2 | 65.33 | 6.31 | 4.62 | 65.17 | 6.32 | 4.58 |
| 6b 2(COOH)2 0.2H2O | 64.94 | 6.34 | 4.59 | 64.73 | 6.23 | 4.54 |
| 7a 2(COOH)2 | 64.85 | 6.12 | 4.73 | 64.75 | 6.20 | 4.71 |
| 7b 2(COOH)2 0.3H2O | 64.27 | 6.17 | 4.68 | 64.24 | 6.21 | 4.73 |
| 7c 2(COOH)2 0.2H2O | 62.57 | 5.81 | 4.56 | 62.48 | 5.87 | 4.60 |
| 7d 2(COOH)2 0.3H2O | 62.39 | 5.82 | 4.55 | 62.29 | 5.85 | 4.59 |
| 11a 2(COOH)2 1.5H2O | 60.82 | 6.19 | 4.30 | 60.43 | 5.95 | 4.23 |
| 11b 2(COOH)2 0.5H2O | 62.55 | 6.04 | 4.42 | 62.54 | 5.94 | 4.33 |
Acknowledgments
This work was supported by the National Institute on Drug Abuse, Grant No. DA 12449 (AKD).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ritz MC, Cone EJ, Kuhar MJ. Cocaine inhibition of ligand binding at dopamine, norepinephrine and serotonin transporters: a structure-activity study. Life Sci. 1990;46:635–45. doi: 10.1016/0024-3205(90)90132-b. [DOI] [PubMed] [Google Scholar]
- 2.Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ. Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science. 1987;237:1219–23. doi: 10.1126/science.2820058. [DOI] [PubMed] [Google Scholar]
- 3.Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–12. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- 4.Witkin JM, Nichols DE, Terry P, Katz JL. Behavioral effects of selective dopaminergic compounds in rats discriminating cocaine injections. J Pharmacol Exp Ther. 1991;257:706–13. [PubMed] [Google Scholar]
- 5.Tilley MR, Cagniard B, Zhuang X, Han DD, Tiao N, Gu HH. Cocaine reward and locomotion stimulation in mice with reduced dopamine transporter expression. BMC Neurosci. 2007;8:42. doi: 10.1186/1471-2202-8-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walsh SL, Cunningham KA. Serotonergic mechanisms involved in the discriminative stimulus, reinforcing and subjective effects of cocaine. Psychopharmacology (Berl) 1997;130:41–58. doi: 10.1007/s002130050210. [DOI] [PubMed] [Google Scholar]
- 7.Glowa JR, Fantegrossi WE, Lewis DB, Matecka D, Rice KC, Rothman RB. Sustained decrease in cocaine-maintained responding in rhesus monkeys with 1-[2-[bis(4-fluorophenyl)methoxy]ethyl]-4-(3-hydroxy-3-phenylpropyl) piperazinyl decanoate, a long-acting ester derivative of GBR 12909. J Med Chem. 1996;39:4689–91. doi: 10.1021/jm960551t. [DOI] [PubMed] [Google Scholar]
- 8.Sogaard U, Michalow J, Butler B, Lund Laursen A, Ingersen SH, Skrumsager BK, Rafaelsen OJ. A tolerance study of single and multiple dosing of the selective dopamine uptake inhibitor GBR 12909 in healthy subjects. Int Clin Psychopharmacol. 1990;5:237–51. doi: 10.1097/00004850-199010000-00001. [DOI] [PubMed] [Google Scholar]
- 9.Carroll FI, Howard JL, Howell LL, Fox BS, Kuhar MJ. Development of the dopamine transporter selective RTI-336 as a pharmacotherapy for cocaine abuse. Aaps J. 2006;8:E196–203. doi: 10.1208/aapsj080124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henry LK, Blakely RD. Distinctions Between Dopamine Transporter Antagonists Could Be Just Around the Bend. Mol Pharmacol. 2007 doi: 10.1124/mol.107.044586. [DOI] [PubMed] [Google Scholar]
- 11.Loland CJ, Desai RI, Zou MF, Cao J, Grundt P, Gerstbrein K, Sitte HH, Newman AH, Katz JL, Gether U. Relationship between conformational changes in the dopamine transporter and cocaine-like subjective effects of uptake inhibitors. Mol Pharmacol. 2007 doi: 10.1124/mol.107.039800. [DOI] [PubMed] [Google Scholar]
- 12.Dutta AK, Zhang S, Kolhatkar R, Reith ME. Dopamine transporter as target for drug development of cocaine dependence medications. Eur J Pharmacol. 2003;479:93–106. doi: 10.1016/j.ejphar.2003.08.060. [DOI] [PubMed] [Google Scholar]
- 13.Dutta AK, Xu C, Reith ME. Structure-activity relationship studies of novel 4-[2-[bis(4-fluorophenyl)methoxy]ethyl]-1-(3-phenylpropyl)piperidine analogs: synthesis and biological evaluation at the dopamine and serotonin transporter sites. J Med Chem. 1996;39:749–56. doi: 10.1021/jm9506581. [DOI] [PubMed] [Google Scholar]
- 14.Dutta AK, Coffey LL, Reith ME. Highly selective, novel analogs of 4-[2-(diphenylmethoxy)ethyl]-1-benzylpiperidine for the dopamine transporter: effect of different aromatic substitutions on their affinity and selectivity. J Med Chem. 1997;40:35–43. doi: 10.1021/jm960638e. [DOI] [PubMed] [Google Scholar]
- 15.Dutta AK, Fei XS, Beardsley PM, Newman JL, Reith ME. Structure-activity relationship studies of 4-[2-(diphenylmethoxy)ethyl]-1-benzylpiperidine derivatives and their N-analogues: evaluation of O-and N-analogues and their binding to monoamine transporters. J Med Chem. 2001;44:937–48. doi: 10.1021/jm000311k. [DOI] [PubMed] [Google Scholar]
- 16.Dutta AK, Davis MC, Reith ME. Rational design and synthesis of novel 2,5-disubstituted cis- and trans-piperidine derivatives exhibiting differential activity for the dopamine transporter. Bioorg Med Chem Lett. 2001;11:2337–40. doi: 10.1016/s0960-894x(01)00443-7. [DOI] [PubMed] [Google Scholar]
- 17.Kolhatkar RB, Ghorai SK, George C, Reith ME, Dutta AK. Interaction of cis-(6-benzhydrylpiperidin-3-yl)benzylamine analogues with monoamine transporters: structure-activity relationship study of structurally constrained 3, 6-disubstituted piperidine analogues of (2,2-diphenylethyl)-[1-(4-fluorobenzyl)piperidin-4-ylmethyl]amine. J Med Chem. 2003;46:2205–15. doi: 10.1021/jm020561w. [DOI] [PubMed] [Google Scholar]
- 18.Kolhatkar R, Cook CD, Ghorai SK, Deschamps J, Beardsley PM, Reith ME, Dutta AK. Further structurally constrained analogues of cis-(6-benzhydrylpiperidin-3-yl)benzylamine with elucidation of bioactive conformation: discovery of 1,4-diazabicyclo[3.3.1]nonane derivatives and evaluation of their biological properties for the monoamine transporters. J Med Chem. 2004;47:5101–13. doi: 10.1021/jm049796t. [DOI] [PubMed] [Google Scholar]
- 19.Ghorai SK, Cook C, Davis M, Venkataraman SK, George C, Beardsley PM, Reith ME, Dutta AK. High affinity hydroxypiperidine analogues of 4-(2-benzhydryloxyethyl)-1-(4-fluorobenzyl)piperidine for the dopamine transporter: stereospecific interactions in vitro and in vivo. J Med Chem. 2003;46:1220–8. doi: 10.1021/jm020275k. [DOI] [PubMed] [Google Scholar]
- 20.Schaus SE, Brandes BD, Larrow JF, Tokunaga M, Hansen KB, Gould AE, Furrow ME, Jacobsen EN. Highly selective hydrolytic kinetic resolution of terminal epoxides catalyzed by chiral (salen)Co(III) complexes. Practical synthesis of enantioenriched terminal epoxides and 1,2-diols. J Am Chem Soc. 2002;124:1307–15. doi: 10.1021/ja016737l. [DOI] [PubMed] [Google Scholar]
- 21.Zhang S, Zhen J, Reith ME, Dutta AK. Discovery of novel trisubstituted asymmetric derivatives of (2S,4R,5R)-2-benzhydryl-5-benzylaminotetrahydropyran-4-ol,exhibiting high affinity for serotonin and norepinephrine transporters in a stereospecific manner. J Med Chem. 2005;48:4962–71. doi: 10.1021/jm049021k. [DOI] [PubMed] [Google Scholar]