

Abstract
Prevention and treatment of cardiovascular disorders by HMG-CoA reductase inhibitors (or statins), beyond their lipid-lowering properties, have been demonstrated including activation of the endothelial nitric oxide synthase (eNOS). Beside endothelial cells, red blood cells (RBCs) possess NOS and produce nitric oxide (NO), which contributes to RBC deformability. The present study tested the capacity of statins to activate NOS in RBCs and subsequently to modulate RBC deformability in vitro. Blood samples of healthy young volunteers were incubated with or without rosuvastatin. Afterwards RBC-NOS activity and RBC deformability were determined. Rosuvastatin incubation significantly increased NOS phosphorylation, NOS dependent NO-formation, and RBC deformability. The NOS inhibitor NG- monomethyl-L-arginine reversed the stimulatory effect of rosuvastatin on RBC-NOS activity. This NO dependent effect of rosuvastatin might have an important influence on microcirculation and may offer new perspectives for the therapeutic use of statins.
Keywords: red blood cell, nitric oxide synthase, red blood cell deformability, statin
Introduction
Beside endothelial cells, monocytes, cardiomyocytes, and platelets (Förstermann et al 1998) also red blood cells (RBCs) (Jubelin and Gierman 1996) carry a constitutive form of nitric oxide synthase (eNOS). We have recently shown an eNOS-like protein in RBC influencing the nitric oxide (NO) production and thereby affecting RBC deformability and platelet aggregation. Apart from its substrate L-arginine, insulin is known to activate NOS in RBCs through a phosphatidylinositol-3 kinase (PI3K)-induced phosphorylation of the residue serine1177. The substrate conversion rate of L-arginine to L-citrulline by RBCs reaches similar levels to that achieved by endothelial cells in healthy volunteers, suggesting that RBCs and endothelial cells contribute likewise to NO metabolism (Kleinbongard et al 2006). However, since under a variety of diseases endothelial cell NOS activity is reduced (“endothelial dysfunction”) (Herman and Moncada 2005), RBC-NOS may appear as an interesting pharmacological target to increase the total NO-pool and blood rheology.
In clinical trials, statins are beneficial in the primary and secondary prevention of coronary heart disease. Statins have been shown to possess several pleiotropic properties independent of cholesterol lowering in experimental settings.
There are various mechanisms whereby statins may alter NO release. Statins regulates the expression of the eNOS and may also increase eNOS activity via post-translational activation of the PI3K/Akt pathway or through an interaction with the molecular chaperone heat-shock protein 90 (Laufs 2003). Statins have been shown to increase NO-bioavailability (Schäfer et al 2005) but the source of NO-production remains to be assessed. Other studies described effects of statins on the blood rheology including RBC deformability (Szapary et al 2004). Our study therefore tested whether or not statins can acutely activate RBC-NOS and if so whether this subsequently affects hemorheological properties.
Materials and methods
The study was performed in accordance with the rules of the internal review board and the tenets of the Helsinki protocol.
Sample preparation
For all experiments blood was taken from the antecubital vein of healthy volunteers. Measurements of RBC deformability and RBC-NOS activity were performed simultaneously (n = 8). Phosphorylation of eNOS in RBCs was measured immunocytochemically in dependence of rosuvastatin in a subset of the mentioned experiments (n = 4).
For measurements of eNOS phosphorylation, NOS activity and RBC deformability, the arginase inhibitor L-valin (30 mM) and the NOS-substrate L-arginine (3 mM) were added to anticoagulated blood samples (Lepirudin 62.5 μg/ml) which were then incubated for 30 minutes at 37 °C with rosuvastatin (20 ng/ml; a concentration achieved in human blood during standard therapy [Cooper et al 2003]) or buffer as control. In a subset of experiments additionally to L-valin and L-arginine the NOS inhibitor NG- monomethyl-L-arginine (L-NMMA, 300 μM) was added and the blood was incubated with or without rosuvastatin (n = 5).
Measurement of eNOS phosphorylation
RBCs incubated with or without rosuvastatin (see above) were fixed as a blood smear with methanol/paraformaldehyd (37%) solution (9:1) and penetrated with PBS/Triton-X-100 (0.1%). The anti-phospho-eNOS (Ser1177) antibody (1:500, rabbit, Upstate, NY) was used as primary antibody; for further details see (Kleinbongard et al 2006; Pott et al 2006).
Measurement of nitrite formation as a marker of NOS activity
Accumulated plasma nitrite was measured after incubation of whole blood using the reductive chemiluminescence technique (Feelisch et al 2002). Nitrate was quantified by flow injection analysis after specific sample processing with prior reduction to nitrite by nitrate reductase (Dejam et al 2003).
Measurement of RBC deformability
The RBC deformability was determined using a modified filtration method (Reid et al 1976). With a negative pressure of 0.98 kPa pure RBCs (hematocrit 35% in buffer) were sucked through a 5 μm pore filter and the flow rate (ml/min) was measured.
Statistical analysis
Data are expressed as mean ± SEM. Two-tailed paired Students t test was used to analyze nitrite, nitrate, and eNOS-phosphorylation data, while the Wilcoxon test was used to compare RBC deformability. For multiple comparisons one-way ANOVA and Bonferoni test were applied. p-values <0.05 were accepted as significant.
Results
RBC-NOS activity is increased by rosuvastatin
A significantly increased phosphorylation of RBC-NOS after incubation with rosuvastatin from 3.76 ± 2.98 arbitrary units (au) to 24.88 ± 4.35 au (p < 0.02) was detected (Figure 1A). The oxidative metabolite of NO, nitrite, has been shown to reflect changes of NOS activity in vivo and in vitro (Kleinbongard et al 2003, 2006). A nitrite accumulation in plasma after incubation of whole blood with rosuvastatin from 55.8 ± 7.9 nmol/l to 98.2 ± 12.4 nmol/l (p < 0.05, Figure 1B) was detectable. Plasma nitrate levels remained unchanged. The specific NOS-inhibitor L-NMMA in addition to rosuvastatin decreased plasma nitrite levels from 64.3 ± 4.5 nmol/l to 46.6 ± 4.0 nmol/l (p < 0.01); incubation with L-NMMA alone decreased plasma nitrite levels to 43.9 ± 2.0 nmol/l (p < 0.01 to control, no significance to inhibition with rosuvastatin).
Figure 1.

Rosuvastatin increases RBC-NOS activity. RBC-NOS activity was measured by the level of eNOS protein phosphorylation and by accumulated plasma nitrite depending on rosuvastatin. (A) Phosphorylation of eNOS at Ser1177 was used to examine the phosphorylation dependent activation status of the eNOS. RBCs incubated with rosuvastatin showed a significant rise of eNOS phosphorylated at Ser1177 as compared with control. (B) Accumulated nitrite was determined in blood samples after incubation under control conditions with and without rosuvastatin. Changes in plasma nitrite reflect the sum of the release (due to NOS-dependent NO-formation) and the reuptake of nitrite by RBC.
Note: *indicates significant differences from control.
Increased RBC deformability and RBC NO-production caused by rosuvastatin
RBC deformability increased significantly after incubation with rosuvastatin from 1.96 ± 0.34 ml/min (controls) to 2.63 ± 0.31 ml/min (p < 0.036). Plasma nitrite levels correlated to RBC deformability (R = 0.59, p < 0.0001; Figure 2).
Figure 2.
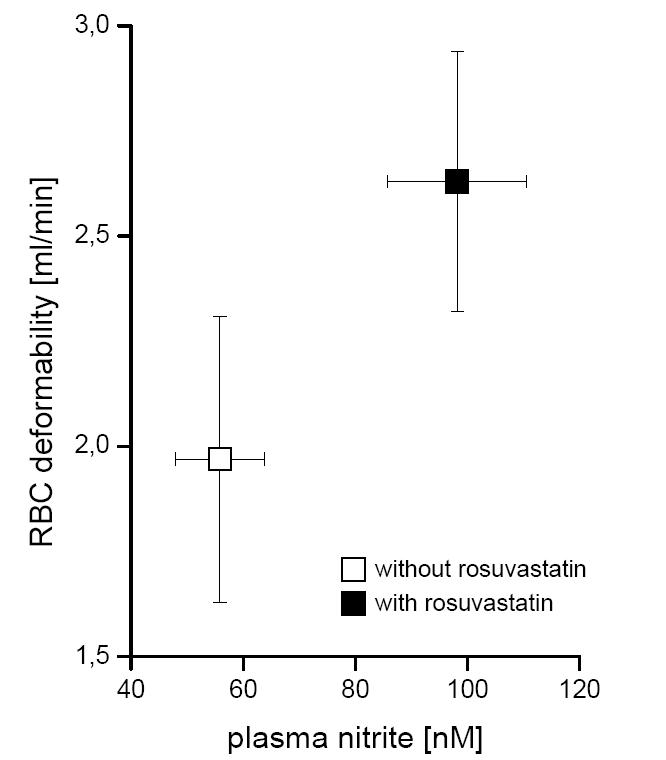
Effect of rosuvastatin on RBC-NOS activity and RBC deformability. Parallel to the RBC-NOS activity (detected as accumulated plasma nitrite concentration) flow rate of RBC through a microfilter (as a measure of RBC deformability) were determined after incubation of whole blood with (black squares) and without rosuvastatin (white squares).
Discussion
The existence of a NO-synthesis in RBC and the fact that it can be modulated raises new perspectives on treatment of vascular diseases. Until now nothing is known about eNOS stimulating therapeutical agents and RBC-NOS activity or function. The novel finding of this study is that statin increases RBC-NOS activity which subjects to an improved RBC deformability.
In endothelial cells statins cause PI3K stimulation recruiting a number of signalling molecules to the membrane, including PI3 dependent kinase. This kinase activates Akt which further phosphorylates eNOS leading to Ca2+ independent enzyme activation (Endres and Laufs 2004). In this study, a significantly increased phosphorylation of RBC-NOS after incubation with rosuvastatin could be shown (Figure 1A). Phoshorylation at Ser1177 of RBC-NOS could also been induced by insulin, which is a known activator of the PI3K pathway (Kleinbongard et al 2006). After incubation of whole blood for a period of 30 minutes, plasma nitrite has been shown to reflect RBC-NOS activity in vitro (Kleinbongard et al 2006). Here we could show a parallel rosuvastatin-dependent increase of plasma nitrite as a marker for RBC-NOS activity, and RBC-NOS phosphorylation (Figure 1B). Rosuvastatin-dependent RBC-NOS activation could be impeded using a specific NOS-inhibitor. Our data, however, imply that RBC-NOS could be regulated through a similar pathway as shown in endothelial cells, since rosuvastatin caused a significant increase in RBC-NOS phosphorylation and NO production. Unknown are the potential upstream pathways of such phosphorylation in RBCs. Described increase of eNOS expression or mRNA stabilization by statins could be excluded in the anucleated RBC. Possible regulatory mechanisms are modification of eNOS interacting proteins like heat shock protein 90 or various protein kinases (Noma et al 2006). Protein kinase B in RBC precursors (Kadri et al 2005) and RBC membrane proteins (α-spectrin and β-spectrin) with more than 50% similarities to the heat shock protein 90 (Bhattacharyya et al 2004) have been identified. Statins may also block proteins interfering with calcium/calmodulin by sequestering eNOS into caveolae (Pelat et al 2003). Calmodulin is present in RBC (Jubelin and Gierman 1996), and RBC NOS activity is calcium-dependent (Kleinbongard et al 2006). Therefore, some of the well known NOS regulatory mechanisms can be influenced by rosuvastatin and are feasible in regulation of RBC-NOS activity.
Vasodilation and particularly RBC deformability (Mchedlishvili and Maeda 2001) are basic needs of microcirculatory blood flow. Reduction of RBC deformability and microcirculatory blood flow has been described during several pathophysiological circumstances such as arterial hypertension (Cicco and Pirrelli 1999) and diabetes mellitus (Symeonidis et al 2001). Statin therapy has been shown to influence RBC deformability after long-term treatment particularly in hypercholesterolemic patients which is thought to relate to a decrease of cholesterol in the erythrocyte lipid double layer (Kohno et al 1997). Up to now this improvement of statin-dependent change in RBC deformability was related to the change in the RBC membrane lipid composition. Here we see an effect of rosuvastatin treatment on RBC deformability and RBC-NOS activity (Figure 2). These data suggest an alternative explanation relating the increased deformability following statin treatment to the increase in RBC-NOS activity. In nearly all blood cells, namely platelets, monocytes, lymphocytes (Förstermann et al 1998) and RBCs (Kleinbongard et al 2006) a constitutive form of NOS has been demonstrated. Neutrophils have been shown to inhibit platelet aggregation after NOS stimulation. Interestingly, the effect was more pronounced after whole blood incubation as compared with incubation with platelets itself or a combination of platelets and neutrophils (De La Cruz et al 1999). Cell-cell interaction with erythrocytes was suggested as possible explanation for this result and, indeed, our data support such hypothesis.
Emerging evidence indicates that the microcirculation is a crucial target for the pleiotropic actions of statins. The major pharmacodynamic actions of statins currently invoked to explain their vascular protective effects are increased blood flow and attenuated inflammatory events in the vessel wall. Effects of statins influencing the microcirculation are anti-adhesive actions by inhibiting leukocyte-endothelium interactions or the effects of cell proliferation an angiogenesis. These effects are related to important functions of the microcirculation under normal conditions and in disease states. In particular, increased bioavailability of NO in the microcirculation by statins could prevent pathophysiological cell-cell interactions and may reduce oxidative stress at the vessel wall. RBC-NOS might appear as an interesting pharmacological target to increase the total NO-pool. Amelioration of blood flow and increased delivery of oxygen and nutrients should be beneficial in pathologically changed circulation for example after myocardial or cerebral ischemia.
Taken together all these findings are of clinical interest because they may uncover new mechanisms of the beneficial action of statins in cardiovascular disease. In vivo experiments are necessary to identify the potential of RBC-NOS in patients with NO deficiency and to develop new strategies to enhance expression and activity of RBC-NOS.
Acknowledgments
This study was supported by the Deutsche Forschungsgemeinschaft, (Ke405/4-3) (MK and PK) and AstraZeneca, Wedel, Germany (RS and BL). The indispensable technical assistance of K. Lysaja and M. Ghilav is gratefully acknowledged.
References
- Bhattacharyya M, Ray S, Bhattacharya S, et al. Chaperone activity and prodan binding at the self-associating domain of erythroid spectrin. J Biol Chem. 2004;31:55080–8. doi: 10.1074/jbc.M406418200. [DOI] [PubMed] [Google Scholar]
- Cicco G, Pirrelli A. Red blood cell (RBC) deformability, RBC aggregability and tissue oxygenation in hypertension. Clin Hemorheol Microcirc. 1999;21:169–77. [PubMed] [Google Scholar]
- Cooper KJ, Martin PD, Dane AL, et al. Lack of effect of ketoconazole on the pharmacokinetics of rosuvastatin in healthy subjects. Br J Clin Pharmacol. 2003;55:94–9. doi: 10.1046/j.1365-2125.2003.01720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Cruz JP, Paez MV, Carmona JA, et al. Antiplatelet effect of the anaesthetic drug propofol: influence of red blood cells and leukocytes. Br J Pharmacol. 1999;128:1538–44. doi: 10.1038/sj.bjp.0702927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejam A, Kleinbongard P, Rassaf T, et al. Thiols enhance NO formation from nitrate photolysis. Free Radic Biol Med. 2003;35:1551–9. doi: 10.1016/j.freeradbiomed.2003.09.009. [DOI] [PubMed] [Google Scholar]
- Endres M, Laufs U. Effects of stains on endothelium and signaling mechanisms. Stroke. 2004;35:1–5. doi: 10.1161/01.STR.0000143319.73503.38. [DOI] [PubMed] [Google Scholar]
- Feelisch M, Rassaf T, Mnaimneh S, et al. Concomitant S-, N-, and heme-nitros(yl)ation in biological tissues and fluids: implications for the fate of NO in vivo. FASEB J. 2002;16:1775–85. doi: 10.1096/fj.02-0363com. [DOI] [PubMed] [Google Scholar]
- Förstermann U, Boissel J-P, Kleinert H. Expressional control of the ‘constitutive’ isoforms of nitric oxide synthase (NOS I and NOS III) FASEB J. 1998;12:773–90. [PubMed] [Google Scholar]
- Herman AG, Moncada S. Therapeutic potential of nitric oxide donors in the prevention and treatment of atherosclerosis. Eur Heart J. 2005;26:1945–55. doi: 10.1093/eurheartj/ehi333. [DOI] [PubMed] [Google Scholar]
- Jubelin BC, Gierman JL. Erythrocytes may synthesize their own nitric oxide. Am J Hypertens. 1996;9:1214–9. doi: 10.1016/S0895-7061(96)00257-9. [DOI] [PubMed] [Google Scholar]
- Kadri Z, Maouche-Chretien L, Rooke HM, et al. Phosphatidylinositol 3-kinase/Akt induced by erythropoietin renders the erythroid differentiation factor GATA-1 competent for TIMP-1 gene transactivation. Moll Cell Biol. 2005;25:7412–22. doi: 10.1128/MCB.25.17.7412-7422.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinbongard P, Dejam A, Lauer T, et al. Plasma nitrite reflects constitutive nitric oxide synthase activity in mammals. Free Radic Biol Med. 2003;35:790–6. doi: 10.1016/s0891-5849(03)00406-4. [DOI] [PubMed] [Google Scholar]
- Kleinbongard P, Schulz R, Rassaf T, et al. Red blood cells express a functional endothelial nitric oxide synthase. Blood. 2006;107:2943–51. doi: 10.1182/blood-2005-10-3992. [DOI] [PubMed] [Google Scholar]
- Kohno M, Murakawa K, Yasunari K, et al. Improvement of erythrocyte deformability by cholesterol-lowering therapy with pravastatin in hypercholesterolemic patients. Metabolism. 1997;46:287–91. doi: 10.1016/s0026-0495(97)90255-9. [DOI] [PubMed] [Google Scholar]
- Laufs U. Beyond lipid-lowering: effects of statins on endothelial nitric oxide. Eur J Clin Pharmacol. 2003;58:719–31. doi: 10.1007/s00228-002-0556-0. [DOI] [PubMed] [Google Scholar]
- Mchedlishvili G, Maeda N. A determinant of blood fluidity in narrow microvessels. Jpn J Physiol. 2001;51:19–30. doi: 10.2170/jjphysiol.51.19. [DOI] [PubMed] [Google Scholar]
- Noma K, Oyama N, Liao JK. Physiological role of ROCKs in the cardiovascular system. Am J Physiol Cell Physiol. 2006;290:C661–C668. doi: 10.1152/ajpcell.00459.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelat M, Dessy C, Massion P, et al. Rosuvastatin decreases caveolin-1 and improves nitric oxide-dependent heart rate and blood pressure variability in apolipoprotein E-/- mice in vivo. Circulation. 2003;107:2480–6. doi: 10.1161/01.CIR.0000065601.83526.3E. [DOI] [PubMed] [Google Scholar]
- Pott C, Steinritz D, Boeleck B, et al. eNOS-translocation but not eNOS-phosphorylation is dependent on intracellular Ca2+ in human atrial myocardium. Am J Physiol Cell Physiol. 2006;290:C1437–C1445. doi: 10.1152/ajpcell.00005.2005. [DOI] [PubMed] [Google Scholar]
- Reid HL, Barnes AJ, Lock PJ, et al. Technical methods. A simple method for measuring erythrocyte deformability. J Clin Path. 1976;29:855–8. doi: 10.1136/jcp.29.9.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer A, Fraccarollo D, Eigenthaler M, et al. Rosuvastatin reduces platelet activation in heart failure; Role of NO bioavailability. Arterioscler Thromb Vasc Bio. 2005;25:1071–7. doi: 10.1161/01.ATV.0000161926.43967.df. [DOI] [PubMed] [Google Scholar]
- Symeonidis A, Athanassiou G, Psiroyannis A, et al. Impairment of erythrocyte viscoelasticity is correlated with levels of glycosylated haemoglobin in diabetic patients. Clin Lab Haematol. 2001;23:103–9. doi: 10.1046/j.1365-2257.2001.00366.x. [DOI] [PubMed] [Google Scholar]
- Szapary L, Horvath B, Marton Z, et al. Short-term effect of low-dose atorvastatin on haemorrheological parameters, platelet aggregation and endothelial function in patients with cerebrovascular disease and hyperlipidaemia. CNS Drugs. 2004;18:165–72. doi: 10.2165/00023210-200418030-00003. [DOI] [PubMed] [Google Scholar]