

Abstract
Tissue repair is a well orchestrated biological process involving numerous soluble mediators, and an imbalance between these factors may result in impaired repair and fibrosis. Transforming growth factor (TGF) β is a key profibrotic element in this process and it is thought that its three isoforms act in a similar way. Here, we report that TGF-β3 administered to rat lungs using transient overexpression initiates profibrotic effects similar to those elicited by TGF-β1, but causes less severe and progressive changes. The data suggest that TGF-β3 does not lead to inhibition of matrix degradation in the same way as TGF-β1, resulting in non-fibrotic tissue repair. Further, TGF-β3 is able to downregulate TGF-β1 induced gene expression, suggesting a regulatory role of TGF-β3. TGF-β3 overexpression results in an upregulation of Smad proteins similar to TGF-β1, but is less efficient in inducing the ALK 5 and TGF-β type II receptor (TβRII). We provide evidence that this difference may contribute to the progressive nature of TGF-β1 induced fibrotic response, in contrast to the limited fibrosis observed following TGF-β3 overexpression. TGF-β3 is important in “normal wound healing”, but is outbalanced by TGF-β1 in “fibrotic wound healing” in the lung.
Introduction
After lung injury, acute inflammation and tissue repair represent key mechanisms engaged to halt the injurious stimulus, remove infectious organisms, and initiate immediate repair to crucial membranes that function to provide gas exchange for survival. In most cases, this results in reconstitution of normal lung function. In chronic tissue injury with repeated episodes of inflammation, however, many of the control mechanisms involved in this otherwise well-orchestrated process are bypassed and injurious stimuli persevere. This leads to remodeling of lung tissue characterized by distorted matrix deposition, mesenchymal cell proliferation, and alteration of normal lung structure with compromised gas exchange, a process known as pulmonary fibrosis (PF) (Noble & Homer, 2004; Selman, King, & Pardo, 2001; Ward & Hunninghake, 1998).
Several growth factors are instrumental to the initiation as well as the termination of such inflammatory and remodeling processes, which have been studied extensively in the context of PF (Kelly, Kolb, Bonniaud, & Gauldie, 2003). It is likely that dysregulations in the balance of these growth factors play a major role in determining the differences between normal and pathologic tissue repair. Among these, transforming growth factor (TGF)-β is one of the key cytokines involved in the pathogenesis of PF (Leask & Abraham, 2004). TGF-β is the prototype of a group of polypeptide growth factors, which exert multiple effects in all cell types studied thus far (Letterio & Roberts, 1998). In mammals, three closely related isoforms with 64–85% amino acid sequence homology are described, which exhibit a substantial overlap in biological functions. Most publications investigating tissue fibrosis have focused on the most prominent isoform, TGF-β1, demonstrating an array of profibrotic functions. It is well established that TGF-β1 promotes differentiation of fibroblasts into activated myofibroblasts, enhances collagen synthesis, and reduces collagen degradation by downregulation of proteases and upregulation of protease inhibitors (Kelly et al., 2003). Only few studies have specifically addressed the role of TGF-β3 in the pathogenesis of pulmonary fibrosis. Moreover, the balance amongst the TGF-β1 and TGF-β3 isoforms is a neglected, but important component in the process of tissue repair. Some of these studies supported no major individual role, and claimed that TGF-β3 acts in concert with TGF-β1(Eickelberg et al., 1999; Khalil, Shing, & Whitman, 1993; Santana, Saxena, Noble, Gold, & Marshall, 1995). In contrast, data from targeted gene knockouts and experimental models of cutaneous wound healing and chronic inflammatory bowel disease suggested distinct features of TGF-β3, when compared with TGF-β1 (Ingman & Robertson, 2002; McKaig, Hughes, Tighe, & Mahida, 2002; Shah, Foreman, & Ferguson, 1995; Van Themsche, Mathieu, Parent, & Asselin, 2007). Fetal wounds, which contain primarily TGF-β3, heal without scars, whereas adult wounds, which contain mainly TGF-β1 and -β2, always exhibit some degree of scarring (Nath, LaRegina, Markham, Ksander, & Weeks, 1994). These observations have raised considerable research efforts in the field of cutaneous wound healing, as well as debates whether TGF-β3 might even carry anti-scarring properties (Shah et al., 1995).
Here, we report that TGF-β3 administered to rat lungs using transient overexpression initiates initial profibrotic effects similar to those elicited by TGF-β1, such as myofibroblast differentiation and increased ECM synthesis. However, TGF-β3 causes a lower intensity and duration of these effects and appears not to inhibit matrix degradation in the same way as TGF-β1. We also show that TGF-β3 overexpression results in an upregulation of Smad proteins similar to TGF-β1, but is less efficient in inducing expression of the ALK 5 and TGF-β type II receptor (TβRII). Examination of Smad-independent pathways shows that TGF-β1 treated tissues have lower levels of phosphorylated AKT compared to TGF-β3 treated animals, indicating a possible protective mechanism. We provide evidence that this difference may contribute to the progressive nature of TGF-β1 induced fibrotic response, which contrasts the limited fibrosis observed following TGF-β3 overexpression. We thus hypothesize that TGF-β3 is important in “normal wound healing”, but is outbalanced by TGF-β1 in “fibrotic wound healing”.
Methods
Recombinant adenovirus
For construction of AdTGFβ3 a mutated mouse TGF-β3 cDNA was used. Cys223, 225Ser mutations were generated using an overlap extension-method. Mutated and wild-type cDNAs were subcloned into a pSG5 vector and subsequently transfected into COS-1 cells. Supernatants were used for a PAI-1/luciferase assay: without heat activation, the mutated TGF-β3 cDNA produced biologically active growth factor several times more active than a wild-type control. With heat activation, no significant differences were found between mutated and wild-type samples (data not shown). The plasmid with the mutation was cloned into a p73 shuttle vector with a human cytomegalovirus (CMV) promoter and co-transfected with a virus-rescuing vector. Similarly, a replication deficient adenovirus carrying a mutated TGF-β1 gene was constructed as previously described (Sime, Xing, Graham, Csaky, & Gauldie, 1997). The resulting replication-deficient virus was amplified and purified by CsCl gradient centrifugation and PD-10 Sephadex chromatography, and plaque titred on 293 cells. The control vectors, AdDL, with no insert in the deleted E1 region were produced in the same way.
Cell Culture and Bioassay for TGF-β
A549 cells were infected with AdTGF-β1223/225, AdTGF-β3223/225 or AdDL70 at a multiplicity of infection (MOI) of 1, 10 and 100 PFU/cell similar to earlier studies. Bioactive TGF-β was detected by an established bioassay using Mink lung epithelial cells (MLEC clone 32, kindly provided by D. Rifkin, New York, NY), with a stable transfection of an 800-bp fragment of the 5′ end of the human plasminogen activator inhibitor 1 (PAI-I) gene fused to the firefly luciferase reporter gene. D-(−)-Luciferin and the firefly luciferase standard (Boehringer, Mannheim, Germany) were used and assayed by luminometer (Lumat LB 9501; Berthold Systems, Pittsburgh, PA). Data are presented in relative light units (RLU).
Animal treatment
Female Sprague Dawley rats (200–250 g bodyweight) received AdTGF-β1 (5 × 108 plaque forming units/ pfu), AdTGF-β3 or AdDL (both 1 × 109 pfu) in a volume of 50 µl phosphate-buffered saline (PBS) by intratracheal administration during a small surgical procedure. Rats were sacrificed at day 3, 7, 14, 28, and 60 after adenoviral administration by abdominal aorta bleeding. In another set of experiments we administered both AdTGF-β1 and AdTGF-β3 concomitantly or each vector combined with AdDL to investigate for potential antifibrotic properties of TGF-β3 in this experimental setting. Bronchoalveolar lavage (BAL) was then performed as described previously. For histological examination, lungs were inflated and fixed by intratracheal instillation of 10% neutral buffered formalin at a constant pressure of 20 cm of water for 5 minutes. For RNA after washing with PBS, the lung was removed and frozen immediately in liquid nitrogen. Frozen tissue samples were ground and stored at −70 °C until further processing. Rodent laboratory food and water were provided ad libitum. The animals were treated in accordance with the guidelines of the Canadian Council of Animal Care. All animal procedures were performed under inhalation anaesthesia with isofluorane (MTC Pharma, Cambridge, ON, Canada).
Determination of cytokine levels in BAL fluid
Bioactive TGF-β1 and TGF-β3 were detected in BALF using the MLEC bioassay as described above. A human TGF-β1 ELISA (R&D Systems, Minneapolis, Minnesota, USA) was used to determine TGF-β1, according to the manufacturer’s protocol.
RNA isolation and RT–PCR
RNA (1 µg) was DNase treated and then reverse transcribed (Invitrogen). Real-time quantitative polymerase chain reaction (PCR) analysis (TaqMan, Applied Biosystems, Foster City, CA) was carried out using an ABI Prism 7700 Sequence Detector. Negative control samples (no template or no reverse transcriptase) were run concurrently. Results were normalized to β-2 microglobulin. Data were normalized to control treatment (AdDL) or naïve animals. The sequences of primers and probes are shown in Table 1.
Table 1.
Taqman primers and probes
| Forward Primer | Reverse Primer | Probe | |
|---|---|---|---|
| TIMP-1 | GAACCGCAGCGAGGAGTTT | GGCAGTGATGTGCAAATTTCC | TCATCGCGGGCCGTTTAAGGAAT |
| TGF-β1 | AAACGGAAGCGCATCGAA | GGGACTGGCGAGCCTTAGTT | CCATCCGTGGCCAGATCCTGTCC |
| β2-microglobulin | CCGATGTATATGCTTGCAGAGTTAA | CCAGATGATTCAGAGCTCCATAGA | CACGTCACTCTGAAGGA |
Western blot analysis
Frozen lung tissues were homogenized in liquid nitrogen and proteins isolated in lysis buffer (20mM Tris pH 7.5, 150mM NaCl, 1mM EDTA, 1mM EGTA, 1%Triton-X100, 2.5mM sodium pyrophosphate and 1mM β-glycerophosphate). The protease inhibitor cocktail CompleteTM at a dilution of 1:25 (Roche Molecular Biochemicals, Indianapolis, IN) and the phosphatase inhibitor Na3VO4 dilution 1:100 were added to the lysis buffer immediately prior to isolation. Protein concentrations in tissue lysates were determined using the Quick Start Bradford Protein Assay (BIO-RAD) and FusionTM Spectrophotometer (Packard). Equal amounts of protein (50µg) from each sample were then separated on 10% polyacrylamide SDS-PAGE gels and transferred to 0.2 µm nitrocellulose membranes (BIO-RAD). Membranes were blocked with 5% nonfat dry milk in PBST (PBS + 0.1% Tween 20) for 1h at room temperature and incubated overnight at 4°C with antibodies listed in Table 2, followed by incubation with HRP-conjugated secondary antibodies at room temperature for 1h. Antibody binding was detected by SuperSignal WestPico chemiluminescent substrate (Pierce Biotechnology, Inc. Rockford, IL, USA).
Table 2.
List of antibodies and dilutions
| Antibody | Company | Dilution |
|---|---|---|
| CDK4 - rabbit polyclonal | Santa Cruz, CA, USA | 1:1000 |
| TGF-βI - rabbit polyclonal | Santa Cruz, CA, USA | 1:500 |
| TGF-βRII - rabbit polyclonal | Santa Cruz, CA, USA | 1.500 |
| P-SMAD2 rabbit monoclonal | Cell Signaling Technology MA, USA | 1:1000 |
| P-SMAD3-rabbit polyclonal | Cell Signaling Technology MA, USA | 1:500 |
| P-AKT - rabbit monoclonal | Cell Signaling Technology MA, USA | 1:1000 |
| AKT - rabbit polyclonal | Cell Signaling Technology MA, USA | 1:1000 |
| P-p44/42 MAPK – mouse polyclonal | Cell Signaling Technology MA, USA | 1:1000 |
| p44/42 MAPK – rabbit polyclonal | Cell Signaling Technology MA, USA | 1:1000 |
Histology
After fixation in 10% buffered formalin for 24 hours, lungs were paraffin embedded, sectioned, and stained with either hematoxylin & eosin, Masson’s trichrome or Picro Sirius Red. Four axial sections of the distal left lung were stained with Picro Sirius Red (PS Red) and 20 microscopically fields were examined for pulmonary fibrosis by three blinded investigators. Each field was individually assessed for the severity of interstitial fibrosis using Ashcroft’s semi quantitative grading previously described (Ashcroft, Simpson, & Timbrell, 1988). Immunohistochemistry was carried out using antibody to αSMA (M0851, Dako Canada Inc., Mississaugua, ON, Canada), phospho-Smad2 Cat no 3101 (Cell Signaling Technology, Inc, Danvers, MA, USA) or TGF beta Receptor I (V-22) Cat No sc-398 (Santa Cruz Biotechnology Inc, Santa Cruz, CA. USA)
Hydroxyproline Assay
Frozen lung samples were homogenized in 5 ml deionized water. The homogenate was diluted (x10) and one ml of this suspension was hydrolyzed in 2 ml 6 N HCl for 16 h at 110°C. Hydroxyproline content was determined by a colorimetric assay described previously (Woessner, 1961). The results were calculated as µg hydroxyproline per mg wet lung weight using hydroxyproline standards (Sigma Chemicals).
Statistical analysis
Data are shown as mean ± SEM. For evaluation of group differences, we used the 2way ANOVA with Bonferroni post-tests (GraphPad Prism® 4.0) and student’s t test. P values less than 0.05 were considered significant.
Results
Bioactivity of TGF-β1 and 3 following transient overexpression in cell culture and in rat lungs
The efficacy of the AdTGF-β3 construct to transfect cells in vitro and lungs was confirmed by Northern blot (Figure 1A). In vitro, AdTGF-β3 needed a higher number of virus particles per cell as quantified by multiplicity of infection to generate the same level of bioactive TGF-β in the PAI-1-luciferase cell assay (Figure 1B). This is probably related to vector specific issues (such as efficacy of gene transfer into the cells, utilization of the CMV promoter, length of inserted cDNA etc), and was the rationale for using 5×108 pfu AdTGF-β1 and comparing it to 1×109 pfu AdTGF-β3 and 1×109 pfu AdDL. These pfu of viral vector resulted in the same levels of bioactive TGF-β in the BALF of both AdTGF-β1 and 3 treated rats (Figure 1C). The bioactive transgenic proteins were undetectable in BALF after day 14 post transfection. There was no TGF-β1 detectable in AdTGF-β3 or AdDL treated animals as measured by ELISA (data not shown).
Figure 1.
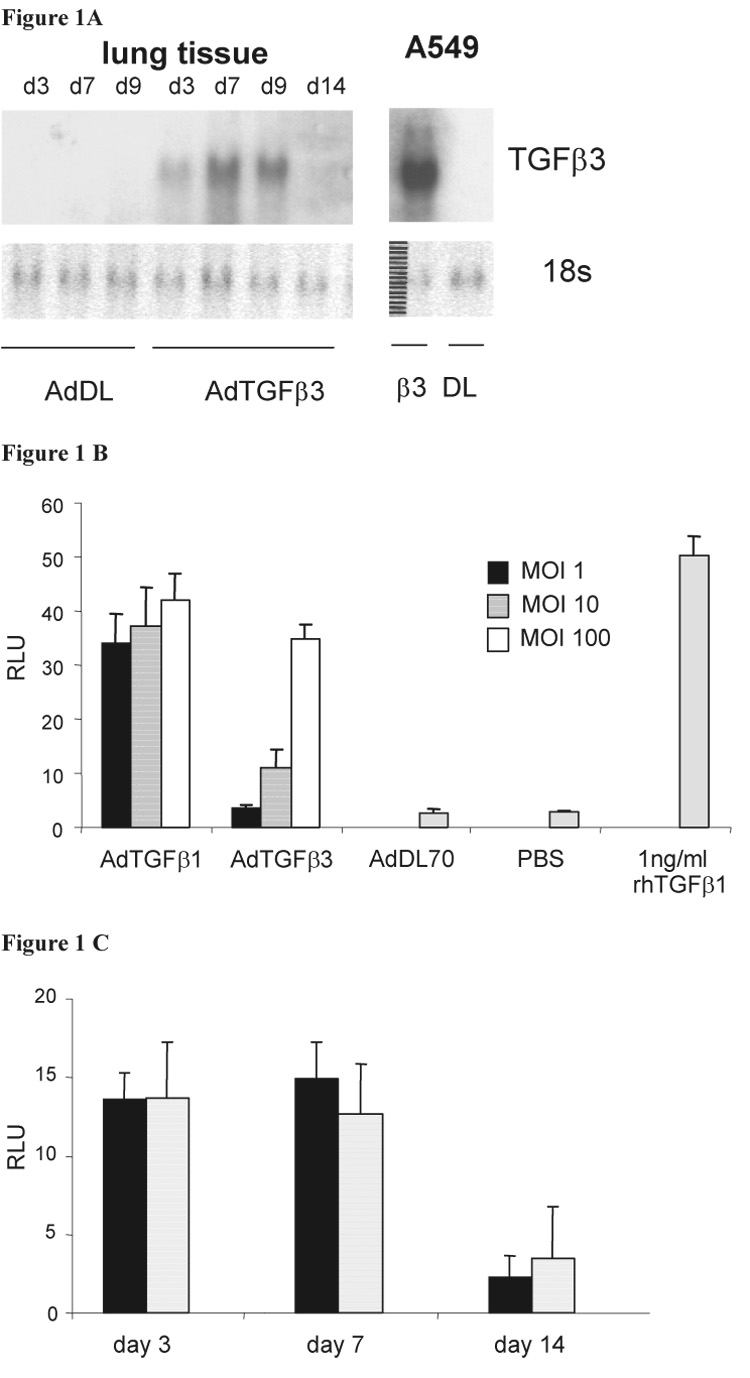
(A) Transgenic TGF-β3 mRNA is detectable by Northern Blot in cell culture (A549 cells) and rat lung tissue after exposure to AdTGFβ3 but not in control AdDL. (B) The bioactivity of transgenic proteins TGF-β1 and -β3 was demonstrated and compared to recombinant TGF-β by assaying supernatant of A459 cells (transfected with different concentration of adenovirus – MOI multiplicity of infection) using mink lung epithelial cells stably transfected with a PAI-1 responsive luciferase gene (RLU = relative light units). (C) Bioactivity of TGF-β1(solid) and -β3 (shaded bars) in BAL fluid of rats exposed to AdTGFβ1 and -β3.
Progressive lung fibrosis in response to TGF-β1 overexpression as opposed to partially reversible fibrosis following TGF-β3
Both AdTGF-β1 and 3 resulted in a similar mild inflammatory response with mainly mononuclear cells by day 7 (not shown). Fibroblast proliferation and accumulation of αSMA positive, spindle shaped cells as indication for myofibroblast differentiation was seen as early as 7 days post injection of adenoviral vectors. There was no obvious difference between AdTGF-β1 and 3. By day 14, collagen deposition was substantial and similar in lungs following overexpression of either TGF-β isoform (Figure 2 A–D). No fibroblast proliferation and collagen deposition was seen in control rat lungs (not shown). By day 28, histology showed severe fibrotic changes in the lungs of rats treated with AdTGF-β1 compared to AdTGF-β3 (Figure 2 E and F). Histological evidence of marked pulmonary fibrosis was still present in AdTGF-β1 exposed rats by day 60, but was almost absent in the AdTGF-β3 group (Figure 2 G and H).
Figure 2.
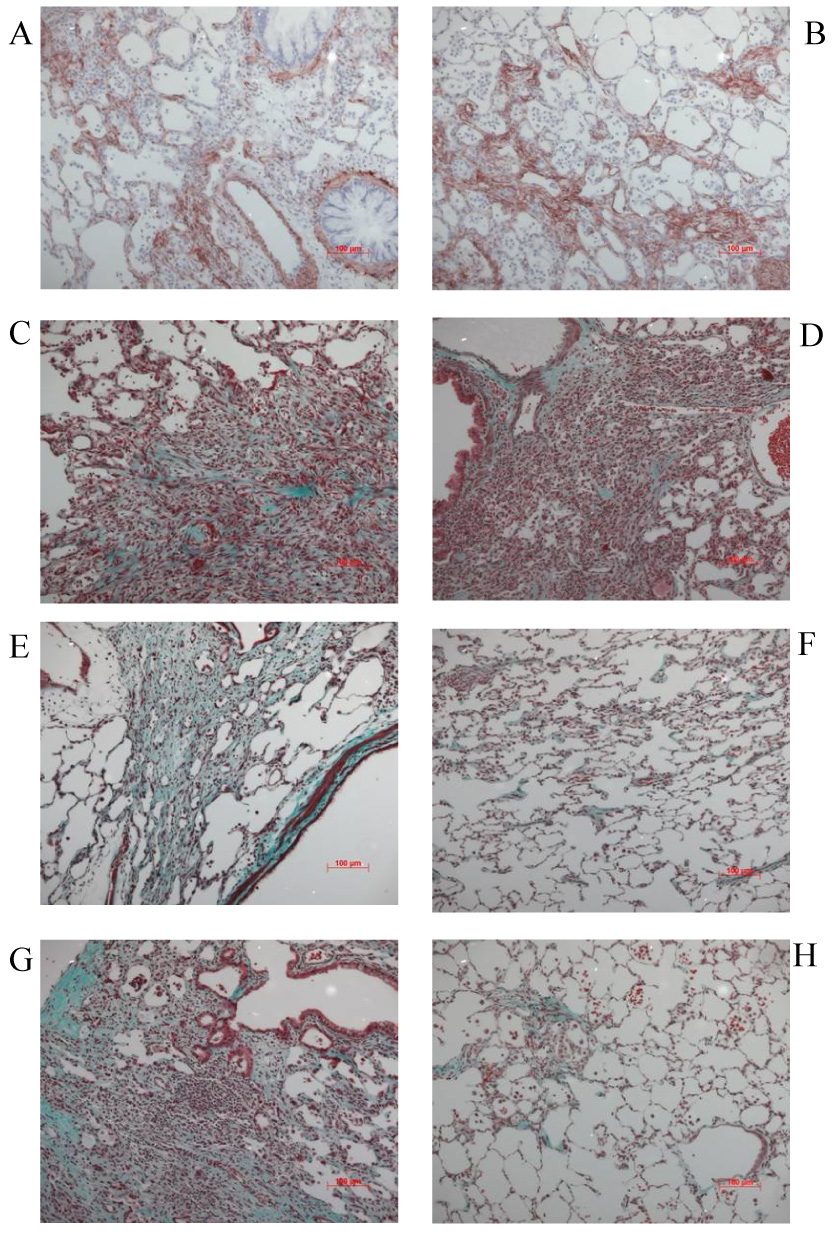
Histology of lungs exposed to AdTGFβ1 (left panel) and AdTGFβ3 (right panel) at different time points. Accumulation of αSMA positive, spindle shaped cells as indication for myofibroblast differentiation (A and B, immunohistochemistry for αSMA) and substantial collagen deposition (C and D, Masson’s trichrome) with no obvious difference between AdTGFβ1 and 3 by day 14. No fibroblast proliferation and collagen deposition was seen in control rat lungs (not shown). (E and F, Masson’s trichrome) By day 28, histology showed similar fibrotic changes in the lungs of rats treated with AdTGFβ1 compared to AdTGFβ3. (G and H, Masson’s trichrome) Marked pulmonary fibrosis was still present in AdTGFβ1 exposed rats by day 60, but was almost absent following AdTGFβ3.
Hydroxyproline analysis of lung homogenates as indication for collagen accumulation show an approximately 2 fold increase of hydroxyproline in AdTGF-β1 and 3 treated rats compared to controls by day 14 and 28 with no difference between the isoforms (p<0.001 vs. AdDL, Figure 3). By 60 days, the hydroxyproline concentration in AdTGF-β1 animals further increased to 5.2±0.3 µg/mg lung tissue, while it decreased to 3.0±0.3 µg/mg in AdTGF-β3 rats (p>0.001).
Figure 3.
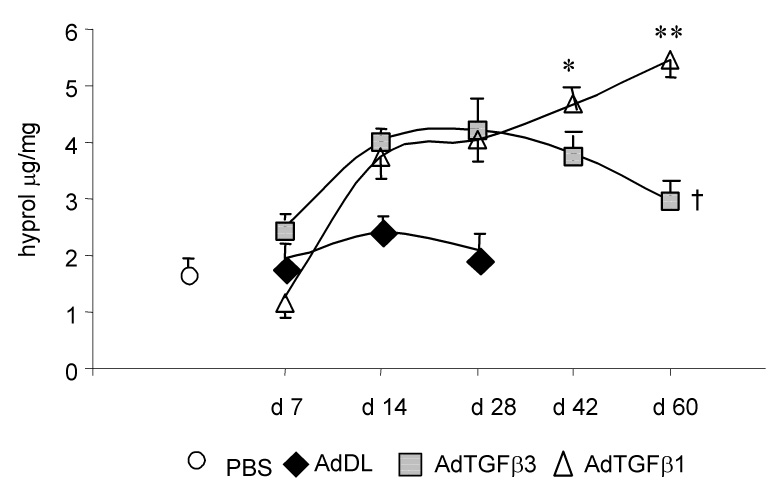
Lung collagen content as measured by hydroxyproline assay in rats exposed to AdTGFβ1, AdTGFβ3, empty vector AdDL and saline (PBS) over a period of 60 days (p < 0.001 for AdTGFβ1, AdTGFβ3, day 14, 28 and 42 vs AdDL; * p < 0.01 and ** p < 0.001 vs AdTGFβ3; † = not significant vs. AdDL day 7–28).
Induction of TGF-β type I and II receptor and non-Smad proteins differ between AdTGF-β1 and AdTGF-β3
Western blot was used to investigate Smad proteins, non-Smad signalling pathways and TGF-β receptors in whole lung tissue by 7 days. There was no difference in the level of phosphorylation of Smad2 and 3 between the isoforms (Figure 4A). Phospho-p44/42 and p44/42 was reduced in AdTGF-β1 and AdTGF-β3 treated animals compared to control (p<0.05, Figure 4B) with a trend to lower level in AdTGF-β1 versus AdTGF-β3. Similarly, phosphorylated AKT appeared to be reduced in animals receiving AdTGF-β1 compared to TGF-β3 and control (densitometry did not reach statistical significance, Figure 4B). The TGF-β receptor I (ALK-5) and TGF-β receptor II were induced 7 days after adenoviral gene transfer, and most importantly, AdTGF-β1 caused a significantly stronger induction of ALK-5 and TGF-β receptor II compared to AdTGF-β3 (Figure 4A). The activation of phospho-Smad 2 and expression of TGFβ RI in lung tissue was also assessed by immunohistochemistry. There was no difference in phospho-Smad 2 between AdTGF-β1 and AdTGF-β3, positive cells were mainly bronchial and alveolar epithelium. TGFβ RI immunostaining was stronger in AdTGF-β1 exposed alveolar epithelial cells (Figure 5).
Figure 4.
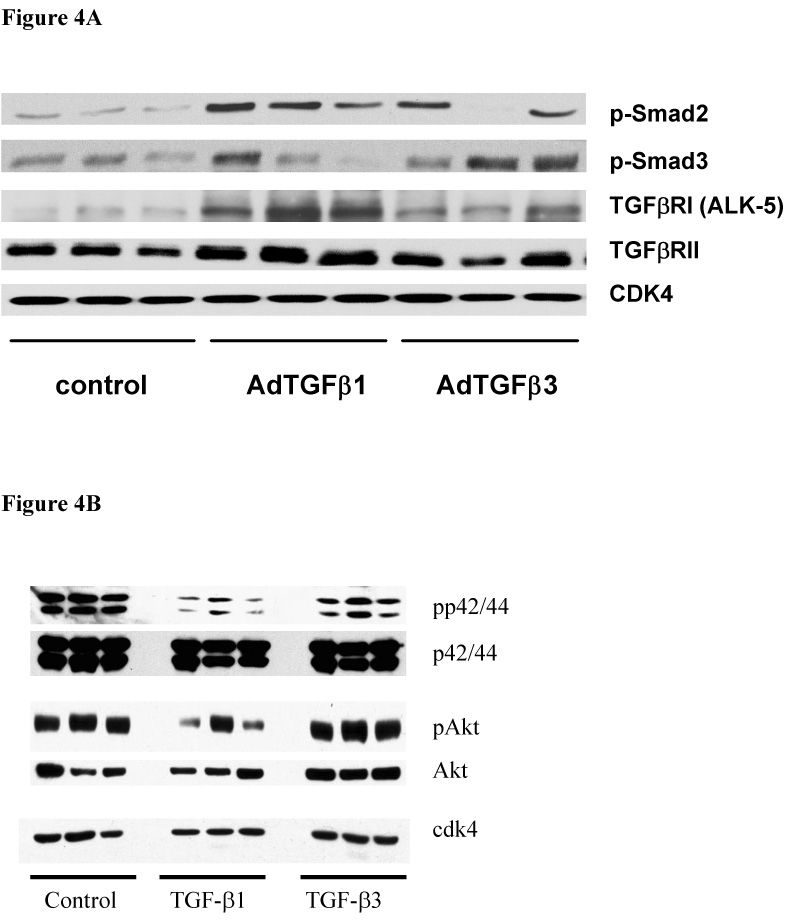
(A) Western Blot analysis confirmed this difference between AdTGF-β1 and -β3 seven days after administration of AdTGF-b1, b3 and AdDL by detection of more TGF-βRI (ALK-5) and TGF-βRII protein in rat lung homogenate (p<0.01 as quantified by band densitometry) (B) Smad-independent pathways were investigated by Western Blots and showed a trend to lower activation levels in the p42/44 and the Akt pathways in AdTGF-β1 treated lungs compared to controls and AdTGF-β3 (bands represent different animals, n=3).
Figure 5.
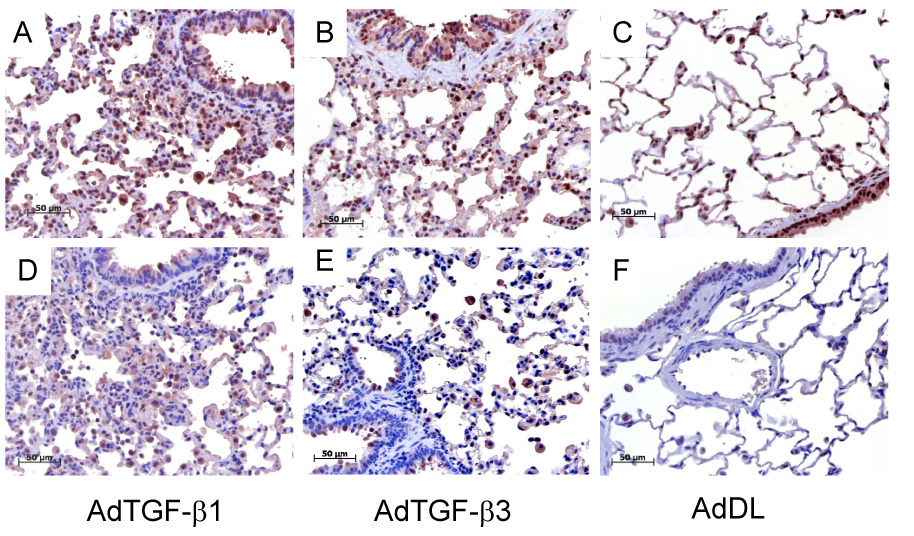
Immunohistochemistry showed no difference for phospho-Smad 2 between AdTGF-β1 and -β3 (A–B) but a stronger positivity for AdTGF-β1 exposed TGFβ RI in alveolar epithelial cells (D–E) at day 7 (C and F: control vector).
AdTGF-β3 reduces AdTGF-β1 induced expression of TGF-β responsive genes
Gene expression of TGF-β responsive genes was examined in lungs following concomitant administration of AdTGF-β1 plus AdTGF-β3 and compared to AdTGF-β1 or 3 plus AdDL. The expression levels of TIMP-1 and TGF-β were elevated in lungs that had received AdTGF-β1 plus AdDL at both day 14 and 28 compared to naïve lungs (Figure 6A and B). The addition of AdTGF-β3 to AdTGF-β1 prevented the increase of TIMP-1, TGFβ1, (Figure 6A and B) and other fibrogenic genes like Col1A2, Fibronectin-1 and CTGF by day 14 (not shown). By day 28, the AdTGF-β1 induced TIMP-1 and TGFβ1 gene expression was still suppressed when AdTGF-β3 was co-administered (Figure 6A and B). Despite this clear negatively regulating effect of AdTGFβ3 on AdTGFβ1 induced gene expression, we were not able to prove a substantial antifibrotic effect of TGF-β3 in this model (Figure 6C).
Figure 6.
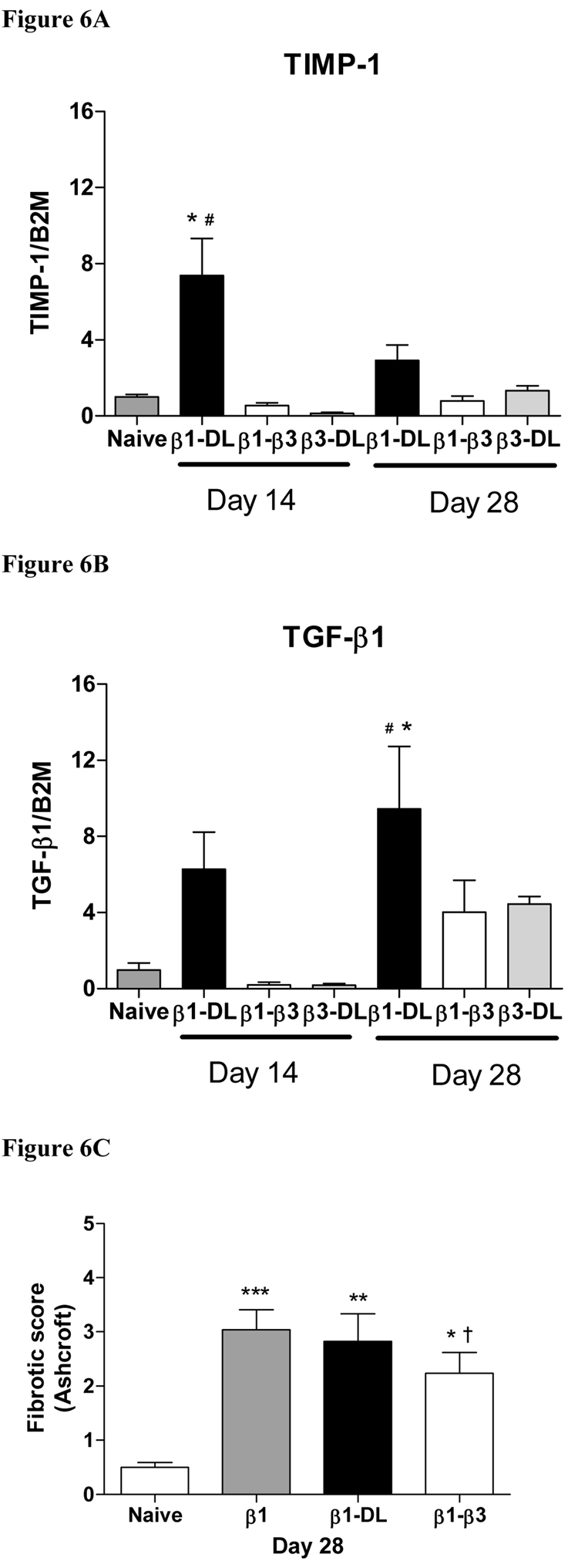
Gene expression of TGF-β1 and the protease inhibitor TIMP-1 in lungs of rats 14 and 28 days after exposure to AdTGF-β1 and AdTGF-β3 in combination or combined with AdDL (measured by real time RT-PCR). AdTGF-β1 induced a strong expression of both these genes when combined with AdDL. However, the gene induction following AdTGF-β1 when combined with AdTGF-β3 was significantly weaker at day 14 and 28 for TIMP-1 (A) and at day 14 for TGF-β1 (B) (* different from naïve p<0.05, and # different to AdTGF-β1 plus AdTGF-β3, day 14, ANOVA). C) Fibrotic score obtained on naïve animals on day 28, AdTGF-β1 (β1), AdTGF-β1 + AdDL (β1-Dl) and AdTGF-β1 + AdTGF-β3 (β1–β3). * p < 0.05, ** p < 0.01, *** vs. naïve † = not significant vs. AdTGF-β1 + AdDL, ANOVA).
Discussion
TGF-β is a key cytokine in the pathogenesis of fibroproliferative disorders of the lung, kidney, liver, or skin (Wynn, 2004). In mammals, three closely related TGF-β isoforms have been cloned and characterized, exhibiting 64–85% amino acid sequence homology and a substantial overlap in biological functions (Leask & Abraham, 2004). Most studies thus far have focussed on the most prominent isoform, TGF-β1, and demonstrated an array of profibrotic functions. Only a limited number of studies have specifically addressed the role of TGF-β3 in the pathogenesis of wound healing and fibrosis (Khalil, O'Connor, Flanders, & Unruh, 1996; Khalil et al., 2001; Santana et al., 1995; Shah et al., 1995). While some of these studies assigned no major individual role to TGF-β3, there is also evidence for distinct and specific features of TGF-β3 that even suggest potential anti-scarring properties. One of the key reports supporting this idea is the observation that cutaneous wounds heal without or only limited scarring, when TGF-β3 is present in the wound bed, either constitutively as found in the embryonic/fetal state or delivered as a therapeutic agent (Nath et al., 1994; Shah et al., 1995). Similarly, cutaneous wounds heal without or only limited scarring when TGF-β1 or - β2 is antagonized by antibodies (Shah, Foreman, & Ferguson, 1992). In contrast, there is marked scarring in the absence of TGF-β3 and presence of high levels of TGF-β1 and -β2 (Hosokawa, Nonaka, Morifuji, Shum, & Ohishi, 2003; Kohama, Nonaka, Hosokawa, Shum, & Ohishi, 2002; Shah et al., 1995). We thus sought to define the role of the TGF-β3 in the development of PF in comparison with TGF-β1, and analyze potential antifibrotic properties of TGF-β3 in this devastating disease.
In this study, we have employed adenoviral gene transfer to induce transient overexpression of TGF-β3 in the lungs of rats after intratracheal injection and compared this to overexpression of TGF-β1. The transgenic protein were routinely detectable in BALF for at least seven days, and the bioactivities of TGF-β1 and -β3 were not found to be different between the two isoforms (Figure 1). TGF-β3 mRNA was not found in lung tissue beyond day 14 in AdTGF-β3 treated animals similar to studies previously with AdTGF-β1. Thus, it is unlikely that differences in the duration of exogenous gene expression are a major contributing factor to the observations reported here (Warshamana et al., 2002). AdTGF-β3 induced a rapid response including hypertrophy of alveolar epithelial cells and appearance of myofibroblasts, as early as four days after instillation of the adenovirus. Of note, this response was devoid of a major inflammatory component as assessed by histological examination. By day 14, we observed substantial fibrosis and collagen accumulation similar to animals exposed to AdTGF-β1. However, in contrast to AdTGF-β1 and despite the comparable bioactivity, the fibroproliferative response in AdTGF-β3 treated rats was transient in nature, and partially resolved after 60 days. We have recently published similar findings after overexpression of CTGF (connective tissue growth factor), which caused marked but transient fibrosis in the lung (Bonniaud et al., 2003). At the level of gene expression, we noticed that the duration of TIMP-1 upregulation following AdTGF-β3 was transient and returned to baseline by day 14, while this gene was persistently induced in AdTGF-β1 treated rats. These findings strongly support previous reports showing that transient fibrosis in the lungs following CTGF overexpression or using bleomycin or AdTGF-β1 in fibrosis resistant mice were accompanied by low levels of TIMP-1 gene induction (Bonniaud et al., 2004; Kolb et al., 2002).
Several studies have examined the ability of TGF-β isoforms to stimulate lung fibroblasts in vitro, but available data comparing the fibrogenic potential of the three TGF-β isoforms are inconclusive. In human fibroblast, both isoforms were reported to equally induce collagen, TIMP-1 and MMP-1 while inhibiting TIMP-2 (Eickelberg et al., 1999). Others have shown a differential expression pattern of protease and antiprotease levels. TGF-β3 alone was not sufficient to induce TIMP-1 expression in intestinal myofibroblasts (McKaig, McWilliams, Watson, & Mahida, 2003) or human endometrial carcinoma cells (Van Themsche et al., 2007). This is in line with a previous report indicating that TGF-β1 is a more potent inductor of TIMP-1 compared to TGF-β3 in human lung fibroblasts, which is consistent with the findings of our study [see figure 8 in (Eickelberg et al., 1999)]. Further, exogenous TGF-β3, but not TGF-β1 increased MMP-9 expression and activity in cancer cells and a lip wound healing model (Hosokawa et al., 2003; Van Themsche et al., 2007), indicating a role in balancing protease/ antiprotease levels. However, there is also a study suggesting that TGF-β3 may be an even more potent inducer of procollagen and inhibitor of matrix degrading activity than TGF-β1 and -β2 (Coker et al., 1997).
In normal lung tissues, TGF-β1 and -β3 are expressed in a variety of cells, most profoundly in bronchial epithelium, alveolar macrophages, mesenchymal, and endothelial cells (Coker et al., 1996). In bleomycin fibrosis, TGF-β1 was shown to be upregulated with maximal expression after 10 days. TGF-β1 mRNA is mainly produced by alveolar macrophages, and to a lesser extent mesenchymal and type-II alveolar epithelial cells (Coker et al., 1997). In these studies, TGF-β3 mRNA was detectable in untreated mice at similar levels as TGF-β1, but in contrast to TGFβ1, bleomycin did not influence expression of TGF-β3. Earlier experiments investigating alveolar macrophages from bleomycin-treated rats have also shown that these cells secrete TGF-β1, but not TGF-β2 or -β3 (Khalil, Shing et al., 1993; Khalil, Whitman, Zuo, Danielpour, & Greenberg, 1993). Another group failed to show differences in the expression of TGF-β isoforms after bleomycin (Santana et al., 1995). So far, only two studies have analyzed TGF-β3 expression in fibrotic human lung tissue, and both supported a more prominent role for TGF-β1 compared with TGF-β3 (Coker et al., 2001; Khalil et al., 1996). More evidence supporting the hypothesis of TGF-β isoform imbalances in “fibrotic” versus “regular wound healing” can be found in other organ systems. The presence of TGF-β3 seems to promote scarless would closure in the fetus, and recombinant TGF-β3 reduces scar formation in adult skin wounds, while TGF-β1 causes increased scarring (Shah et al., 1995). Pre-irradiation of wound beds results in diminished expression of TGF-β3, suggesting that a relative deficiency of TGF-β3 might be responsible for impaired wound healing in irradiated tissue (Schultze-Mosgau et al., 2003). Some reports, however, have been unable to demonstrate differences between the effects of TGF-β isoforms in cutaneous wounds (Wu et al., 1997). Investigations on intestinal myofibroblasts from patients with chronic inflammatory bowel disease have shown that TGF-β3 may be protective (McKaig et al., 2002; McKaig et al., 2003). Intestinal myofibroblasts express mRNA of all three TGF-β isoforms, but secret mainly active TGF-β3, which enhances the migration of epithelial cells from the edges of wounds. Myofibroblasts derived from normal colon express mainly TGF-β3, those from patients with ulcerative colitis TGF-β1 and -β3 and are growth-inhibited by antibody-mediated depletion of TGF-β. In contrast, myofibroblasts from patients with Crohn’s disease express primarily TGF-β2 and almost no TGF-β3, and these cells are not growth-inhibited by antibodies to TGF-β (McKaig et al., 2002). In conclusion, these observations from other organ systems provide further support for the anti-scarring role of TGF-β3 in chronic inflammatory disease.
The hypothesis that the balance between TGF-β isoforms is an important component in wound healing was further addressed in this study by an experiment in which both TGF-Β1 and TGF-β3 were administered to rat lungs concomitantly. Addition of AdTGF-β3 blocked the AdTGF-β1 induced upregulation of TGF-β1, TIMP-1, fibronectin and CTGF by day 14, with sustained inhibition of TIMP-1 gene. This suggests a modulator role of TGF-β3 on TGF-β1 in this model. It is well established that TGF-β1 is capable of activating its own mRNA expression (Kim et al., 1990) which might explain the transient reduction of TGF-β1 responsive genes by 14 days, when TGF-β1 was still under “control” of TGF-β3. However, we were not able to demonstrate a substantial antifibrotic effect of TGF-β3 in the examined timeframe. This might be due to the complexity of the changes induced in the animal model and the fact that we are not able to accurately titrate the concentration and spatial distribution of transgenic TGF-β when using adenoviral gene transfer. We believe, however, that this data nevertheless strengthens the idea that a TGF-β isoform balance skewed towards higher amounts of TGF-β3 might be of beneficial effect regarding wound healing.
It may be difficult to imagine why TGF-β1 and -β3 exhibit such distinct features in vivo, considering that both are supposed to similarly bind to the type II TGF-β receptor and transduce signals to various intracellular pathways (Eickelberg, 2001; Leask & Abraham, 2004). Ligand binding studies are usually performed in vitro, and may vary widely depending on the cell line examined, and do not necessarily reflect the in vivo situation. Studies of TGF-β isoform knockouts strongly suggest distinct biological actions due to different phenotypes [Review in (Ingman & Robertson, 2002)]. TGF-β1 null mutations show yolk sac defects with early embryonic death and chronic inflammation leading to perinatal mortality. In contrast, TGF-β3 null animals develop better, but die within few weeks after birth because of failure of palate fusion and bronchial branching abnormalities.
Recent research has demonstrated a wide range of TGF-β receptor subtypes (Derynck & Zhang, 2003). The binding of TGF-β is complicated by modulation through additional proteins – betaglycan, endoglin, and a pseudoreceptor named BAMBI. They all can bind TGF-β and either present it to signal transducing receptors or prevent signalling by sequestering TGF-β. TGF-β signals through various intracellular pathways, the profibrotic mainly through Smad proteins. Recent evidence showed that non Smad signalling proteins, such as members of the activating protein-1 (AP-1 and JunD) and mitogen-activated protein kinase (MAPK) are also important for fibrogenesis (Eickelberg, 2001). In this study we analyzed expression of Smad proteins and found similarly increased phosphorylation of Smad2 and 3 upon overexpression of TGF-β1 and -β3 as expected. In contrast, the TGF-β type I receptor ALK 5 and type II receptor were more strongly induced following AdTGF-β1 expression. In line with the data presented in this paper, it has already been suggested that an increased TGFβ RI/RII ratio could promote fibrosis (Bock et al., 2005). The observation that TGF-β isoforms affect receptors differently, but not phosphorylation of Smad proteins suggests that non-Smad signalling pathways may be involved in determining the final fibrotic response to TGF-β. Further analysis of Smad-independent pathways indicated reduced phosphorylation of AKT in AdTGF-β1 treated animals compared to AdTGF-β3. This pathway has been shown to be involved in the fibrotic response in other models (Martinez-Salgado, Fuentes-Calvo, Garcia-Cenador, Santos, & Lopez-Novoa, 2006), and a relative downregulation of AKT might promote fibrotic tissue repair. Similarly, we observed some isoform related differences in the levels of phospho-p44/42, which is part of another signalling cascade with cross talk with the Smad pathway (Martinez-Salgado et al., 2006). We acknowledge that the differences in induction and phosphorylation of non-Smad signalling proteins are subtle, but the observation nevertheless suggests that TGF-β isoforms 1 and 3 might affect tissue repair in a different manner due to influences on non-Smad signalling pathways and TGF-β receptors. Our hypothesis for future studies is that the presence of a non-Smad dependent mechanism in TGF-β1 signalling causes upregulation of TGF-β receptors, which is not manifest in TGF-β3 treated tissues. The progressive nature of TGF-β1-induced pulmonary fibrosis may be dependent on this regulatory loop rather than on differences in the Smad pathway.
In summary, we demonstrated that overexpression of TGF-β3 in rat lungs causes a transient and non-progressive fibrogenic response, which is strikingly different from TGF-β1. The progressive fibrogenesis in the lungs of AdTGF-β1-treated animals is associated with a prolonged upregulation of the TIMP-1 gene, which may result in reduced matrix degradation. We believe that these findings together with evidence from human tissue analysis and more extensive experimental work with skin wounds and chronic inflammatory bowel disease support our hypothesis that the balance between TGF-β isoforms is an important component in the progression of fibrotic wound healing. We further provide mechanistic explanations for this biological difference between TGF-β1 and -β3 by showing that TGF-β1 induces a stronger expression of the TGF-β type I receptor ALK 5 and the TGF-β type II receptor compared to TGF-β3, both on gene and protein levels. This suggests that the progressive fibroproliferation in is determined by the TGF-β type II and ALK 5 receptor, and less by changes in the Smad signalling pathway.
Acknowledgements
The authors are grateful for the invaluable technical help of Jane Ann Smith, Mary Bruni, Fuqin Duan, Tom Galt, Bozena Nejman and Daniela Farkas. The research was funded by the Canadian Institute for Health Research and NHLBI Program Project Grant H L60231. P.J. M. has a Clinician Scientist Award through CIHR; M.K. is a Parker B Francis Fellow and supported by a career award from McMaster University, Department of Medicine.
Non-standard Abbreviations
- aSMA
alpha smooth muscle actin
- BALF
bronchoalveolar lavage fluid
- CTGF
connective tissue growth factor
- ECM
Extracellular Matrix
- MMP
matrix metalloproteinase
- PF
Pulmonary Fibrosis
- RLU
relative light units
- TβRII
TGF-β type II receptor
- TIMP
tissue inhibitor of matrix metalloproteinase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ashcroft T, Simpson JM, Timbrell V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J Clin Pathol. 1988;41(4):467–470. doi: 10.1136/jcp.41.4.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock O, Yu H, Zitron S, Bayat A, Ferguson MW, Mrowietz U. Studies of transforming growth factors beta 1–3 and their receptors I and II in fibroblast of keloids and hypertrophic scars. Acta Derm Venereol. 2005;85(3):216–220. doi: 10.1080/00015550410025453. [DOI] [PubMed] [Google Scholar]
- Bonniaud P, Kolb M, Galt T, Robertson J, Robbins C, Stampfli M, et al. Smad3 null mice develop airspace enlargement and are resistant to TGF-beta-mediated pulmonary fibrosis. J Immunol. 2004;173(3):2099–2108. doi: 10.4049/jimmunol.173.3.2099. [DOI] [PubMed] [Google Scholar]
- Bonniaud P, Margetts PJ, Kolb M, Haberberger T, Kelly M, Robertson J, et al. Adenoviral gene transfer of connective tissue growth factor in the lung induces transient fibrosis. Am J Respir Crit Care Med. 2003;168(7):770–778. doi: 10.1164/rccm.200210-1254OC. [DOI] [PubMed] [Google Scholar]
- Coker RK, Laurent GJ, Jeffery PK, du Bois RM, Black CM, McAnulty RJ. Localisation of transforming growth factor beta1 and beta3 mRNA transcripts in normal and fibrotic human lung. Thorax. 2001;56(7):549–556. doi: 10.1136/thorax.56.7.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coker RK, Laurent GJ, Shahzeidi S, Hernandez-Rodriguez NA, Pantelidis P, du Bois RM, et al. Diverse cellular TGF-beta 1 and TGF-beta 3 gene expression in normal human and murine lung. Eur Respir J. 1996;9(12):2501–2507. doi: 10.1183/09031936.96.09122501. [DOI] [PubMed] [Google Scholar]
- Coker RK, Laurent GJ, Shahzeidi S, Lympany PA, du Bois RM, Jeffery PK, et al. Transforming growth factors-beta(1), -beta(2), and -beta(3) stimulate fibroblast procollagen production in vitro but are differentially expressed during bleomycin-induced lung fibrosis. American Journal of Pathology. 1997;150(3):981–991. [PMC free article] [PubMed] [Google Scholar]
- Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425(6958):577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- Eickelberg O. Endless healing: TGF-beta, SMADs, and fibrosis. FEBS Lett. 2001;506(1):11–14. doi: 10.1016/s0014-5793(01)02875-7. [DOI] [PubMed] [Google Scholar]
- Eickelberg O, Kohler E, Reichenberger F, Bertschin S, Woodtli T, Erne P, et al. Extracellular matrix deposition by primary human lung fibroblasts in response to TGF-beta1 and TGF-beta3. Am J Physiol. 1999;276(5 Pt 1):L814–L824. doi: 10.1152/ajplung.1999.276.5.L814. [DOI] [PubMed] [Google Scholar]
- Hosokawa R, Nonaka K, Morifuji M, Shum L, Ohishi M. TGF-beta 3 decreases type I collagen and scarring after labioplasty. J Dent Res. 2003;82(7):558–564. doi: 10.1177/154405910308200714. [DOI] [PubMed] [Google Scholar]
- Ingman WV, Robertson SA. Defining the actions of transforming growth factor beta in reproduction. Bioessays. 2002;24(10):904–914. doi: 10.1002/bies.10155. [DOI] [PubMed] [Google Scholar]
- Kelly M, Kolb M, Bonniaud P, Gauldie J. Re-evaluation of fibrogenic cytokines in lung fibrosis. Curr Pharm Des. 2003;9(1):39–49. doi: 10.2174/1381612033392341. [DOI] [PubMed] [Google Scholar]
- Khalil N, O'Connor RN, Flanders KC, Unruh H. TGF-beta 1, but not TGF-beta 2 or TGF-beta 3, is differentially present in epithelial cells of advanced pulmonary fibrosis: an immunohistochemical study. Am J Respir Cell Mol Biol. 1996;14(2):131–138. doi: 10.1165/ajrcmb.14.2.8630262. [DOI] [PubMed] [Google Scholar]
- Khalil N, Parekh TV, O'Connor R, Antman N, Kepron W, Yehaulaeshet T, et al. Regulation of the effects of TGF-beta 1 by activation of latent TGF-beta 1 and differential expression of TGF-beta receptors (T beta R-I and T beta R-II) in idiopathic pulmonary fibrosis. Thorax. 2001;56(12):907–915. doi: 10.1136/thorax.56.12.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil N, Shing W, Whitman C. Differential Secretion of Transforming Growth Factor-Beta(1–3) by Pulmonary Cells During Bleomycin-Induced Lung Injury. Am Rev Resp Dis. 1993;(147):A757. [Google Scholar]
- Khalil N, Whitman C, Zuo L, Danielpour D, Greenberg A. Regulation of alveolar macrophage transforming growth factor-beta secretion by corticosteroids in bleomycin-induced pulmonary inflammation in the rat. J Clin Invest. 1993;92(4):1812–1818. doi: 10.1172/JCI116771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Angel P, Lafyatis R, Hattori K, Kim KY, Sporn MB, et al. Autoinduction of transforming growth factor beta 1 is mediated by the AP-1 complex. Mol. Cell. Biol. 1990;10(4):1492–1497. doi: 10.1128/mcb.10.4.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohama K, Nonaka K, Hosokawa R, Shum L, Ohishi M. TGF-beta-3 promotes scarless repair of cleft lip in mouse fetuses. J Dent Res. 2002;81(10):688–694. doi: 10.1177/154405910208101007. [DOI] [PubMed] [Google Scholar]
- Kolb M, Bonniaud P, Galt T, Sime PJ, Kelly MM, Margetts PJ, et al. Differences in the fibrogenic response after transfer of active transforming growth factor-beta1 gene to lungs of "fibrosis-prone" and "fibrosis-resistant" mouse strains. Am J Respir Cell Mol Biol. 2002;27(2):141–150. doi: 10.1165/ajrcmb.27.2.4674. [DOI] [PubMed] [Google Scholar]
- Leask A, Abraham DJ. TGF-{beta} signaling and the fibrotic response. FASEB J. 2004;18(7):816–827. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- Letterio JJ, Roberts AB. Regulation of immune responses by TGF-beta. Annu Rev Immunol. 1998;16:137–161. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- Martinez-Salgado C, Fuentes-Calvo I, Garcia-Cenador B, Santos E, Lopez-Novoa JM. Involvement of H- and N-Ras isoforms in transforming growth factor-[beta]1-induced proliferation and in collagen and fibronectin synthesis. Experimental Cell Research. 2006;312(11):2093–2106. doi: 10.1016/j.yexcr.2006.03.008. [DOI] [PubMed] [Google Scholar]
- McKaig BC, Hughes K, Tighe PJ, Mahida YR. Differential expression of TGF-beta isoforms by normal and inflammatory bowel disease intestinal myofibroblasts. Am J Physiol Cell Physiol. 2002;282(1):C172–C182. doi: 10.1152/ajpcell.00048.2001. [DOI] [PubMed] [Google Scholar]
- McKaig BC, McWilliams D, Watson SA, Mahida YR. Expression and regulation of tissue inhibitor of metalloproteinase-1 and matrix metalloproteinases by intestinal myofibroblasts in inflammatory bowel disease. Am J Pathol. 2003;162(4):1355–1360. doi: 10.1016/S0002-9440(10)63931-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath RK, LaRegina M, Markham M, Ksander GA, Weeks PM. The expression of transforming growth factor type beta in fetal and adult rabbit skin wounds. J Pediatr Surg. 1994;29(3):416–421. doi: 10.1016/0022-3468(94)90582-7. [DOI] [PubMed] [Google Scholar]
- Noble PW, Homer RJ. Idiopathic pulmonary fibrosis: new insights into pathogenesis. Clin Chest Med. 2004;25(4):749–758. doi: 10.1016/j.ccm.2004.04.003. vii. [DOI] [PubMed] [Google Scholar]
- Santana A, Saxena B, Noble NA, Gold LI, Marshall BC. Increased expression of transforming growth factor beta isoforms (beta 1, beta 2, beta 3) in bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 1995;13(1):34–44. doi: 10.1165/ajrcmb.13.1.7541221. [DOI] [PubMed] [Google Scholar]
- Schultze-Mosgau S, Wehrhan F, Amann K, Radespiel-Troger M, Rodel F, Grabenbauer GG. In Vivo TGF-beta 3 expression during wound healing in irradiated tissue. An experimental study. Strahlenther Onkol. 2003;179(6):410–416. doi: 10.1007/s00066-003-1049-5. [DOI] [PubMed] [Google Scholar]
- Selman M, King TE, Jr, Pardo A. Idiopathic Pulmonary Fibrosis: Prevailing and Evolving Hypotheses about Its Pathogenesis and Implications for Therapy. Ann Intern Med. 2001;134(2):136–151. doi: 10.7326/0003-4819-134-2-200101160-00015. [DOI] [PubMed] [Google Scholar]
- Shah M, Foreman DM, Ferguson MW. Control of scarring in adult wounds by neutralising antibody to transforming growth factor beta. Lancet. 1992;339(8787):213–214. doi: 10.1016/0140-6736(92)90009-r. [DOI] [PubMed] [Google Scholar]
- Shah M, Foreman DM, Ferguson MW. Neutralisation of TGF-beta 1 and TGF-beta 2 or exogenous addition of TGF-beta 3 to cutaneous rat wounds reduces scarring. J Cell Sci. 1995;108(Pt 3):985–1002. doi: 10.1242/jcs.108.3.985. [DOI] [PubMed] [Google Scholar]
- Sime PJ, Xing Z, Graham FL, Csaky KG, Gauldie J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J Clin Invest. 1997;100(4):768–776. doi: 10.1172/JCI119590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Themsche C, Mathieu I, Parent S, Asselin E. Transforming Growth Factor-beta3 Increases the Invasiveness of Endometrial Carcinoma Cells through Phosphatidylinositol 3-Kinase-dependent Up-regulation of X-linked Inhibitor of Apoptosis and Protein Kinase C-dependent Induction of Matrix Metalloproteinase-9. J. Biol. Chem. 2007;282(7):4794–4802. doi: 10.1074/jbc.M608497200. [DOI] [PubMed] [Google Scholar]
- Ward PA, Hunninghake GW. Lung inflammation and fibrosis. Am J Respir Crit Care Med. 1998;157(4 Pt 2):S123–S129. doi: 10.1164/ajrccm.157.4.nhlbi-10. [DOI] [PubMed] [Google Scholar]
- Warshamana GS, Pociask DA, Fisher KJ, Liu J-Y, Sime PJ, Brody AR. Titration of non-replicating adenovirus as a vector for transducing active TGF-beta1 gene expression causing inflammation and fibrogenesis in the lungs of C57BL/6 mice. International Journal of Experimental Pathology. 2002;83(4):183–202. doi: 10.1046/j.1365-2613.2002.00229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woessner JF., Jr The determination of hydroxyproline in tissue and protein samples containing small proportions of this imino acid. Arch Biochem Biophys. 1961;93:440–447. doi: 10.1016/0003-9861(61)90291-0. [DOI] [PubMed] [Google Scholar]
- Wu L, Siddiqui A, Morris DE, Cox DA, Roth SI, Mustoe TA. Transforming growth factor beta 3 (TGF beta 3) accelerates wound healing without alteration of scar prominence. Histologic and competitive reverse-transcription-polymerase chain reaction studies. Arch Surg. 1997;132(7):753–760. doi: 10.1001/archsurg.1997.01430310067014. [DOI] [PubMed] [Google Scholar]
- Wynn TA. Fibrotic disease and the Th1/Th2 paradigm. Nature Reviews Immunology. 2004;4(8):583. doi: 10.1038/nri1412. [DOI] [PMC free article] [PubMed] [Google Scholar]