

Abstract
Dephosphorylated and activated glycogen synthase kinase (GSK) 3β hyperphophorylates β-catenin, leading to its ubiquitin-proteosome-mediated degradation. β-catenin-knockdown increases while β-catenin overexpression prevents neuronal death in vitro; in addition, protein levels of β-catenin are reduced in the brain of Alzheimer’s patients. However, whether β-catenin degradation is involved in stroke-induced brain injury is unknown. Here we studied activities of GSK3 β and β-catenin, and the protective effect of moderate hypothermia (30 °C) on these activities after focal ischemia in rats. The results of Western blot showed that GSK3 β was dephosphorylated at 5 and 24 hours after stroke in the normothermic (37 °C) brain; hypothermia augmented GSK3β dephosphorylation. Because hypothermia reduces infarction, these results contradict with previous studies showing that GSK3β dephosphorylation worsens neuronal death. Nevertheless, hypothermia blocked degradation of total GSK3β protein. Corresponding to GSK3β activity in normothermic rats, β-catenin phosphorylation transiently increased at 5 hours in both the ischemic penumbra and core, and the total protein level of β-catenin degraded after normothermic stroke. Hypothermia did not inhibit β-catenin phosphorylation, but it blocked β-catenin degradation in the ischemic penumbra. In conclusion, moderate hypothermia can stabilize β-catenin, which may contribute to the protective effect of moderate hypothermia.
Keywords: Focal ischemia, hypothermia, GSK-3β, β-catenin
1. Introduction
After stroke, abnormal activities of glycogen synthase kinase 3 β (GSK 3 β) and β-catenin, two molecules that are downstream of Akt and Wnt survival pathways, are implicated in ischemic injury. In the Akt pathway, activated-Akt phosphorylates and inactivates GSK 3 β, thereby stabilizing β-catenin. Activated GSK 3 β, on the other hand, phosphorylates β-catenin, resulting its proteosomal degradation [13]. Stabilized β-catenin translocates into the nuclei, activating gene expression that supports cell survival [9,11]. β-catenin has been shown to decrease in brains of patients with Alzheimer’s disease [5]. In addition, β-catenin knockdown results in apoptosis, whereas β-catenin overexpression prevents neuronal death in vitro [7]. Thus, we hypothesize that β-catenin degradation is also complicated in brain injury after stroke.
Mild (33–36°C) or moderate (28–32°C) hypothermia is one of the strongest neuroprotectants identified to date that protect against cerebral ischemia [1,3,14], but the exact mechanisms for its protective effects are not fully understood [15]. We have previously reported that intra-ischemic moderate hypothermia (30 °C) reduces infarct size in focal ischemia by regulating the Akt survival pathways [14]. Moderate hypothermia attenuates reductions in Akt activity after stroke [14] but does not attenuate reduction in GSK 3 β phosphorylation, suggesting that it may not inhibit GSK3β activity [14]. Since GSK3β activity is known to exacerbate ischemic injury, and its inhibition reduces infarction [2,6,8], it is puzzling that hypothermia can inhibit ischemic injury without blocking GSK 3 β activity. To address this paradox, we further examined the protective effects of moderate hypothermia on β-catenin phosphorylation and total protein level of β-catenin (molecules downstream of GSK3β), in the same focal ischemia model with rats we used before for studying the Akt/GSK3β pathway.
2. Results
We have previously reported that moderate hypothermia (30°C) reduced infarct size from 62.6 ± 3.3 (n=7) to 3.4 ± 4.4 % (n=7) (P<0.001) [14]. In the current study, representative infarcts from rats subjected to normothermic and hypothermic stroke under exact same conditions are shown (Fig. 1)
Fig.1.
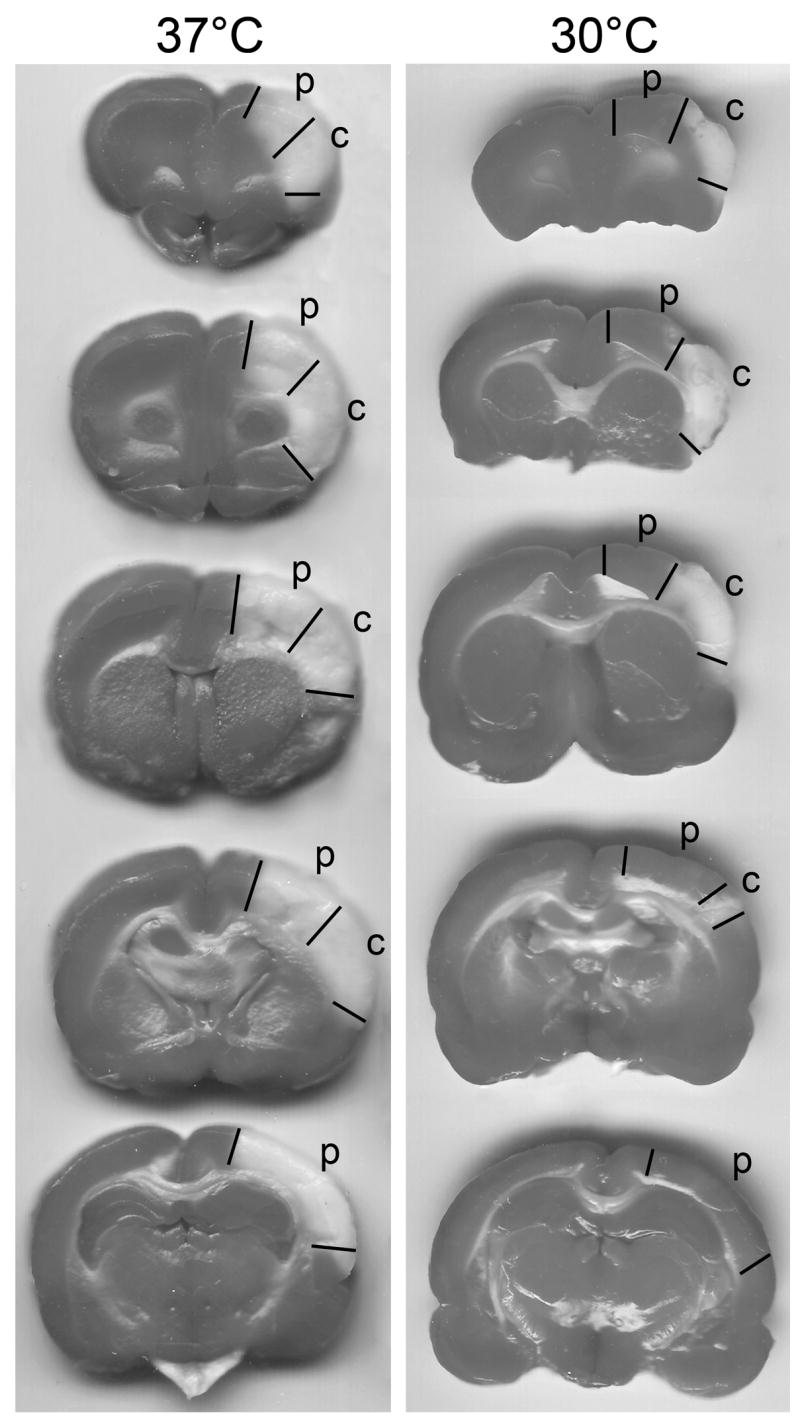
A. Representative infarcts by TTC staining from normothermic and hypothermic rats and tissues corresponding to the ischemic penumbra and core were dissected for Western blot. The rats were euthanized 2 days after stroke, and brains were cut into 5 blocks and stained with TTC. Hypothermia (30 °C) reduced infarct compared with normothermia (37 °C). Region P and C represent the ischemic penumbra and core, respectively. The penumbra is defined as the area spared by hypothermia.
Levels of phosphorylated GSK 3β decreased at 5 and 24h after stroke in the ischemic penumbra and core of normothermic rats; moderate hypothermia augmented such decrease at 5 and 48 h in both the penumbra and core (Fig. 2). In addition, total protein level of GSK3 β also decreased from 5 to 48h after stroke in both the penumbra and core; moderate hypothermia blocked such decreases (Fig.3).
Fig. 2.

The effect of hypothermia on GSK3β phosphorylation. A. Representative protein bands of phosphorylated GSK3β from Western blot. β-actin was probed to show equal loading of protein. Rats subjected to normothermic or hypothermic stroke were euthanized at 5, 24 and 48 h later; ischemic penumbra and core were dissected for Western blot. B. Bar graphs show optical densities of protein bands of phosphorylated GSK3 β. In the penumbra, * p=0.043, ** P=0.006, vs sham. † vs 48h con, p=0.004; in the core, # vs sham and 5h con , P<0.05; & vs 48h con , P<0.05. n=5/group.
Fig. 3.

Hypothermia blocked reduction in protein levels of total GSK3β. A. Representative protein bands of GSK3β and β-actin from Western blot. B. Statistical results of optical densities of GSK3 β. In the penumbra, * P<0.05, *** P<0.001, vs sham. Two-way ANOVA, 30 °C vs 37 °C, p<0.001;+ vs 48h, p=0.021, ++ vs 24h, p<0.001. In the core, two-way ANOVA, P<0.001, con vs 30 °C;& vs 24h, p=0.003. ### vs sham, P<0.001. n=5/group.
β-catenin phosphorylation was increased at 5h in normothermic brain after stroke, then it returned to near normal levels at 24 and 48h; it increased from 5 to 48 h in hypothermic brains after stroke (Fig.4). Nevertheless, protein levels of total β-catenin decreased in normothermic brains from 5 to 48 h after stroke; moderate hypothermia blocks its degradation (Fig.5).
Fig. 4.

Hypothermia augmented phosphorylation of β-catenin after stroke. A. Representative protein bands of phosphorylated β-catenin. B. Optical densities of protein bands of phosphorylated β-catenin. In the penumbra, * p<0.05, ** P<0.01, *** p<0.001, all vs sham, + vs 24h 30 °C. Two-way ANOVA, overall, hypothermia increased β-catenin phosphorylation in the penumbra. In the core, ### vs sham, p<0.001, & vs con, p=0.009. n=5/group.
Fig. 5.

Hypothermia blocks β-catenin degradation. A. A representative Western blot indicates that the amount of β-catenin decreased after stroke in normothermic rats and hypothermic blocked such decreases. B. Bar graphs represent optical densities of β-catenin. In the penumbra, * vs con 5, 24, 48 h, respectively, P<0.001. Two-way ANOVA, Fisher LSD. n=5/group.
3. Discussion
We found for the first time that β-catenin phosphorylation was transiently increased, and total protein of β-catenin degraded after stroke in both the penumbra and core of the normothermic brain; moderate hypothermia increased β-catenin phosphorylation at 24 h to 48 h after stroke, and blocked degradation of total β-catenin in the penumbra.
We have demonstrated in this study that hypothermia prevented stroke-induced β-catenin degradation in the ischemic penumbra but not in the core; this action might contribute to the protective effect of hypothermia. β-catenin is not only a transcription factor in the Akt pathway [9], but is also associated with multiple adhesion molecules such as α-catenin, p-catenin and cadherin to form cell-cell adhesion, which is fundamental for constructing and maintaining multicellular organisms [10]. In the brain, β-catenin plays a critical role in the formation of synapses by associating with the cell adhesion proteins [12]. Therefore, β-catenin degradation after stroke may down-regulate pro-survival gene expression activated by β-catenin and dismantle cell-cell adhesion and connection, thus aggravating ischemic injury. Indeed, β-catenin degradation has been shown to increase while its overexpression blocks apoptosis in vitro [7], and protein levels of β-catenin decrease in patient brains with Alzheimer’s disease [4] [5]. β-catenin degradation may also play critical roles in pathological processes after stroke. We found in this study that β-catenin phosphorylation transiently increased at 5h in the normothermic brain, and this hyper-phosphorylation was accompanied by a later degradation of total β-catenin. This correlation suggests that β-catenin phosphorylation progresses to degradation [9,11]. Furthermore, β-catenin might be phosphorylated by GSK3 β activity, as levels of phosphorylated GSK3 β transiently decreased from 5h to 24h, indicating increased-activity during this period after stroke.
We have previously reported that hypothermia blocks ischemic injury without inhibiting increased-GSK3β activity [14]; this result contradicts with the earlier results showing that GSK3β activity leads to neuronal death [2,6,8]. In our prior study, however, hypothermia blocks nuclear translocation of β-catenin [14], suggesting that hypothermia may block molecular signaling downstream of GSK3β. The current study further supports this conclusion by showing that hypothermia could stop the degradation of β-catenin. Nevertheless, it is yet not clear how hypothermia retarded β-catenin’s degradation. As we have discussed, in the penumbra of normothermic brain, transient increases in β-catenin phosphorylation at 5h appear to correlate with β-catenin degradation at later time points, which is consistent with the notion that β-catenin phosphorylation results its degradation [9,11]. However, in the penumbra of hypothermic brains, phosphorylated β-catenin accumulated from 5 to 48 h after stroke while total β-catenin was not degraded, which contradicts the aforementioned hypothesis. A possible reason is that hypothermia blocks transportation of phosphorylated β-catenin into the proteosome and thus stops β-catenin degradation. Nevertheless, in the ischemic core where infarction was not attenuated by hypothermia, β-catenin degradation was not blocked by hypothermia either, suggesting that β-catenin degradation correlates with ischemic injury.
Transient reduction in GSK3β phosphorylation after stroke in normothermic brains appears to contradict the result of our previous study [14], in which GSK3 β phosphorylation persistently decreased. This inconsistency might be due to sample collection. In the previous study, the sample was dissected from a region lateral to the midline of the brain; by contrast, in the current study, the sample was collected from a more dorsal region. Nevertheless, hypothermia showed a similar effect by reducing additional GSK3β phosphorylation.
Intra-ischemic hypothermia has long term effects on multiple cell signaling pathways after stroke. As we recently reviewed [16], intra-ischemic hypothermia not only blocks excitatory neurotransmitter, glutamate release and inhibits intracellular Calcium increase during stroke, it also improves ATP recovery after reperfusion, inhibits free radical generation, inflammatory response and blood–brain barrier permeability. In addition, intra-ischemic hypothermia has multiple effects on necrotic and apoptotic pathways. As consistent with previous investigations, here we provide further evidence that intra-ischemic hypothermia enhances β-catenin phosphorylation and protein levels from 5 to 48 h after stroke.
In conclusion, we demonstrated that β-catenin degraded after stroke in normothermic rats, whereas moderate hypothermia inhibited its degradation. Inhibiting β-catenin degradation might contribute to the protective effect of moderate hypothermia.
4. Experimental Procedure
Experimental protocols were approved by the Administrative Panel on Laboratory Animal Care of Stanford University.
Focal Cerebral Ischemia and moderate hypothermia
Focal ischemia was generated as described previously [14]. Anesthesia was induced by 5% isoflurane and maintained with 2–3% isoflurane during surgery in male Sprague-Dawley rats (350–390g). Core body temperatures were monitored with a rectal probe and were maintained at 37°C throughout the experiment. The distal middle cerebral artery (MCA) was exposed and cauterized permanently above the rhinal fissure. The bilateral common carotid arteries (CCA) were occluded for 1h. Moderate hypothermia (30 °C) was induced by spraying 100% alcohol on the rat body. Body temperature was adjusted to 30 ± 0.5 °C (at 10 min before ischemia onset and maintained for 1h during CCA occlusion; temperature was controlled by a heating pad underneath the rat combined with an overhead light over the rat, as described previously [18]. After CCA release, the wound was sutured and body temperature was increased to 37 ± 0.5°C [14].
Western blots
To determine how hypothermia affects protein levels of phosphorylated and total protein of GSK 3-β and of β-catenin, brain tissues from three groups were prepared: 1) Sham surgery without ischemia; 2) normothermic ischemia (control ischemia), 3) hypothermic ischemia. In groups 2 and 3, brains were harvested at 5, 24 and 48 hours after stroke onset, and tissue corresponding to the ischemic penumbra was dissected for Western blot. The ischemic penumbra was defined as the tissue saved by hypothermia 2 days post-stroke, and the corresponding region from the control ischemic brain was dissected for comparison. Whole cell protein was extracted from the fresh brain tissue, and Western blot was performed as described with modification [14,17,18]. In each lane 20 μg protein was subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using 4–15% Ready Gel (BIO-RAD Laboratories, Cat# L050505A2) for 1.5 hours. Protein bands were transferred from the gel to Hybond-LFP membranes (GE Healthcare, USA) for 1 hour.
To determine phosphorylated GSK 3 β or total protein levels of GSK 3 β and phosphorylated β-catenin, primary antibodies of rabbit anti-phosphorylated GSK3 β (Ser9) (1:1000, Cat# 9336, Cell signaling, MA), rabbit anti-GSK3 β (1:1000, Cat# 9332, Cell signaling, MA), and rabbit anti-phosphorylated β-catenin (1:1000, Cat# 9561, Cell signaling, MA) were incubated overnight and followed by a horseradish peroxidase-conjugated anti-rabbit secondary antibody (1:2000 for GSK 3 β, 1:1000 for the other two primary antibodies, Cell Signaling Technology, MA) for 1 hour, incubated with ECL plus solution (GE Healthcare) for 5 min. The membranes were scanned using Typhoon trio (GE Health, Sunnyvale, CA).
To determine β-catenin and β-actin, the membranes were incubated overnight at 4°C with gentle agitation in a mixture of primary antibodies consisting of anti-β-catenin (1:1000, Upstate, NY, cat. 05-482) and β-actin (1:20000, rabbit, Bethyl, cat. A300-491A), then incubated with secondary antibodies of Alexa Fluor®647 donkey anti-mouse (1:1500, cat. A31573) for detecting β-catenin, and Alexa Fluor®488 donkey anti-rabbit (1:5000, cat. A21206 ) for detecting β-actin, for 1h at room temperature and shielded from light. Protein bands of β-catenin and β-actin were scanned simultaneously by the Typhoon trio.
Statistics
For Western blot, two-way ANOVA was used to analyze on various protein bands between hypothermia and control ischemia; one-way ANOVA was used to compare protein bands after ischemia with sham; tests were followed by Fisher LSD post-hoc test. All were considered statistically significant for p values ≤ 0.05. Data are presented as means ± SEM.
Acknowledgments
The authors wish to thank Elizabeth Hoyte for preparing the figures, and Felicia Beppu for manuscript editing. This study was supported by AHA National Scientist Development Grant 0730113N (HZ), NINDS grants R01 NS27292 (GKS) and P01 NS37520 (GKS and RMS).
Abbreviations
- GSK 3 β
glycogen synthase kinase 3 β
- MCA
middle cerebral artery
- CCA
common carotid arteries
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature References
- 1.Busto R, Dietrich WD, Globus MY, Valdes I, Scheinberg P, Ginsberg MD. Small differences in intraischemic brain temperature critically determine the extent of ischemic neuronal injury. J Cereb Blood Flow Metab. 1987;7:729–738. doi: 10.1038/jcbfm.1987.127. [DOI] [PubMed] [Google Scholar]
- 2.Cimarosti H, Zamin LL, Frozza R, Nassif M, Horn AP, Tavares A, Netto CA, Salbego C. Estradiol protects against oxygen and glucose deprivation in rat hippocampal organotypic cultures and activates Akt and inactivates GSK-3beta. Neurochem Res. 2005;30:191–199. doi: 10.1007/s11064-004-2441-y. [DOI] [PubMed] [Google Scholar]
- 3.Colbourne F, Corbett D, Zhao Z, Yang J, Buchan AM. Prolonged but delayed postischemic hypothermia: a long-term outcome study in the rat middle cerebral artery occlusion model. J Cereb Blood Flow Metab. 2000;20:1702–1708. doi: 10.1097/00004647-200012000-00009. [DOI] [PubMed] [Google Scholar]
- 4.De Ferrari GV, Inestrosa NC. Wnt signaling function in Alzheimer’s disease. Brain Res Brain Res Rev. 2000;33:1–12. doi: 10.1016/s0165-0173(00)00021-7. [DOI] [PubMed] [Google Scholar]
- 5.Fuentealba RA, Farias G, Scheu J, Bronfman M, Marzolo MP, Inestrosa NC. Signal transduction during amyloid-beta-peptide neurotoxicity: role in Alzheimer disease. Brain Res Brain Res Rev. 2004;47:275–289. doi: 10.1016/j.brainresrev.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 6.Kelly S, Zhao H, Hua Sun G, Cheng D, Qiao Y, Luo J, Martin K, Steinberg GK, Harrison SD, Yenari MA. Glycogen synthase kinase 3beta inhibitor Chir025 reduces neuronal death resulting from oxygen-glucose deprivation, glutamate excitotoxicity, and cerebral ischemia. Exp Neurol. 2004;188:378–386. doi: 10.1016/j.expneurol.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 7.Li HL, Wang HH, Liu SJ, Deng YQ, Zhang YJ, Tian Q, Wang XC, Chen XQ, Yang Y, Zhang JY, Wang Q, Xu H, Liao FF, Wang JZ. Phosphorylation of tau antagonizes apoptosis by stabilizing beta-catenin, a mechanism involved in Alzheimer’s neurodegeneration. Proc Natl Acad Sci U S A. 2007;104:3591–3596. doi: 10.1073/pnas.0609303104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martinez A, Castro A, Dorronsoro I, Alonso M. Glycogen synthase kinase 3 (GSK-3) inhibitors as new promising drugs for diabetes, neurodegeneration, cancer, and inflammation. Med Res Rev. 2002;22:373–384. doi: 10.1002/med.10011. [DOI] [PubMed] [Google Scholar]
- 9.Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science. 2004;303:1483–1487. doi: 10.1126/science.1094291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perez-Moreno M, Fuchs E. Catenins: keeping cells from getting their signals crossed. Dev Cell. 2006;11:601–612. doi: 10.1016/j.devcel.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rasola A, Fassetta M, De Bacco F, D’Alessandro L, Gramaglia D, Di Renzo MF, Comoglio PM. A positive feedback loop between hepatocyte growth factor receptor and beta-catenin sustains colorectal cancer cell invasive growth. Oncogene. 2007;26:1078–1087. doi: 10.1038/sj.onc.1209859. [DOI] [PubMed] [Google Scholar]
- 12.Takeichi M, Abe K. Synaptic contact dynamics controlled by cadherin and catenins. Trends Cell Biol. 2005;15:216–221. doi: 10.1016/j.tcb.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 13.Zhao H, Sapolsky RM, Steinberg GK. PI3K/Akt survival signal pathways are implicated in neuronal survival after stroke. Mol Neurobiol. 2006 doi: 10.1385/MN:34:3:249. in press. [DOI] [PubMed] [Google Scholar]
- 14.Zhao H, Shimohata T, Wang JQ, Sun G, Schaal DW, Sapolsky RM, Steinberg GK. Akt contributes to neuroprotection by hypothermia against cerebral ischemia in rats. J Neurosci. 2005;25:9794–9806. doi: 10.1523/JNEUROSCI.3163-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao H, Steinberg GK, Sapolsky RM. General versus specific actions of mild-moderate hypothermia in attenuating cerebral ischemic damage. J Cereb Blood Flow Metab. 2007 doi: 10.1038/sj.jcbfm.9600540. [DOI] [PubMed] [Google Scholar]
- 16.Zhao H, Steinberg GK, Sapolsky RM. General versus specific actions of mild-moderate hypothermia in attenuating cerebral ischemic damage. J Cereb Blood Flow Metab. 2007;27:1879–1894. doi: 10.1038/sj.jcbfm.9600540. [DOI] [PubMed] [Google Scholar]
- 17.Zhao H, Yenari MA, Cheng D, Barreto-Chang OL, Sapolsky RM, Steinberg GK. Bcl-2 transfection via herpes simplex virus blocks apoptosis inducing factor translocation after focal ischemia in rat. J Cereb Blood Flow Metab. 2004 doi: 10.1097/01.WCB.0000127161.89708.A5. in press. [DOI] [PubMed] [Google Scholar]
- 18.Zhao H, Yenari MA, Cheng D, Sapolsky RM, Steinberg GK. Biphasic cytochrome c release after transient global ischemia and its inhibition by hypothermia. J Cereb Blood Flow Metab. 2005;25:1119–1129. doi: 10.1038/sj.jcbfm.9600111. [DOI] [PubMed] [Google Scholar]