

Abstract
A non-integrating mutant, SIVsmD116N, was derived from the infectious pathogenic SIVsmE543-3 clone by introducing an Asp (D) to Asn (N) mutation into the catalytic domain of integrase. Although SIVsmD116N generated all viral proteins following transfection, cell free virus did not productively infect CEM×174 cells, macaque peripheral blood mononuclear cells (PBMCs) or monocyte-derived macrophages (MDM). Viral DNA and transcripts were observed transiently in SIVsmD116N-infected CEM×174 cells and macaque PBMC but persisted in MDM for as long as 20 days. Circular forms of viral DNA were detected but there was no evidence of integration detected by Alu PCR. We found that SIV D116N mutant remained transcriptionally active and expressed low levels of viral proteins persistently in MDM. These data are consistent with a role for macrophages as a persistent latent reservoir for AIDS viruses. The capacity of non-integrating SIV to persistently generate viral products in macrophages suggests that non-integrating lentiviral vectors could be engineered to efficiently and safely express proteins for vaccine purposes.
Keywords: Simian immunodeficiency virus, Non-integrating mutant, Transcription, Macrophage
Introduction
As retroviruses, human immunodeficiency virus (HIV) and simian immunodeficiency virus (SIV) must accomplish sequential events to achieve successful infection. Following viral entry and reverse transcription, the newly synthesized viral DNA integrates into the host chromatin (Bukrinsky et al., 1992). Integration is mandatory for viral production, but HIV/SIV infection in vivo results in high levels of non-integrated DNA (Wu, 2004). Prior work has demonstrated transient expression of both early and late message by non-integrated HIV DNA in CD4 T cells (Stevenson et al., 1990; Wiskerchen and Muesing, 1995). The transcription from non-integrated DNA is a normal and early step in HIV replication. Non-integrated DNA has the capacity to synthesize all classes of viral transcripts, early, multiply-spliced, late, singly-spliced, and non-spliced transcripts, prior to integration (Wu and Marsh, 2003a). The capacity of pre-integration HIV DNA to transcribe is of interest, since about half of the reverse transcribed DNA from HIV infection does not integrate into the cellular DNA. Previous report shows that about 45% of HIV DNA is not integrated (Zennou et al., 2000), but the biological relevance and possible pathogenic role of non-integrated viral DNA is not clear (Wu and Marsh, 2003b). The non-integrated viral DNA has been shown to carry transcriptional activity in human CD4 T cells and it can direct limited syntheses of viral early proteins such as Nef. It has been demonstrated that Nef synthesized from non-integrated DNA can promote T cell activity and down regulate CD4 molecules (Gillim-Ross, Cara, and Klotman, 2005b). These observations prompted us to evaluate SIV viral gene expression in non-dividing macaque macrophages infected with a non-integrated mutant SIV virus. In this study, we constructed a non-integrated mutant, SIVsmD116N (D116N), and evaluated its ability to replicate, transcribe and express viral proteins following infection of macrophages in vitro.
Results
Non-integrating SIV mutant and reverse transcriptase assay for its infectivity
In previous research, HIV-1IN/D116N mutant was derived by introducing an Asp (D) to Asn (N) mutation within the integrase D(35)E motif. This single point mutation has been shown to completely abolish viral DNA integration of HIV without affecting other known viral functions such as reverse transcription and nuclear targeting (Engelman et al., 1995). In this study, we constructed a non-integrating mutant of SIV, using the infectious molecular clone, SIVsmE543-3 as a parent. SIVsmE543-3, derived from a biological isolate obtained late in disease from an immunodeficient rhesus macaque (E543) with SIV-induced encephalitis (Hirsch et al., 1997) replicated efficiently in macaque peripheral blood mononuclear cells (PBMCs) and monocyte-derived macrophages (MDM) in vitro and induced AIDS when inoculated intravenously into macaques (Dehghani et al., 2003; Hirsch et al., 1997; Hirsch et al., 2004). The non-integrating mutant, SIVsmD116N, was derived from SIVsmE543-3 by a “G” to “A” substitution at site 4877, resulting in an Asp (D) to Asn (N) substitution into the integrase catalytic domain. The wild type (WT) and mutant plasmids were transfected to 293T cells and produced comparable levels of cell free reverse transcriptase activity (data not shown). Western blot analysis of cell free virus supernatants revealed a similar pattern of viral proteins (Fig. 1). This suggests that the SIVsmD116N mutation had no effect on viral protein synthesis, processing and assembly. Infectivity of the WT and SIVsmD116N mutant was evaluated in CEM×174 cells, macaque PBMC and macaque MDM using equivalent virus inoculum size based on RT activity. As expected, the WT virus replicated well in CEM×174 cells, macaque PBMCs as well as in MDM (Fig. 2 A, B, and C). In contrast, no RT activity was observed in cell-free media following infection with the integrase mutant SIVsmD116N in any of these cell types (Fig. 2). This was consistent with previous findings demonstrating an essential role for integration in HIV/SIV infection (Englund et al., 1995).
Figure 1.
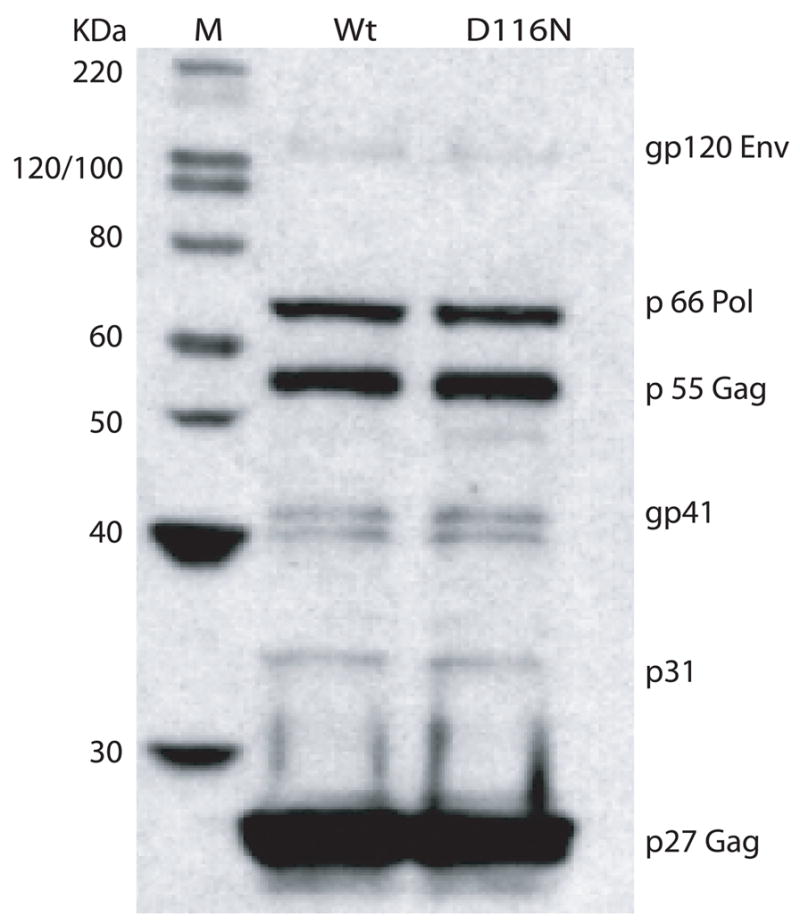
SIVsmD116N virus can generate all the viral proteins at the same level as WT SIVsmE543. The SIVsmD116N and WT SIVsmE543 clones were transfected into 293T cells to generate cell free viruses. Viruses were concentrated and western blotting was performed to analyze the viral proteins. The proteins shown here are gp120, p66, p55, gp41, gp31, p27 (from top to bottom).
Figure 2.
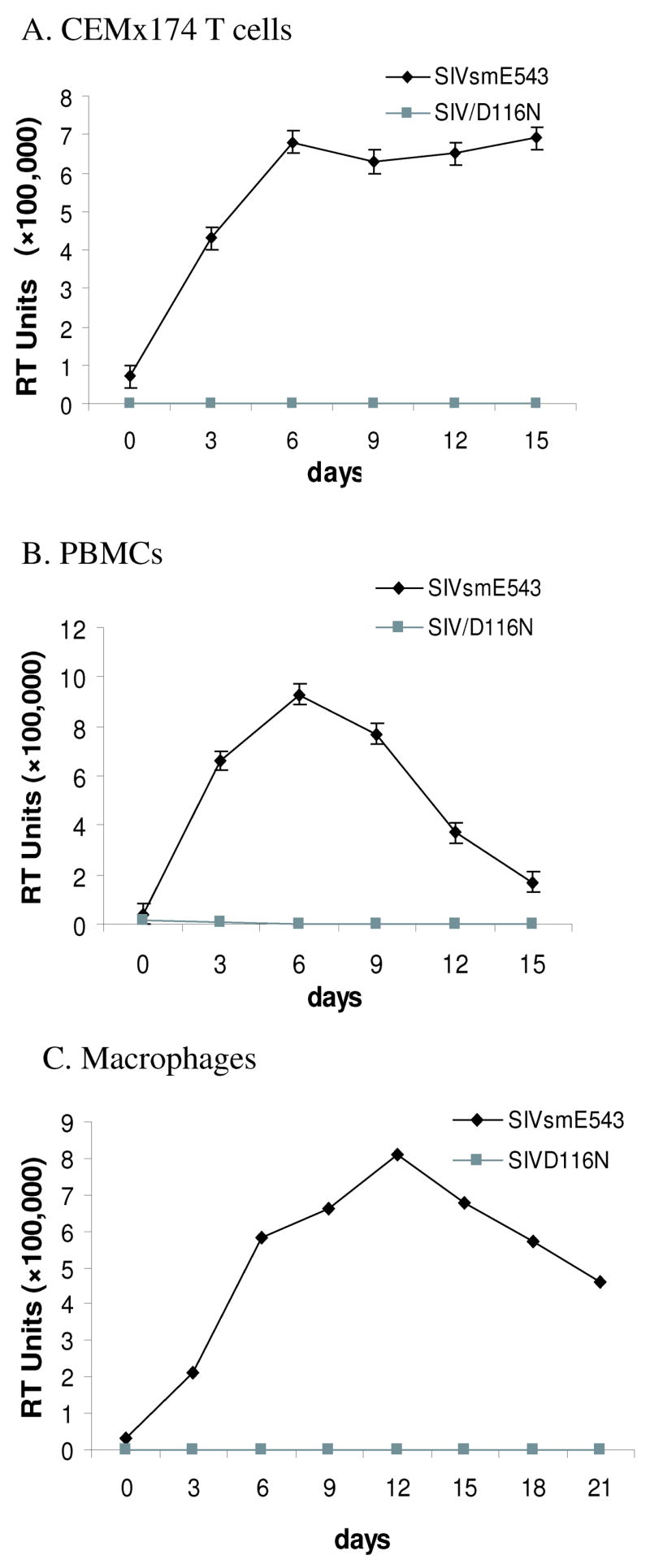
Non-integrated mutant virus SIVsmD116N does not replicate in host cells. Reverse transcriptase assay was carried out to evaluate infectivity of the WT and SIVsmD116N mutant in CEM×174 cells (2A), macaque PBMC (2B) and macaque MDM (2C). The WT virus replicated well in CEM×174 cells, macaque PBMCs as well as in MDM. In contrast, no RT activity was observed in cell-free media following infection with the integrase mutant SIVsmD116N in any of these cell types.
Persistence of non-integrated viral DNA in macrophages
Since previous studies of HIVD116N demonstrated persistence and expression in resting T cells, we chose to focus on macrophages, another non-dividing target of HIV/SIV. PCR was used to investigate whether SIVsmD116N was capable of entering and replicating in monocyte derived macrophages (MDM) in vitro. Total cellular DNA from WT and SIVsmD116N infected macrophages was isolated at different times post infection and PCR amplified with primers specific for SIV late DNA, 1-LTR-circles and 2-LTR-circles DNA. Production of SIV full length DNA was indicated by the detection of a LTR product using SIV/DNA-5 and SIV/DNA-3 primers (see Table 1). Circular viral DNA forms were also detected using SIV/Circle-5′ primer and SIV/Circle-3′ primer. Based on detection of full length viral DNA, the SIVsmD116N mutant was able to synthesize viral DNA in MDM with equal efficiency to the WT virus. Viral DNA synthesized from SIVsmD116N persisted for 20 days (the termination of culture) at a level comparable with the WT virus (Fig. 3A). In addition, 1-LTR-circles and 2-LTR circles were found to be present and persisted in both WT and SIVsmD116N infections, and 1-LTR circles were much more abundant than 2-LTR circles. Although this assay was not quantitative, the SIVsmD116N infected cells appeared to have significantly higher levels of 2-LTR-circles than the WT infected cells (Fig. 3B). This result is consistent with previous research in HIV-1 non-integrating mutant (Wiskerchen and Muesing, 1995). To confirm that the PCR products came from SIV viral DNA rather than residual plasmid DNA, we used cells infected with heat-inactivated (56°C for 60 min) as a control. As shown in Fig. 3C, no products were detected in PCR reactions for SIV DNA or LTR circles from cells infected with heat-inactivated virus at 2 days post-infection, whereas these products were readily detected in the untreated virus infection. These results indicated that the viral DNA detected by PCR came from replicating virus rather than residual plasmid DNA from the transfection.
Table 1.
Sequences of Used Primers
| Primer | Sequence | Position* |
|---|---|---|
| Subclone-1 | 5′-GGTATTACCTCAAGGGTGGAAGGG-3′ | 3295–3318 |
| Subclone-2 | 5′-TCGGCAGGTGATTTACTGCTG-3′ | 5918–5938 |
| Mutagenic-1 | 5′-CCATCACACATCTGCATACTAATAATGGTGCCAATTTCACAT-3′ | |
| Mutagenic-2 | 5′-ATGTGAAATTGGCACCATTATTAGTATGCAGATGT GTGATGG-3′ | |
| SIV/DNA-5′ | 5′-CCGAACAGGGACTTGAAGG AGGTGA-3′ | 708–731 |
| SIV/DNA-3′ | 5′-TTTGTTCCTGCCGCCCTTACTG-3′ | 894–915 |
| SIV/Circle-5′ | 5′-GCAAGTGTGTGTTCCCATCTCTCC-3′ | 9310–9331 |
| SIV/Circle-3′ | 5′-AGCTCTCCGTCGTGGTTTGTTC-3′ | 909–930 |
| Alu1-1 | 5′-TAGTCGGGAGGCTGAGGCAGGAGAA-3′ | 172–196 (human Alu) |
| Alu1-2 | 5′-GTCATCCCACTGATGAGTCTGTGC-3′ | 207– 230 (LTR U3) |
| Alu2-1 | 5′-TGGAAGGGATTTATTACAATGAGAAAAGAC-3′ | 1–30 |
| Alu2-2 | 5′-CTCGTCTTCCTGGGCTTCATCT-3′ | 164–185 |
| SIV/5′ | 5′-CCGAACAGGGACTTGAAGGAGGTGA-3′ | 842–866 |
| SIV/nef-tat-rev | 5′-CTCTGGAGCACTGGTTGGAGGATCTG-3′ | 9011–9036 |
| SIV/3′ env | 5′-GCAAGAGCGCGATAAGCA GCTGATTC-3′ | 6616–6641 |
| SIV/3′ vif | 5′-CTGCTGCAAGTCCACCATGCCC ATC-3′ | 5485–5509 |
| SIV/3′ gagpol | 5′-CTGAACCTGTCGGAACTAATGGAGC-3′ | 1257–1281 |
They are referred to positions in SIVsmE543 sequence except Alu1-1 and mutagenic primers.
Figure 3.

Persistence of unintegrated viral DNA in macrophages. A. Total cellular DNA from WT and D116N infected cells was extracted at different times post infection and PCR amplified. The non-integrated DNA persisted for 20 days and the viral DNA synthesized from SIVsmD116N was at a comparable level with the WT virus. B. Two of the non-integrated viral DNA forms, both the 1-LTR-circle and 2-LTR-circle were found to be present and persist with viral infection. 1-LTR circles were much more abundant than 2-LTR circles. The D116N infected cells had higher levels of 2-LTR-circles than the wild type infection. Cellular actin DNA was co-amplified from each sample to ensure equal amount of cellular DNA was used. C. Uninfected cells and heat-inactivated virus infected cells were used as control to compare with WT or D116N infected cells.
SIVsmD116N did not integrate into genomic DNA in either T cells or macrophages
To determine whether SIVsmD116N was capable of integration into the host genome, we used an Alu sequence-based PCR strategy (Zhuge et al., 2001). The Alu sequence is a ubiquitous repeat element found in the human/monkey genome but absent in the HIV/SIV genomes which can be used to specifically demonstrate whether viral DNA is integrated into the host chromosome. As expected, DNA was amplified from as few as 103 CEM×174 cells infected with SIVsmE543-3, consistent with the presence of proviral DNA in these cells (185 bp product). In contrast, no product was detected in up to 2.5×104 CEM×174 T cells infected with SIVsmD116N virus (Fig. 4A). Similarly, no integrated viral DNA was observed in SIVsmD116N infected macrophages, while in WT infected macrophages, integrated viral DNA persisted until the termination of the cell culture at 20 days (Fig. 4B). These results suggested that SIVsmD116N is not capable of integration into genomic DNA in either T cells or macrophages.
Figure 4.
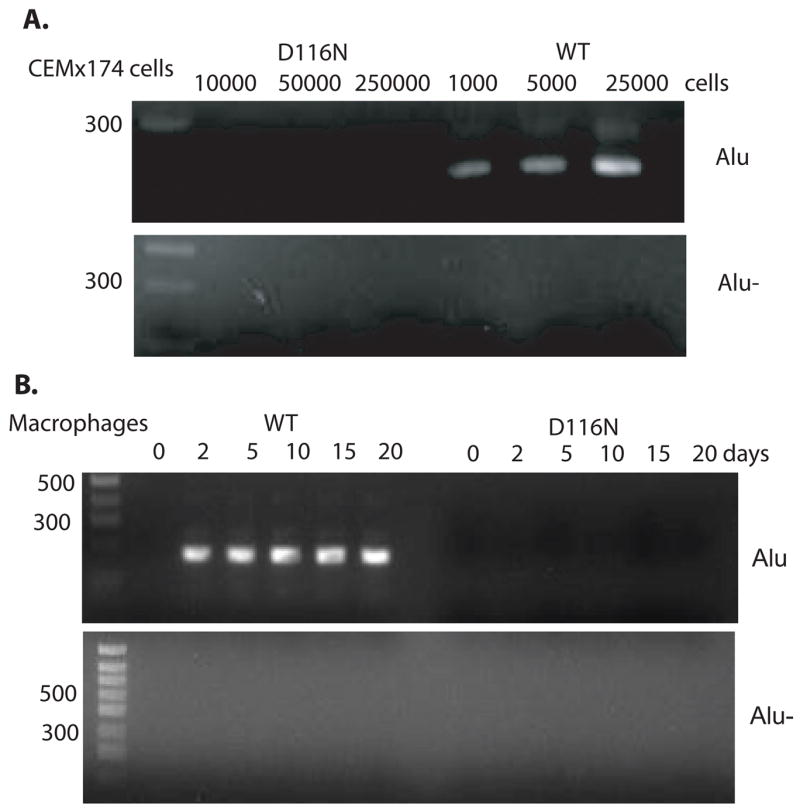
The SIVsmD116N mutant does not integrate into cell chromosome. CEM×174 cells and macaque macrophages were infected with SIVsmE543 and SIVsmD116N viruses. Total cellular DNA harvested was subjected to Alu-PCR amplification. A. No integrated viral DNA was amplified in up to 25×104 CEM×174 T cells infected with integrase mutant SIVsmD116N, while integrated DNA was amplified well in just 1×103 CEM×174 T cells infected with SIVsmE543. B. In D116N infected macrophages, Alu-PCR demonstrated that no integrated viral DNA persisted for 20 days until the termination of the cell culture. While in WT infected macrophages, integrated DNA amplified well from day 1 to day 20. For the “Alu-” controls without first round PCR, no product was amplified.
Persistence of transcriptional activity from non-integrated viral DNA in macrophages
To determine transcriptional activity of non-integrated viral DNA in macrophages, total cellular RNA was isolated and amplified by semi-quantitative RT-PCR at different times post infection (Wu and Marsh, 2001). Briefly, reverse transcription of cellular RNA was accomplished with random decamers as the first-strand primers. Following cDNA synthesis, PCR was carried out using primers to detect the specific messages of SIV using the primers detailed in Table 1. Primer combinations used the same forward primer, SIV/5′ and four different reverse primers, SIV/nef-tat-rev, SIV/3′env, SIV/3′vif and SIV/3′gag-pol to detect multiply, singly and unspliced messages. For relative quantification of RT-PCR, cellular β-actin transcripts (294 bp) were co-amplified using QuantumRNA β-actin Internal Standards (Ambion, Austin, TX), with a ratio of 3/7 for actin primer/competitor. The PCR results were compared between WT and SIVsmD116N based upon the reference of co-amplified cellular β-actin transcripts. Consistent with the persistence of non-integrated DNA, the viral transcription in SIVsmD116N infected macrophages was also persistent. In contrast to the transient presence of viral DNA and its transcripts in T cells (Wu and Marsh, 2003b), the non-integrated viral DNA in macrophages remained transcriptionally active until the termination of the cultures at 20 days. However, RNA expression levels of the SIVsmD116N mutant were much lower than that of WT virus. As shown in Figure 5A, PCR amplification with primers specific for different viral transcripts demonstrated that Nef, Tat and Env were the predominant transcripts in SIVsmD116N infection. Expression level of the Rev transcript was very low and Vif message also declined over time in SIVsmD116N infected macrophages. To make sure the RT-PCR products came from SIV viral gene expression, we used uninfected cells and heat-inactivated virus infected cells as controls. As shown in Fig. 5B, these controls were negative in contrast to amplification from cells infected with untreated virus. These results indicated that the viral gene expression detected by RT-PCR came from viruses instead of any residual DNA plasmid.
Figure 5.
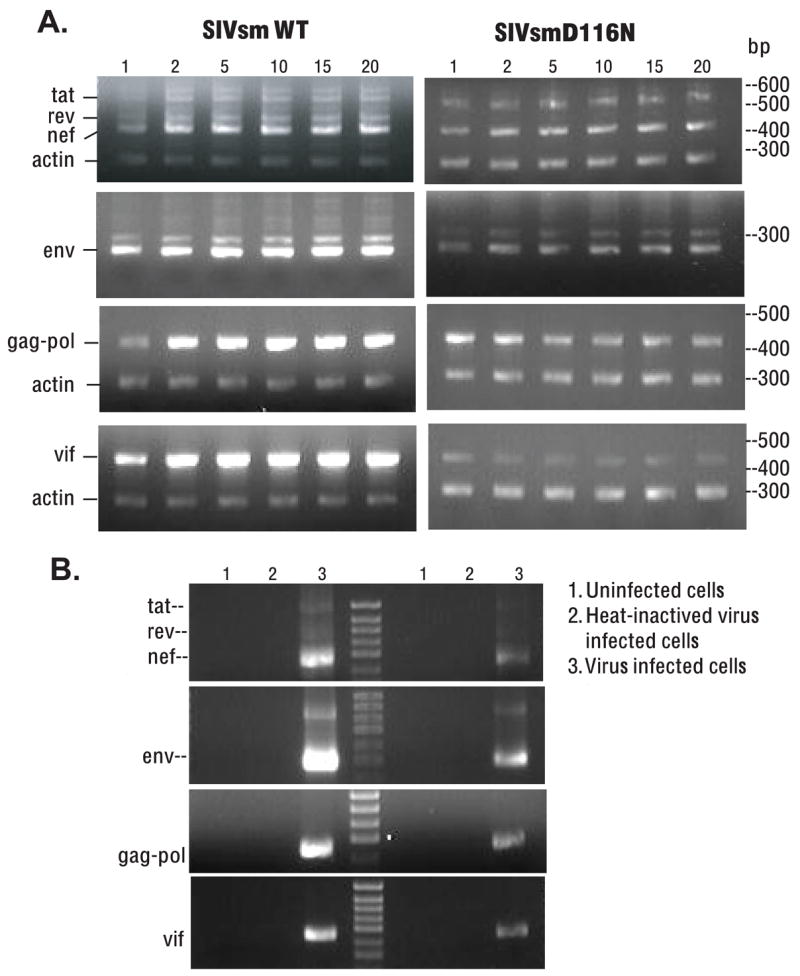
Persistence of transcriptional activity from non-integrated viral DNA in macrophages. A. Total cellular mRNA from WT or D116N infected macrophages was purified and amplified with RT-PCR. Multiple viral transcripts including all classes of viral splicing isoforms were detected. The non-integrated viral DNA in macrophages remained transcriptionally active for 20 days. RNA expression level of non-integrated D116N mutant was much lower than that of wild type. PCR products from RT-PCR were shown in the figure using primers specific for Tat/Rev/Nef, Env, Gag-pol, and Vif (from top to bottom). Nef, Tat and Env were the predominant transcripts in D116N infection. Expression level of Rev transcript was very low and Vif diminished in D116N infected macrophages over time. The PCR results were compared between WT and SIVsmD116N based upon the reference of co-amplified cellular β-actin transcripts. B. To confirm that the RT-PCR products were the result of SIV viral gene expression, we add uninfected cells and heat-inactivated virus infected cells as controls. No SIV transcripts were amplified from uninfected cells or heat-inactivated virus infected cells. The appropriate transcripts were detected in WT and D116N infected cells, but not in uninfected cells or cells infected with heat-inactivated virus.
Protein synthesis from SIVsmE543 and SIVsmD116N viruses in macrophages
The capacity of non-integrated viral DNA to direct viral protein synthesis in infected macrophages was examined by western blot analysis of cell lysates. Macrophages were cultured and divided into 3 groups: infected with SIVsmE543 or SIVsmD116N, and uninfected. Cells were harvested at different times and total cellular proteins were extracted and resolved in 4–12% Bis-Tris gel (Invitrogen, Carlsbad, CA) for western blot detection of viral proteins using plasma from a SIV-infected rhesus macaque E544 as a primary antibody. As shown in Fig. 6A, viral proteins including gp160, gp120, p66, p55, gp41, p31, p27 were strongly and persistently expressed in WT infected macrophages from day 1 to day 18. In contrast, the expression level of viral proteins was much lower in D116N infected cells. However, some of the viral proteins were expressed including, Pol-p66, Gag-p55, and Nef-p31. In order to confirm Gag expression in D116N infected macrophages, SIV polyclonal anti-p27 antibody generated in rabbit was used for western blotting. As shown in Fig. 6B, p27 was detected in both D116N and WT infected cells at day 3 and 5 post infection, but the expression level in D116N was much lower than that in WT infection. No p27 protein was detected in heat-inactivated D116N or WT infected cells and uninfected cells. This result confirmed de novo, low level expression of p27 (Gag) in D116N infected macrophages.
Figure 6.

Proteins synthesis from SIVsmE543 and SIVsmD116N viruses in macrophages detected by western blotting. A. Macrophages were cultured and divided into 3 groups, infected with SIVsmE543 or SIVsmD116N, and uninfected cells. Cells were harvested at different times and western blot was performed using plasma from a SIV-infected rhesus macaque E544 as primary antibody. In WT infected cells, viral proteins including gp160, gp120, p66, p55, gp41, p31 and p27 were strongly and persistently expressed from day 1 to day 18. In D116N infected cells, expression of viral p66, p55, p31 proteins were also observed, although the expression levels were much lower than those in wild type infection. The proteins also expressed persistently for 18 days in D116N infected cells. B. Macrophages were infected with WT SIVsmE543, SIVsmD116N, heat-inactivated WT, or heat-inactivated D116N viruses and harvested at day 3 and 5 for western blotting with a SIV anti-p27 antibody. P27 was detected in both D116N and WT infected cells at day 3 and 5 post infection, with much higher levels in the WT infection.
Discussion
In this study, using cultured macaque macrophages as an in vitro model, we demonstrated that non-integrated viral DNA can transcribe persistently in infected macrophages. In contrast to the transient presence of non-integrated viral DNA in T cells, the non-integrated SIV DNA was found to have a remarkable capacity to persist and remain transcriptionally active in macrophages. Non-integrated viral DNA and its transcripts persisted for 20 days, with minimal decay of the transcriptional activity. Additionally, some viral proteins were found to be synthesized and persisted in infected macrophages. These data suggested non-integrated viral DNA could support long-term, low level viral protein production in non-divided macrophages with the potential as a therapeutic vaccine.
The nature of the transcribing, non-integrated HIV/SIV DNA in macrophages has not yet been determined. Non-integrated DNA has three different forms: linear, 1-LTR-circle, and 2-LTR-circle DNA (Pauza and Galindo, 1989). Previous studies showed that HIV 2-LTR-circles are rapidly lost in dividing cell populations, such as primary CD4+ T lymphocytes (Pauza et al., 1994). Recent work demonstrated that 2-LTR-circles persist out up to 21 days post-infection in macrophages, a non-dividing target of HIV-1 (Gillim-Ross, Cara, and Klotman, 2005a). 2-LTR-circles have been detected in the PBMCs of HIV-1 infected patients on suppressive therapy (Cara et al., 2002; Sharkey et al., 2000). They may also persist in non-dividing cells of PBMC such as the resting memory CD4+ T lymphocytes (Chun et al., 2000). Extra-chromosomal circular DNA in non-dividing cells such as macrophages could be a source of persistent viral protein expression. Also, the macrophage population utilized in our study served as a model of a primary non-dividing cell population, non-integrated HIV/SIV may persist in other non-dividing cell populations such as memory CD4+ T lymphocytes (Chun and Fauci, 1999; Chun et al., 1997). Persistent transcription ability of non-integrated SIV DNA in non-dividing cells may have unknown functions and be important for SIV pathogenesis.
Previous research of D116N mutant HIV-1 in CD4+ T cells, demonstrated transient expression of both early and late messages by nonintegrated HIV DNA. However, only the early multiply spliced transcripts were measurably translated (Wu and Marsh, 2003a). The selective expression of early proteins from unintegrated HIV-1 templates has been hypothesized to result from low levels of Rev expression (Wu and Marsh, 2003a). During the late phase of infection, Rev promotes nuclear export and enables the cytoplasmic expression of single spliced and unspliced transcripts and therefore the translation of structural proteins. In contrast with these findings with HIV-1 integrase mutants, we observed persistent low level synthesis of both early and structural proteins from nonintegrated SIV DNA in macrophages. The transcription profile of the SIV D116N mutant was consistent with the hypothesis that low levels of Rev were responsible for the low level expression of structural proteins. In addition, low level expression of structural proteins of SIVsmD116N could be due to the difference in target cells used in the two studies (T cells versus macrophages) or to a difference in the amount of SIV Rev required for structural protein synthesis as compared to HIV. However, the HIV and SIV D116N mutants are similar in all other respects including the lack of detectable virus production. Our SIV mutant also expressed diminishing levels of the Vif transcript. The major cell types for infection, CD4 T cells and macrophages, are restrictive for HIV/SIV replication and require Vif for maintaining viral infectivity (Goncalves et al., 1996). Thus the Vif protein of HIV is required during late stages of viral production to counter the antiviral activity of APOBEC3G, a protein expressed notably in human T lymphocytes. When produced in the presence of APOBEC3G, vif-defective virus is non-infectious (Mangeat et al., 2003).
An effective vaccine is urgently needed to stop the continuing spread of HIV worldwide. However, the development of an AIDS vaccine is proving to be an unprecedented challenge (Nabel, 2002). The lack of functional virus-specific effector T lymphocytes and neutralizing antibodies is a key immunogical feature of chronic HIV and SIV infections (Singh, Jeang, and Smith, 2005). The path to an HIV vaccine has been fraught with difficulties mainly because of the virulent nature of the virus and its ability to thwart the immune system over time (Letvin and Walker, 2003). Clearly an integrase mutant such as used in the present study did not express viral proteins efficiently as required for an immunogen and would have an inherent risk of reversion. However, the capacity of a non-integrating SIV to persistently generate viral proteins in macrophages implies that non-integrating lentiviral vectors could be engineered to more efficiently express proteins for use as a therapeutic vaccine, without the permanency of an integrated retrovirus or disruption of normal cellular genes.
Materials and Methods
Construction of Non-integrating mutant of SIVsmD116N clone
The SIVsmE543-3 DNA sequence was changed from “GAT” to “AAT”, by substitution of “G” at site 4877 with “A” using the QuikChange site-directed mutagenesis kit (Stratagene, La Jalla, CA). Initially we derived a 2644 nt subclone of SIVsmE543 by PCR with Subclone-1 and Subclone-2 primers (Table 1) using the Top10 cloning kit (Invitrogen, Carlsbad, CA) and QuikChange PCR used to derive subSIVD116N according to the manufactures protocols. Mutagenic-1 and mutagenic-2 primers (Table 1) were designed according to Stratagene primer design software. SubSIVsmD116N and SIVsmE543 were digested by BstE II/BstB I, and the 2561bp fragment with the correct mutation was ligated into the 12Kb SIVsmE543 vector to construct the SIVsmD116N plasmid. The SIVsmD116N plasmid was sequenced to confirm that it contained only the desired mutation.
Viruses, cells, and replication assay
The SIVsmD116N and WT SIVsmE543-3 clones were transfected into 293T cells to generate cell free viruses using FuGENE transfection reagent (Roche, Indianapolis, IN) according to the manufacturer’s instructions. Cell free virus supernatants used for subsequent infections were treated with DNase I (Invitrogen, Carlsbad, CA) at 25°C for 10 min. to remove contaminating plasmid DNA. CEM×174 T cells, PBMC, and MDM were infected with cell free supernatant normalized for p27 antigen content from 293T cells transfected with SIVsmD116N or SIVsmE543 viruses (a multiplicity of infection 0.2). Cells were incubated with virus for 2–3 hours, then pelleted and resuspended in fresh culture medium. Virus replication was detected by 32P reverse transcriptase (RT) assay as previously reported (Sears, Repaske, and Khan, 1999). For experiment control, SIVsmD116N or SIVsmE543 viruses were heated on 56°C for 60 min to be inactivated and then were used to infect cells.
Culturing of Monocyte-derived Macrophages (MDMs)
To obtain MDMs, PBMCs were separated from whole blood by Percoll density centrifugation and were cultured in 48-well plates in RPMI 1640 medium (Life Technologies, Rockville, MD) containing 15% heat-inactivated fetal bovine serum (FBS, Hyclone, Logan, Utah), 10% human type AB serum (Sigma, St.Louis, MO) and 500 units/ml macrophage colony-stimulating factor (M-CSF, R&D systems, Minneapolis, MN) for five days. Cells were washed 3–5 times with pre-warmed RPMI 1640 medium to remove non-adherent cells. Adherent cells were maintained in fresh RPMI 1640 medium containing 15% heat-inactivated FBS, 5% human type AB serum and 200 units/ml M-CSF for two days, after which they were infected with viruses.
DNA and RNA isolation and detection
Total cellular DNA and RNA were purified using SV total RNA isolation kit (Promega, Madison, WI, USA), as recommended by the manufacturer. The primer sequences used to detect viral RNA and DNA are listed in Table 1. For the detection of full-length viral DNA by PCR, forward SIV/DNA-5 primer and anti-sense SIV/DNA-3 primer were used. PCR was carried out in 1 x Ambion PCR buffer, 125μM dNTP, 50 pmol each primer, 1U SuperTaq Plus (Ambion, Austin, TX) with 30 cycles of 94°C for 20s, 60°C for 30s, 68°C for 40s. Both the 1-long terminal repeat (LTR) circles and 2-LTR-circles, SIV/Circle-5′ primer and SIV/Circle-3′ primer were used for detection of circular viral DNA forms. For detection of viral RNA species, reverse transcription of cellular RNA was accomplished with random decamers as the first-strand primers. Following cDNA synthesis, PCR was performed using SIV/5′ primer and SIV/nef-tat-rev primer to amplify doubly spliced viral transcripts Nef, Tat and Rev. For relative quantification of RT-PCR, cellular β-actin transcripts were co-amplified using QuantumRNA β-actin Internal Standards (Ambion, Austin, TX), with a ratio of 3/7 for actin primer/competitor. For analysis of singly spliced viral transcripts such as the Env transcript, SIV/5′ primer and SIV/3′ env primer were used, whereas for the Vif transcript, SIV/5′ primer and SIV/3′ vif primer were used. For analysis of Gag-Pol transcript, SIV/5′ primer and SIV/3′ gag-pol primer were used.
Assay for integration
To detect SIV integration into the host genome, we used an Alu sequence-based PCR strategy to amplify the junction between the nearest Alu sequence and the SIV LTR. Nested PCR was used for Alu-PCR, with Alu 1-1 primer corresponding to human Alu sequence position 172–196, and Alu 1–2 primer located in the U3 region (bp230-207) of SIV LTR (Zhuge et al., 2001). Following the first round of Alu-PCR, a second round of PCR was carried out by using a 1:500 dilution of the first round PCR product as a template and the LTR specific primers Alu 2-1 and Alu 2-2 (Table 1). The amplification conditions for Alu-PCR were 35 cycles of 94°C for 20s, 65 ° C for 30s, 68° C for 40s. To ensure that the second round PCR products were derived from first round PCR product (integrated DNA) rather than non-integrated DNA carry-over, a 1:500 dilution of the original DNA samples were used as templates for second round PCR as a negative control.
Immunodetection of viral protein
Western blotting was performed for detecting viral protein expression. Samples were run on 4–12% Bis-Tris gel (Invitrogen, Carlsbad, CA) and transferred to 0.2um nitrocellulose membrane in a Novex Mini-Cell unit (Invitrogen, Carlsbad, CA) as suggested by manufacturer. For figure 1 and figure 6A, anti-SIV antiserum from SIVsmF236-infected rhesus E544 was used as a primary antibody. This antiserum reacts with the Env, Gag, Pol and Nef proteins of SIV. A 1:1000 dilution of primary antibody was incubated with the membrane overnight, followed by 3 washes. The membrane was then incubated with a secondary 1:10,000 diluted sheep anti-human Ig whole antibody conjugated with horseradish peroxidase (HRP, Amersham, Piscataway, NJ) for 1 hr. Proteins on membrane were detected by ECL plus western blotting detection reagents (Amersham, Piscataway, NJ). MagicMark™ XP Western Protein Standard (Invitrogen, Carlsbad, CA) was used as marker. SIV anti-p27 antibody was a gift from Dr. Raoul Benveniste in NCI, NIH. It is a polyclonal antibody generated in rabbit immunized with p27 protein of SIVmneE11S virus. The SIV anti-p27 antibody was used as primary antibody for the western blotting shown in figure 6B, and anti-rabbit IgG whole antibody conjugated with horseradish peroxidase (HRP, Amersham, Piscataway, NJ) was used as secondary antibody.
Acknowledgments
We thank Yuntao Wu for help in experimental design, Robert Goeken and Charles Brown for technical assistance, Russell Byrum for providing macaque blood samples, and Takeo Kuwata for beneficial discussion. This work was supported by the intramural program of NIAID, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bukrinsky MI, Sharova N, Dempsey MP, Stanwick TL, Bukrinskaya AG, Haggerty S, Stevenson M. Active nuclear import of human immunodeficiency virus type 1 preintegration complexes. Proc Natl Acad Sci U S A. 1992;89(14):6580–4. doi: 10.1073/pnas.89.14.6580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cara A, Vargas J, Jr, Keller M, Jones S, Mosoian A, Gurtman A, Cohen A, Parkas V, Wallach F, Chusid E, Gelman IH, Klotman ME. Circular viral DNA and anomalous junction sequence in PBMC of HIV-infected individuals with no detectable plasma HIV RNA. Virology. 2002;292(1):1–5. doi: 10.1006/viro.2001.1243. [DOI] [PubMed] [Google Scholar]
- Chun TW, Davey RT, Jr, Ostrowski M, Shawn Justement J, Engel D, Mullins JI, Fauci AS. Relationship between pre-existing viral reservoirs and the re-emergence of plasma viremia after discontinuation of highly active anti-retroviral therapy. Nat Med. 2000;6(7):757–61. doi: 10.1038/77481. [DOI] [PubMed] [Google Scholar]
- Chun TW, Fauci AS. Latent reservoirs of HIV: obstacles to the eradication of virus. Proc Natl Acad Sci U S A. 1999;96(20):10958–61. doi: 10.1073/pnas.96.20.10958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JA, Baseler M, Lloyd AL, Nowak MA, Fauci AS. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci U S A. 1997;94(24):13193–7. doi: 10.1073/pnas.94.24.13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehghani H, Puffer BA, Doms RW, Hirsch VM. Unique pattern of convergent envelope evolution in simian immunodeficiency virus-infected rapid progressor macaques: association with CD4-independent usage of CCR5. J Virol. 2003;77(11):6405–18. doi: 10.1128/JVI.77.11.6405-6418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman A, Englund G, Orenstein JM, Martin MA, Craigie R. Multiple effects of mutations in human immunodeficiency virus type 1 integrase on viral replication. J Virol. 1995;69(5):2729–36. doi: 10.1128/jvi.69.5.2729-2736.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englund G, Theodore TS, Freed EO, Engelman A, Martin MA. Integration is required for productive infection of monocyte-derived macrophages by human immunodeficiency virus type 1. J Virol. 1995;69(5):3216–9. doi: 10.1128/jvi.69.5.3216-3219.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillim-Ross L, Cara A, Klotman ME. HIV-1 extrachromosomal 2-LTR circular DNA is long-lived in human macrophages. Viral Immunol. 2005a;18(1):190–6. doi: 10.1089/vim.2005.18.190. [DOI] [PubMed] [Google Scholar]
- Gillim-Ross L, Cara A, Klotman ME. Nef expressed from human immunodeficiency virus type 1 extrachromosomal DNA downregulates CD4 on primary CD4+ T lymphocytes: implications for integrase inhibitors. J Gen Virol. 2005b;86(Pt 3):765–71. doi: 10.1099/vir.0.80570-0. [DOI] [PubMed] [Google Scholar]
- Goncalves J, Korin Y, Zack J, Gabuzda D. Role of Vif in human immunodeficiency virus type 1 reverse transcription. J Virol. 1996;70(12):8701–9. doi: 10.1128/jvi.70.12.8701-8709.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch V, Adger-Johnson D, Campbell B, Goldstein S, Brown C, Elkins WR, Montefiori DC. A molecularly cloned, pathogenic, neutralization-resistant simian immunodeficiency virus, SIVsmE543-3. J Virol. 1997;71(2):1608–20. doi: 10.1128/jvi.71.2.1608-1620.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch VM, Santra S, Goldstein S, Plishka R, Buckler-White A, Seth A, Ourmanov I, Brown CR, Engle R, Montefiori D, Glowczwskie J, Kunstman K, Wolinsky S, Letvin NL. Immune failure in the absence of profound CD4+ T-lymphocyte depletion in simian immunodeficiency virus-infected rapid progressor macaques. J Virol. 2004;78(1):275–84. doi: 10.1128/JVI.78.1.275-284.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letvin NL, Walker BD. Immunopathogenesis and immunotherapy in AIDS virus infections. Nat Med. 2003;9(7):861–6. doi: 10.1038/nm0703-861. [DOI] [PubMed] [Google Scholar]
- Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424(6944):99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- Nabel GJ. HIV vaccine strategies. Vaccine. 2002;20(15):1945–7. doi: 10.1016/s0264-410x(02)00074-9. [DOI] [PubMed] [Google Scholar]
- Pauza CD, Galindo J. Persistent human immunodeficiency virus type 1 infection of monoblastoid cells leads to accumulation of self-integrated viral DNA and to production of defective virions. J Virol. 1989;63(9):3700–7. doi: 10.1128/jvi.63.9.3700-3707.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauza CD, Trivedi P, McKechnie TS, Richman DD, Graziano FM. 2-LTR circular viral DNA as a marker for human immunodeficiency virus type 1 infection in vivo. Virology. 1994;205(2):470–8. doi: 10.1006/viro.1994.1667. [DOI] [PubMed] [Google Scholar]
- Sears JF, Repaske R, Khan AS. Improved Mg2+-based reverse transcriptase assay for detection of primate retroviruses. J Clin Microbiol. 1999;37(6):1704–8. doi: 10.1128/jcm.37.6.1704-1708.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharkey ME, Teo I, Greenough T, Sharova N, Luzuriaga K, Sullivan JL, Bucy RP, Kostrikis LG, Haase A, Veryard C, Davaro RE, Cheeseman SH, Daly JS, Bova C, Ellison RT, 3rd, Mady B, Lai KK, Moyle G, Nelson M, Gazzard B, Shaunak S, Stevenson M. Persistence of episomal HIV-1 infection intermediates in patients on highly active anti-retroviral therapy. Nat Med. 2000;6(1):76–81. doi: 10.1038/71569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Jeang KT, Smith SM. HIV vaccine development. Front Biosci. 2005;10:2064–81. doi: 10.2741/1682. [DOI] [PubMed] [Google Scholar]
- Stevenson M, Haggerty S, Lamonica CA, Meier CM, Welch SK, Wasiak AJ. Integration is not necessary for expression of human immunodeficiency virus type 1 protein products. J Virol. 1990;64(5):2421–5. doi: 10.1128/jvi.64.5.2421-2425.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiskerchen M, Muesing MA. Human immunodeficiency virus type 1 integrase: effects of mutations on viral ability to integrate, direct viral gene expression from unintegrated viral DNA templates, and sustain viral propagation in primary cells. J Virol. 1995;69(1):376–86. doi: 10.1128/jvi.69.1.376-386.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y. HIV-1 gene expression: lessons from provirus and non-integrated DNA. Retrovirology. 2004;1(1):13. doi: 10.1186/1742-4690-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Marsh JW. Selective transcription and modulation of resting T cell activity by preintegrated HIV DNA. Science. 2001;293(5534):1503–6. doi: 10.1126/science.1061548. [DOI] [PubMed] [Google Scholar]
- Wu Y, Marsh JW. Early transcription from nonintegrated DNA in human immunodeficiency virus infection. J Virol. 2003a;77(19):10376–82. doi: 10.1128/JVI.77.19.10376-10382.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Marsh JW. Gene transcription in HIV infection. Microbes Infect. 2003b;5(11):1023–7. doi: 10.1016/s1286-4579(03)00187-4. [DOI] [PubMed] [Google Scholar]
- Zennou V, Petit C, Guetard D, Nerhbass U, Montagnier L, Charneau P. HIV-1 genome nuclear import is mediated by a central DNA flap. Cell. 2000;101(2):173–85. doi: 10.1016/S0092-8674(00)80828-4. [DOI] [PubMed] [Google Scholar]
- Zhuge W, Jia F, Mackay G, Kumar A, Narayan O. Antibodies that neutralize SIV(mac)251 in T lymphocytes cause interruption of the viral life cycle in macrophages by preventing nuclear import of viral DNA. Virology. 2001;287(2):436–45. doi: 10.1006/viro.2001.1053. [DOI] [PubMed] [Google Scholar]