

Abstract
JC virus (JCV) is the causative agent of progressive multifocal leukoenchaphalopathy (PML). The disease develops when JCV gains access to the central nervous system, infects and destroys oligodendrocytes. The disease is rapidly progressing, typically fatal and no effective therapies exist to treat or prevent PML. The recent occurrence of PML in multiple sclerosis patients being treated with Avonex (IFNβ1a) and Tysabri (Natalizumab) and the recent reports linking JCV infection to the 5HT2a serotonin receptor led us to evaluate the effects of IFNβ1-a and a panel of 5HT2a receptor antagonists for their ability to modulate virus infection. IFNβ1-a was found to be a potent inhibitor of both virus infection and viral early and late gene expression. In addition, several 5HT2a receptor antagonists inhibited initial infection of cells by JCV but were less effective and reducing viral loads in an already established infection.
Keywords: Polyomavirus, JCV, PML, Natalizumab, Interferons
INTRODUCTION
Seroepidemiological studies indicate that 70% of the human population worldwide are infected with JCV (Bofill-Mas et al., 2001; Bofill-Mas, Pina, and Girones, 2000; Gardner, 1973; Gardner et al., 1971; Hogan et al., 1980; Imperiale, 2000; Shah, Daniel, and Warszawski, 1973). The mode of virus transmission is unknown, and no clinical illness has been associated with primary infection. Like all polyomaviruses, infection with JCV is associated with the establishment of lifelong persistent infection. In immunosuppressed patients, JCV causes a fatal demyelinating disease known as progressive multifocal leukoencephalopathy (PML). PML occurs predominately in immunosuppressed patients with the majority of cases occurring in the setting of HIV infection (Holman et al., 1998). Recently, PML has also been reported in patients being treated with Natalizumab, a drug designed to inhibit leukocyte trafficking into inflamed tissue (Kleinschmidt-DeMasters and Tyler, 2005; Langer-Gould et al., 2005; Van Assche et al., 2005). PML is thought to develop following reactivation of the virus and dissemination from peripheral sites to the CNS, where the primary targets are astrocytes and oligodendrocytes (Achim and Wiley, 1992; Fotiadis, Kilpatrick, and Lipton, 1991; Telenti et al., 1992). Others have suggested that reactivation of latent JCV within the CNS can also contribute to the development and progression of PML (Boone et al., 1992; Elsner and Dorries, 1992; Kestler Harry et al., 1991). The mechanism by which JCV becomes reactivated and traffics to the CNS is unclear.
Cell surface receptors have been described for several polyomaviruses including JCV, BKV, SV40, and the mouse polyomavirus. All of these polyomaviruses with the exception of SV40 initially interact with sialic acid containing glycoproteins or glycolipids on the cell surface. Infection of glial cells by JCV is dependent on virus binding to a receptor complex that includes alpha (2,3) or alpha (2,6)-linked sialic acid and the 5HT2a serotonin receptor (Fonseca-Elphick et al., 2004; Liu, Wei, and Atwood, 1998). Recently human brain microvascular endothelial cells were shown to be susceptible to JCV infection independent of the 5HT2a receptor component indicating that at least some cell types do not require this receptor (Chapagain et al., 2007).
In this report, the impact of IFNβ1-a and selective 5HT2a receptor antagonists on JCV infection was investigated. IFNβ1-a potently inhibited virus infection and virus early and late gene expression. The 5HT2a receptor antagonists ritanserin, ketanserin, mianserin, and to a lesser extent mirtazapine, cyproheptadine, risperidone, and ziprasedone reduced initial virus infection but failed to significantly reduce viral loads or viral spread in cells already infected.
MATERIALS and METHODS
Cells and virus
SVG-A cells are a subclone of the SVG human glial cell line established by transformation of human fetal glial cells with an origin-defective SV40 mutant (Major et al., 1985). Cells were cultured in EMEM supplemented with 10% fetal calf serum (Mediatech, Inc.) and maintained in a humidified 37°C CO2 incubator. The production of the Mad-1SVEΔ strain of virus has been previously described (Liu and Atwood, 2001).
Infection and indirect immunofluoresence
Cells were grown to ~70% confluency in a 6-well dish, and infected for 1 hr with JCV (512HAU/105 cells) in the presence of EMEM plus 2% serum in a total volume of 100 µl. At 3 days post-infection (p.i.), cells were fixed in ice-cold acetone for 10mins. Infected cells were detected using a monoclonal antibody (PAB597) against the major capsid protein VP1. PAB597 targets the SV40 major capsid protein VP1 and has been previously shown to cross-react with JCV VP1 (Atwood et al., 1995). Primary antibody was detected with an Alexa Fluor 488-labeled goat anti-mouse secondary antibody (Molecular Probes). Cells were counter-stained with Evans Blue. Positive cells were visualized on a Nikon epifluorescence microscope (Eclipse E800; Nikon Inc.).
Quantitative PCR measurement of viral load
SVG-A cells were infected with JCV and treated with or without drugs beginning at 24 hours post-infection. Fresh drug was added every three days. At 15 days post-infection virus was harvested from the infected cells, treated with Dnase to remove non-virion associated DNA, and then with proteinase K to expose the virion DNA. Virion associated DNA was then quantitated by real time PCR using the protocol, primers, and probes described by McNees et al. on a Biorad IQ5 system (Biorad) (McNees et al., 2005). Primers and probes were designed using primer express software (Applied Biosystems). The iScript one-step RT-PCR kit was used for amplification (Biorad). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the internal standard.
Transfection and luciferase assays
pJCV-MadE and L-luc have been previously described (Okada et al., 2000). The pRL-TK is a construct that expresses Renilla luciferase under the control of the thymidine kinase promoter of HSV, and was purchased from Promega. Transient transfections were carried out using Fugene as described by the manufactures protocol (Roche). Vectors were co-transfected with the Renilla luciferase control vector. Renilla and Firefly luciferase activity were measured 48 h post-transfection using the Promega Dual-Luciferase Reporter Assay System. Relative luciferase units were measured using a Berthoid Lumat LB9501 luminometer.
Infection inhibition studies
Ritanserin, ketanserin, mianserin, mirtazapine, cyproheptadine, nefazadone, and trazadone were obtained from Sigma-Aldrich and dissolved according to the manufacturers recommendations. Risperidone and ziprasidone were obtained from Toronto Research Chemicals, Inc. IFN β1-a was obtained from Biogen-Idec, Inc. For the anti-5HT2a antagonist studies cells were pre-treated for 24 hours at 37°C to maximize receptor down-regulation. The cells were maintained in drug throughout the experiment. For the long term inhibition experiment fresh drug was added to the media every third day.
Viability assays
Cell viability was measured using the CellTiter 96 Cell Proliferation MTS Assay (Promega) according to the manufacturers protocol. Briefly, the MTS assay is a colorimetric method for determining the number of viable cells in culture. The MTS tetrazolium compound [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] is bioreduced by cells into a colored formazan product that is soluble in tissue culture media. This conversion is accomplished by NADPH/NADH produced in metabolically active cells. The quantity of formazan product is proportional to the number of living cells. To determine toxicity, cells were plated at a density of 5,000 cells/well and incubated with drug for a duration of 15 days. Results were assayed by measuring A490 on a Spectramax M5 microplate reader.
RESULTS
Interferon β1-a inhibits viral infection and viral early and late gene expression
The SVG-A human glial cell line was treated with increasing doses of IFNβ1-a (5IU-500IU) and challenged 24 hours later with JCV (Mad-1SVEΔ). The cells were scored at 72 hours post-infection for expression of the late viral protein V antigen. IFNβ1-a inhibited the percentage of V antigen positive cells and the inhibition was dose dependent (Fig. 1A). None of the concentrations of IFNβ1-a were toxic as determined by a MTS assay (Fig. 1C). To determine the mechanism of inhibition SVG-A cells were transfected with JCV (Mad-1) promoter constructs driving the expression of luciferase in both the early and late orientation. Following transfection the cells were treated with the same doses of IFNβ1-a as used for the infection experiments and luciferase activity measured at 48 hours post-transfection. Transfection efficiency was normalized to an internal Renilla luciferase expressed from a heterologous promoter that was not sensitive to IFNβ1-a treatment. IFNβ1-a inhibited both the early and late promoters of JCV indicating that IFNβ1-a is a potent inhibitor of viral gene expression (Fig. 1B).
Figure 1. Inhibition of JCV infection and gene expression by IFNβ 1-a.
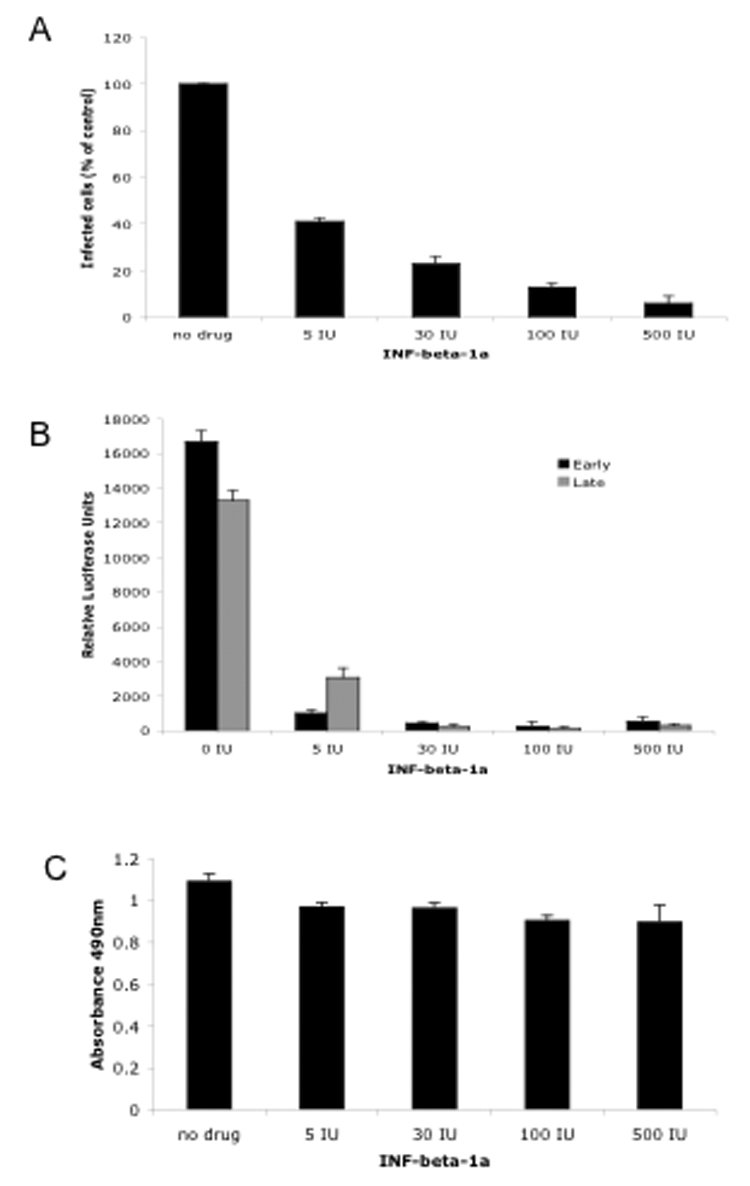
A. SVG- cells were either untreated or treated with increasing concentrations of IFNβ 1-a as indicated and challenged with JCV. Infected cells were scored by indirect immunofluoresence analysis of V antigen positive cells and plotted as a percentage of control untreated cells. The experiment was performed in triplicate and a minimum of 10 microscopic fields (20X) were counted per coverslip. B. SVG-A cells were transfected with luciferase reporter vectors under the control of the JCV early or late promoter as indicated. At 48 hours post-transfection luciferase activity was measured. The experiment was done in triplicate. C. Toxicity of IFNβ 1-a as measured by an MTS assay. Error bars are standard deviations.
Selective 5HT2a receptor antagonists inhibit infection by JCV
SVG-A cells were pre-incubated for 24 hours at 37°C with a panel of drugs known to antagonize the 5HT2a receptor. The cells were then challenged with JCV (Mad-1SVEΔ) and cultured in the presence of drug for an additional 72 hours. At 72 hours post-infection the cells were scored for expression of the late viral protein, V antigen. The drugs ritanserin, ketanserin, and mianserin were most effective at inhibiting virus infection in a dose dependent manner (Fig. 2, A–C). Trazadone and nefazadone were least effective and cyproheptadine was only effective at a relatively high dose (Fig. 2, D–F). Mirtazapine, risperidone, and ziprasidone were moderately efficacious (Fig.2, G–I).
Figure 2. 5HT2a receptor antagonists inhibit JCV infection.
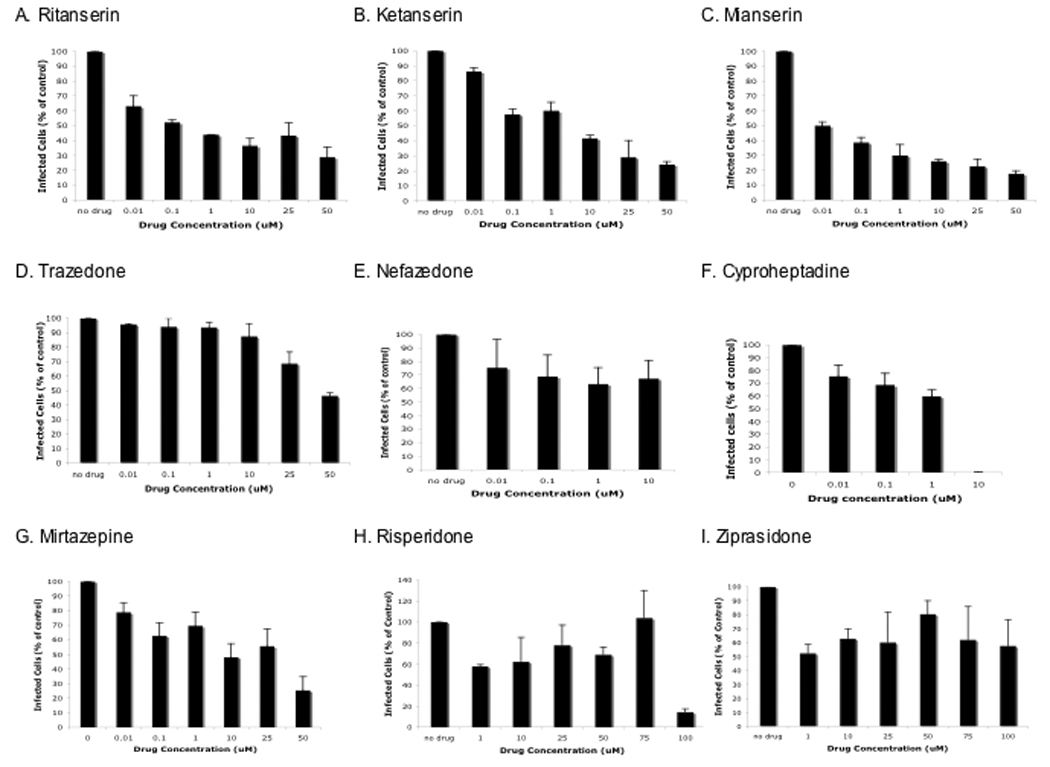
A–I. SVG-A cells were pretreated with drugs as indicated for 24 hours and then challenged with JCV. Infected cells were scored at 72 hours post-infection by indirect immunofluoresence analysis of V antigen positive cells and plotted as a percentage of control untreated cells. The experiment was performed in triplicate and a minimum of 10 microscopic fields (20X) were counted per coverslip.
We next asked whether any of a subset of these drugs would be capable of inhibiting an already established infection. To do this SVG-A cells were infected with JCV (Mad-1SVEΔ) and cultured in the absence of drug for 24 hours. Drugs were then added and cells scored for infection every three days for 15 days. At each time point fresh media and drug were added to the cultures. The presence of these drugs in the media only moderately inhibited the spread of infection (Fig. 3). In addition viral loads were not appreciably affected as determined by quantitative real time PCR (Fig. 4). Maintaining the cells in these concentrations of drug was not toxic as measured by a MTS assay (data not shown).
Figure 3. Effect of 5HT2a receptor antagonists on inhibiting viral spread in an already established infection.
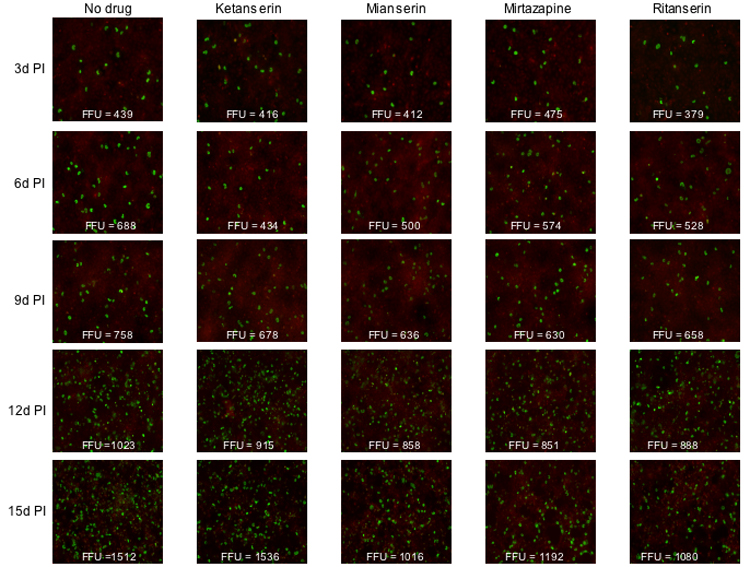
SVG-A cells were infected with JCV and then treated with drug as indicated beginning at 24 hours post-infection. The spread of infection over time was quantitated by staining the cells with anti-V antigen antibodies. FFU= fluorescent forming units.
Figure 4. JCV viral loads in drug treated cells.
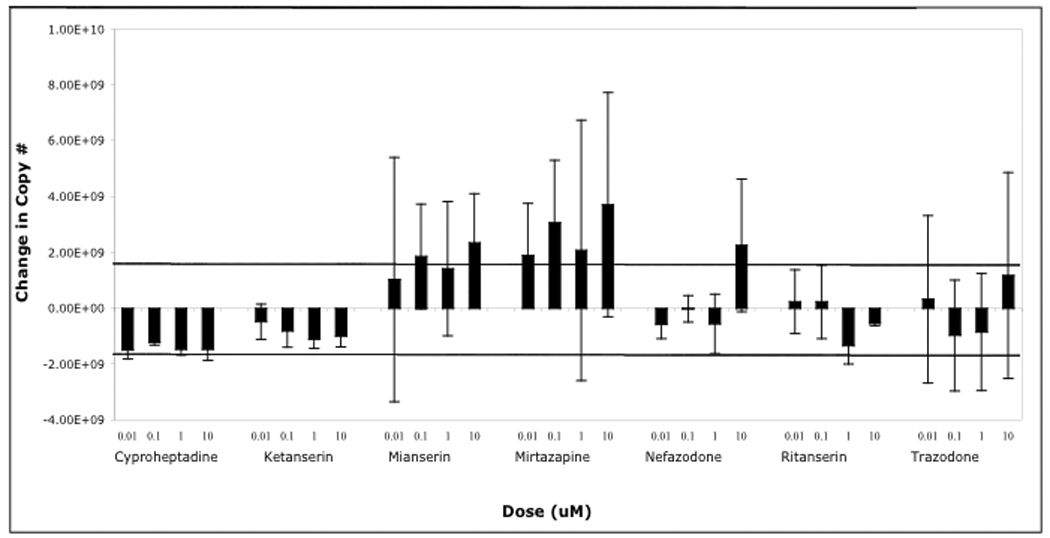
SVG-A cells were infected with JCV and treated 24 hours post-infection with or without drugs, as indicated. At 15 days post-infection viral loads were measured in lysates that had been treated with Dnase to eliminate non-encapsidated viral DNA. Virion associated DNA was then exposed by treatment with proteinase K and amplified as described in materials and methods. The change in viral DNA copy number is compared to the untreated samples that we set to 0. The dark horizontal lines indicate the average standard deviation in copy number from the untreated samples.
JCV does not spread by direct cell-cell contact
One possibility to account for the poor efficacy of these drugs at inhibiting an already established infection is that JCV might be capable of direct cell-cell spread thereby by-passing a requirement for cellular receptors. To ask whether this was the case cells were transfected with viral DNA and the cultures were treated with rabbit anti-JCV neutralizing sera beginning two days post-transfection. The presence of neutralizing JCV antisera significantly limited the spread of JCV in the cultures indicating that direct cell-cell spread is not a major mechanism of virus spread in tissue culture (Fig. 5).
Figure 5. Anti-JCV sera inhibits virus spread.
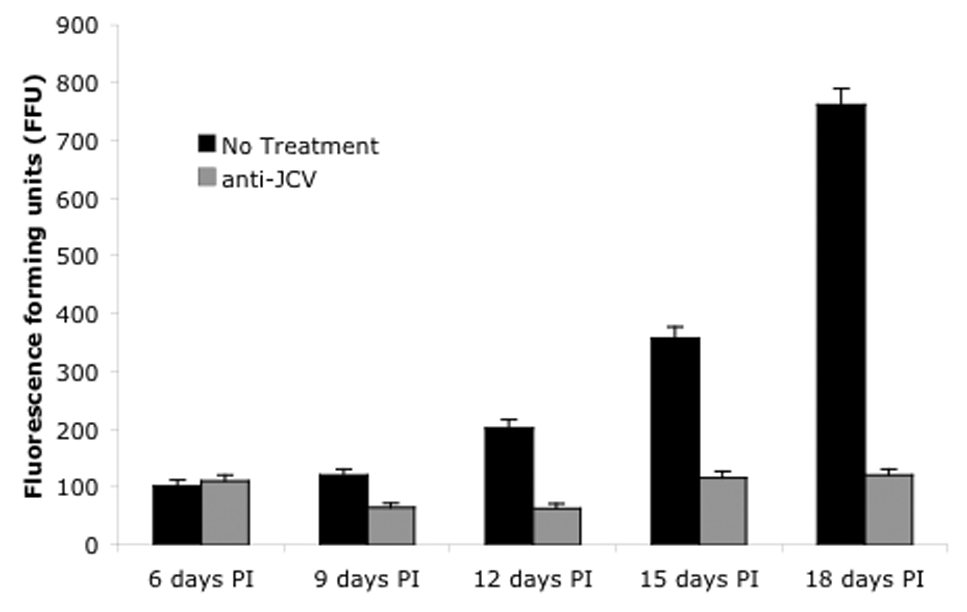
To determine whether virus was capable of direct cell-to-cell spread neutralizing anti-JCV sera was added to the cultures at 24 hours post-transfection and the spread of infected cells scored every three days beginning at 6 days post-transfection. FFU=fluorescent forming units.
DISCUSSION
The incidence of Progressive Multifocal Leukoencephalopathy (PML) in the United States increased in parallel with the AIDS epidemic from 1 in 10 million to over 1 in 500,000 between 1979 and 1992 (Holman et al., 1998). The introduction of highly active antiretroviral therapies (HAART) has increased the mean survival time of AIDS patients with PML from 2 months to one year but has had no effect at reducing the overall incidence of PML in the HIV infected population (Koralnik, 2006). Recently, PML emerged in two multiple sclerosis patients undergoing treatment with IFNβ-1a (Avonex) and Natalizumab (Tysabri) and one Crohn’s disease patient undergoing treatment with Natalizumab (Tysabri) (Berger and Koralnik, 2005; Kleinschmidt-DeMasters and Tyler, 2005; Langer-Gould et al., 2005; Van Assche et al., 2005). IFNβ-1a (Avonex) is approved for the treatment of multiple sclerosis. Its mechanism of action involves inhibiting the expression of inflammatory cytokines that contribute to numerous autoimmune diseases. Natalizumab (Tysabri) is a humanized monoclonal antibody that binds to α4β1 and α4β7 integrins and blocks leukocyte trafficking into inflamed tissues (Rice, Hartung, and Calabresi, 2005).
In this report, the impact of IFNβ-1a on JCV replication and gene expression was investigated. IFNβ1-a inhibited viral infection and viral early and late gene expression in a dose dependent manner. This is consistent with earlier reports demonstrating no increased JC viral loads in multiple sclerosis patients being treated IFNβ1-a (Alvarez-Lafuente et al., 2007; Delbue et al., 2007). It is also consistent with two recent reports documenting induction of an interferon response in JCV infected cells and inhibition of JCV infection by type 1 interferons (Co et al., 2007; Verma et al., 2006). The mechanism of inhibition of JCV gene expression is not known but may be related to the induction of immediate response genes that bind to specific sites in the JCV promoter (Raj and Khalili, 1994). As IFNβ-1a treatment would be expected to inhibit viral multiplication outside of the CNS it is reasonable to suspect that JCV was already present in the CNS of these two patients who developed PML. IFNβ-1a does not cross the blood brain barrier efficiently so it would not be expected to be useful at inhibiting viral replication in CNS parenchyma.
A panel of drugs that target the cellular receptor for JCV was also investigated. The drugs chosen all antagonize the 5HT2a receptor that is a critical component of the JCV receptor complex on glial cells (Fonseca-Elphick et al., 2004). All of these drugs are known to cross the blood brain barrier and have been approved as anti-psychotics either in the US or in Europe. Ketanserin, ritanserin, and mianserin were most effective at inhibiting virus infection. Mirtazapine, cyprohepatadine, risperidone, and ziprasedone showed some efficacy and nefazadone and trazadone were the least effective at inhibiting infection. The different effects that each of the drugs has on infection could be related to how effectively the individual compounds down-regulate the 5HT2a receptor on this cell type.
As these drugs are likely to be considered for therapeutic treatment of patients with active lesions a subset of these drugs were tested for their ability to inhibit an already established virus infection. Ritanserin, mianserin, and mirtazapine modestly reduced the percentage of infected cells over the course of a two-week infection but had no significant effect on viral loads as measured by real time PCR of culture superntants. In contrast, administration of neutralizing antisera significantly reduced the infection indicating that direct cell to cell spread is not a major mechanism of virus spread in tissue culture. It is unclear why these drugs are less effective at inhibiting an already established infection but it could relate to drug turnover or resistance to receptor down-regulation which is currently under investigation. Regardless, several anecdotal reports have suggested that mirtazapine and risperidone may be effective in the treatment of PML so these compounds or related compounds may be somewhat efficaceous (Altschuler and Kast, 2005; Focosi et al., 2007; Lima et al., 2007).
Acknowledgements
We thank all members of the Atwood lab for critical discussion during the course of this work. We thank Hirofumi Sawa for kindly providing JCV early and late luciferase reporter constructs and Susan Goelz at Biogen-Idec, Inc. for IFNβ1-a. We thank Tammy Glass, Wendy Virgadamo, and Heather Forand for administrative assistance. Work in our laboratory was supported by a grant from the National Cancer Institute, R01 CA71878, by a grant from the National Institute of Neurological Disorders and Stroke, R01 NS43097 to W.A., and by a grant from Biogen-Idec, Inc. to W.A.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- Achim C, Wiley C. Expression of major histocompatibility complex antigens in the brains of patients with progressive multifocal leukoencephalopathy. J.Neuropathol.Exp.Neurol. 1992;51:257–263. doi: 10.1097/00005072-199205000-00003. [DOI] [PubMed] [Google Scholar]
- Altschuler EL, Kast RE. The atypical antipsychotic agents ziprasidone [correction of zisprasidone], risperdone and olanzapine as treatment for and prophylaxis against progressive multifocal leukoencephalopathy. Med Hypotheses. 2005;65(3):585–586. doi: 10.1016/j.mehy.2005.01.037. [DOI] [PubMed] [Google Scholar]
- Alvarez-Lafuente R, Garcia-Montojo M, De Las Heras V, Bartolome M, Arroyo R. Interferon-beta treatment and active replication of the JC virus in relapsing-remitting multiple sclerosis patients. Eur J Neurol. 2007;14(2):233–236. doi: 10.1111/j.1468-1331.2006.01638.x. [DOI] [PubMed] [Google Scholar]
- Atwood WJ, Wang L, Durham LC, Amemiya K, Traub RG, Major EO. Evaluation of the role of cytokine activation in the multiplication of JC virus (JCV) in human fetal glial cells. J.Neurovirol. 1995;1:40–49. doi: 10.3109/13550289509111009. [DOI] [PubMed] [Google Scholar]
- Berger JR, Koralnik IJ. Progressive multifocal leukoencephalopathy and natalizumab--unforeseen consequences. N Engl J Med. 2005;353(4):414–416. doi: 10.1056/NEJMe058122. [DOI] [PubMed] [Google Scholar]
- Bofill-Mas S, Formiga-Cruz M, Clemente-Casares P, Calafell F, Girones R. Potential transmission of human polyomaviruses through the gastrointestinal tract after exposure to virions or viral DNA. J Virol. 2001;75(21):10290–10299. doi: 10.1128/JVI.75.21.10290-10299.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bofill-Mas S, Pina S, Girones R. Documenting the epidemiologic patterns of polyomaviruses in human populations by studying their presence in urban sewage. Appl Environ Microbiol. 2000;66(1):238–245. doi: 10.1128/aem.66.1.238-245.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone K, Miller B, Lesser I, Mehringer C, Hill-Gutierrez E, Goldberg M, Berman N. Neuropsychological correlates of white-matter lesions in healthy elderly. Arch.Neurol. 1992;49:549–554. doi: 10.1001/archneur.1992.00530290141024. [DOI] [PubMed] [Google Scholar]
- Chapagain ML, Verma S, Mercier F, Yanagihara R, Nerurkar VR. Polyomavirus JC infects human brain microvascular endothelial cells independent of serotonin receptor 2A. Virology. 2007;364(1):55–63. doi: 10.1016/j.virol.2007.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Co JK, Verma S, Gurjav U, Sumibcay L, Nerurkar VR. Interferon-alpha and -beta restrict polyomavirus JC replication in primary human fetal glial cells: implications for progressive multifocal leukoencephalopathy therapy. J Infect Dis. 2007;196(5):712–718. doi: 10.1086/520518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delbue S, Guerini FR, Mancuso R, Caputo D, Mazziotti R, Saresella M, Ferrante P. JC virus viremia in interferon-beta -treated and untreated Italian multiple sclerosis patients and healthy controls. J Neurovirol. 2007;13(1):73–77. doi: 10.1080/13550280601094563. [DOI] [PubMed] [Google Scholar]
- Elsner C, Dorries K. Evidence of human polyomavirus BK and JC infection in normal brain tissue. Virology. 1992;191:72–80. doi: 10.1016/0042-6822(92)90167-n. [DOI] [PubMed] [Google Scholar]
- Focosi D, Fazzi R, Montanaro D, Emdin M, Petrini M. Progressive multifocal leukoencephalopathy in a haploidentical stem cell transplant recipient: a clinical, neuroradiological and virological response after treatment with risperidone. Antiviral Res. 2007;74(2):156–158. doi: 10.1016/j.antiviral.2006.10.011. [DOI] [PubMed] [Google Scholar]
- Fonseca-Elphick G, Querbes W, Jordan JA, Gee GV, Eash S, Manley K, Dugan A, Stanifer M, Bhatnagar A, Kroeze WK, Roth BL, Atwood WJ. The human polyomavirus, JCV, uses serotonin receptors to infect cells. Science. 2004;306:1241–1420. doi: 10.1126/science.1103492. [DOI] [PubMed] [Google Scholar]
- Fotiadis C, Kilpatrick DR, Lipton HL. Comparison of the binding characteristics to BHK-21 cells of viruses representing the two Theiler's virus neurovirulence groups. Virology. 1991;182(1):365–370. doi: 10.1016/0042-6822(91)90683-3. [DOI] [PubMed] [Google Scholar]
- Gardner SD. Prevalence in England of antibody to human polyomavirus (BK) Br. Med. J. 1973;1:77. doi: 10.1136/bmj.1.5845.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner SD, Field AM, Coleman DV, Hulme B. New human papovavirus (BK) isolated from urine after renal transplantation. Lancet. 1971;i:1253–1257. doi: 10.1016/s0140-6736(71)91776-4. [DOI] [PubMed] [Google Scholar]
- Hogan TF, Borden EC, McBain JA, Padgett BL, Walker DL. Human polyomavirus infections with JC virus and BK in renal transplant patients. Ann. Intern. Med. 1980;92:373–378. doi: 10.7326/0003-4819-92-3-373. [DOI] [PubMed] [Google Scholar]
- Holman RC, Torok TJ, Belay ED, Janssen RS, Schonberger LB. Progressive multifocal leukoencephalopathy in the United States, 1979–1994: increased mortality associated with HIV infection. Neuroepidemiology. 1998;17:303–309. doi: 10.1159/000026184. [DOI] [PubMed] [Google Scholar]
- Imperiale MJ. The human polyomaviruses, BKV and JCV: Molecular pathogensis of acute disease and potential role in cancer. Virology. 2000;267:1–7. doi: 10.1006/viro.1999.0092. [DOI] [PubMed] [Google Scholar]
- Kestler Harry W, III, Ringler Douglas J, Mori Kazuyasu, Panicali Dennis L, Sehgal Prabhat K, Daniel Muthiah D, Desrosiers Ronald Importance of the nef gene for maintenance of high virus loads and for. Cell. 1991;65:651–662. doi: 10.1016/0092-8674(91)90097-i. [DOI] [PubMed] [Google Scholar]
- Kleinschmidt-DeMasters BK, Tyler KL. Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta-1a for multiple sclerosis. N Engl J Med. 2005;353(4):369–374. doi: 10.1056/NEJMoa051782. [DOI] [PubMed] [Google Scholar]
- Koralnik IJ. Progressive multifocal leukoencephalopathy revisited: Has the disease outgrown its name? Ann Neurol. 2006;60(2):162–173. doi: 10.1002/ana.20933. [DOI] [PubMed] [Google Scholar]
- Langer-Gould A, Atlas SW, Green AJ, Bollen AW, Pelletier D. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N Engl J Med. 2005;353(4):375–381. doi: 10.1056/NEJMoa051847. [DOI] [PubMed] [Google Scholar]
- Lima MA, Katz-Brull R, Lenkinski RE, Nunez R, Feinrider D, Koralnik IJ. Remission of progressive multifocal leukoencephalopathy and primary central nervous system lymphoma in an HIV-infected patient. Eur J Neurol. 2007;14(6):598–602. doi: 10.1111/j.1468-1331.2007.01820.x. [DOI] [PubMed] [Google Scholar]
- Liu CK, Atwood WJ. Propagation and assay of the JC virus. Methods Mol Biol. 2001;165:9–17. doi: 10.1385/1-59259-117-5:9. [DOI] [PubMed] [Google Scholar]
- Liu CK, Wei G, Atwood WJ. Infection of glial cells by the human polyomavirus JC is mediated by an N-linked glycoprotein containing terminal alpha 2–6 linked sialic acids. J. Virol. 1998;72:4643–4649. doi: 10.1128/jvi.72.6.4643-4649.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major EO, Miller AE, Mourrain P, Traub RG, de Widt E, Sever J. Establishment of a line of human fetal glial cells that supports JC virus multiplication. Proc.Natl.Acad.Sci.U.S.A. 1985;82:1257–1261. doi: 10.1073/pnas.82.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNees AL, White ZS, Zanwar P, Vilchez RA, Butel JS. Specific and quantitative detection of human polyomaviruses BKV, JCV, and SV40 by real time PCR. J Clin Virol. 2005;34(1):52–62. doi: 10.1016/j.jcv.2004.12.018. [DOI] [PubMed] [Google Scholar]
- Okada Y, Sawa H, Tanaka S, Takada A, Suzuki S, Hasegawa H, Umemura T, Fujisawa J, Tanaka Y, Hall WW, Nagashima K. Transcriptional activation of JC virus by human T-lymphotropic virus type I Tax protein in human neuronal cell lines. J Biol Chem. 2000;275(22):17016–17023. doi: 10.1074/jbc.275.22.17016. [DOI] [PubMed] [Google Scholar]
- Raj GV, Khalili K. Identification and characterization of a novel GGA/C-binding protein, GBP-i, that is rapidly inducible by cytokines. Mol Cell Biol. 1994;14(12):7770–7781. doi: 10.1128/mcb.14.12.7770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GP, Hartung HP, Calabresi PA. Anti-alpha4 integrin therapy for multiple sclerosis: mechanisms and rationale. Neurology. 2005;64(8):1336–1342. doi: 10.1212/01.WNL.0000158329.30470.D0. [DOI] [PubMed] [Google Scholar]
- Shah KV, Daniel R, Warszawski R. High prevalence of antibodies to BK virus, a SV40 related papovavirus, in residents of Maryland. J. Infect. Dis. 1973;128:784. doi: 10.1093/infdis/128.6.784. [DOI] [PubMed] [Google Scholar]
- Telenti A, Marshall W, Aksamit A, Smilack J, Smith T. Detection of JC virus by polymerase chain reaction in cerebrospinal fluid. Eur.J.Clin.Microbiol.Infect.Dis. 1992;11:253–254. doi: 10.1007/BF02098091. [DOI] [PubMed] [Google Scholar]
- Van Assche G, Van Ranst M, Sciot R, Dubois B, Vermeire S, Noman M, Verbeeck J, Geboes K, Robberecht W, Rutgeerts P. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn's disease. N Engl J Med. 2005;353(4):362–368. doi: 10.1056/NEJMoa051586. [DOI] [PubMed] [Google Scholar]
- Verma S, Ziegler K, Ananthula P, Co JK, Frisque RJ, Yanagihara R, Nerurkar VR. JC virus induces altered patterns of cellular gene expression: interferon-inducible genes as major transcriptional targets. Virology. 2006;345(2):457–467. doi: 10.1016/j.virol.2005.10.012. [DOI] [PubMed] [Google Scholar]