

Abstract
Sulindac has been reported to be effective in suppressing tumor growth through the induction of p21WAF1/cip1 in human, animal models of colon cancer and colon cancer cells. In this study, we treated human breast cancer cell line MCF-7 and lung cancer cell line A549 as well as colon cancer cell line SW620 with sulindac to observe the effects of sulindac in other tissue sites. In all cell lines, proliferation was significantly inhibited by sulindac after 24 and 72 h of treatment. Apoptosis was induced by sulindac in both lung cancer cells and colon cancer cells but was not induced in breast cancer cells. Western blots showed that p21 protein level were induced by sulindac in lung cancer cells and colon cancer cells, but not in breast cancer cells. Most importantly, the suppression of β-catenin, a key mediator of Wnt signaling pathway, was seen in all three cell lines with sulindac administration. Further studies revealed that transcriptional activities of β-catenin were significantly inhibited by sulindac and that the inhibition was sulindac dosage-dependent. The transcriptional targets of β-catenin, c-myc, cyclin D1 and cdk 4 were also dramatically downregulated. In conclusion, our data demonstrated that the efficacy of sulindac in the inhibition of cell proliferation (rather than the induction of apoptosis) might be through the suppression of β-catenin pathway in human cancer cells.
Keywords: sulindac, β-catenin, p21WAF1/cip1, proliferation, apoptosis, cancer
1 Introduction
Sulindac, a nonsteroidal anti-inflammatory drug (NSAID), is known to prevent recurrence and reduce colorectal polyps in number and size in patients with familial adenomatous polyposi (Clevers, 2006; Kelloff et al., 2006; Szabo, 2006). The molecular mechanisms underlying these biological effects are not completely understood. Inhibition of cyclooxygenase-2 (COX-2) underlies part of this effect. However, COX-independent mechanisms may also play a role, since NSAIDs inhibit the growth of colon cancer cell lines lacking COX-2 expression (Smith et al., 2000; Zhang et al., 1999). Our previous study showed that cyclin dependent kinase (CDK) inhibitor p21WAF/cip1 was an important determinant of intestinal cell response to sulindac in vitro and in vivo (Yang et al., 2005a; Yang et al., 2001). Our recent study demonstrated that c-jun NH2-terminal kinase 1 (JNK1) was synergistic with p21WAF1 to inhibit cell proliferation and induced apoptosis in vitro and in vivo by sulindac (Song et al., 2007). Other studies have shown that β-catenin could be a target for NSAIDs in colorectal adenomas of patients and colorectal cancer cell lines (Boon et al., 2004; Bordonaro et al., 1999; Dihlmann et al., 2001; Gardner et al., 2004).
The Wnt/β-catenin signaling pathway is tightly regulated and has important functions in development, tissue homeostasis, and regeneration. Oncogenic activation of the Wnt-signalling pathway by mutations in Adenomatous polyposis coli (APC) or β-catenin, which results in the accumulation and nuclear translocation of β-catenin and in β-catenin/T-cell factor (TCF) 4 - regulated transcription of TCF target genes such as cyclin D1 and c-MYC, is mandatory for the initial neoplastic transformation of intestinal epithelium (Wong and Pignatelli, 2002). Recent studies also found that activation of Wnt/β-catenin signaling is important for both initiation and progression of cancers of different tissues/organs, including liver (Lee et al., 2006), prostate (Terry et al., 2006), breast (Turashvili et al., 2006), esophagus (Clement et al., 2007) and lung (Mazieres et al., 2005). Thus, Wnt/β-catenin signaling is becoming a promising target for chemoprevention and chemotherapy (Herbst and Kolligs, 2007; Luo et al., 2007).
In the present study, we determined the effects of sulindac on breast and lung cancer cells as well as colorectal cancer cells. Our results demonstrated that sulindac inhibited human cancer cell proliferation in breast, lung and colon cancer cells, which was associated with suppression of β-catenin expression and decrease of transcriptional activities and its transcriptional targets cyclin D1, c-myc and cdk4, and that sulindac-induced apoptosis in cancer cells was mainly associated with induction of p21WAF1/cip1.
2. Materials and methods
2. 1 Cell lines and cell culture
Colon cancer cell line SW620, breast cancer cell line MCF7 and lung cancer cell line A549 were purchased from the American Type Culture Collection (Manassas, VA). All cells were maintained in MEM medium. The medium was supplemented with 10% (v/v) fetal bovine serum (FBS), 1 × antibiotic/antimycotic (100 units/ml streptomycin, 100 units/ml penicillin, and 0.25 μg/ml amphotericin B). All cell lines were cultured in humidified incubator at 37 °C with 5% CO2. Sulindac (Sigma, St. Louis, MO) was dissolved in dimethyl sulfoxide (DMSO) and diluted in a series concentration.
2.2 Cell proliferation assay
As described previously (Song et al., 2007), 5 ×103 cells were seeded in each well of 96-well plate and incubated overnight. The medium was removed. 100 μl of full assay medium with the final concentration of sulindac from 0 to 3.2 mM was added to each well, DMSO was used as control. All groups were triplicated. After 24 h and 72 h exposure to sulindac, cell proliferation was determined by 3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay (CellTiter 96 Non-Radioactive Cell Proliferation Assay Kit, Promega Corporation, Madison, WI).
2.3 Analysis of apoptosis
6 × 105 cells were seeded in 6-well plates and incubated overnight or till 50-60% confluence. Sulindac was added to medium at a final concentration of 1.6mM. DMSO was used as control. The cells were treated with sulindac and harvested at different time point of 0, 8, 24 and 48 h, washed in cold PBS, then fixed with 80% ethanol for 8 h at 4 °C, then stained with propidium iodide buffer (50 μg/ml propidium iodide, 0.1% sodium citrate, and 0.1% Triton X-100) for 3 h at 4 °C. 10,000 cells were analyzed for apoptosis using a Becton Dickinson FACScan (Becton Dickinson Immunocytometry Systems, San Jose, CA). The percentage of apoptotic cells were quantified using Cell Quest software. Same experiments were triplicated independently.
2.4 Western blot analysis
At different time point, cells treated with sulindac at the final concentration of 1.6mM were harvested. The cell pellet was washed twice with ice-cold PBS and lysed with lysis buffer. 30 μg of protein was loaded and separated in 12% SDS-PAGE gel and transferred to polyvinylidine difluoride membranes (Millipore, Bedford, MA). The following antibodies were used to probe the alterations of protein: anti-β-catenin, anti-p21WAF1/cip1, anti-p27/kip1 (Santa Cruz Biotechnology, Santa Cruz, CA), anti-CDK4 (BD PharMingen, San Diego, CA), anti-c-myc (Sigma, St. Louis, MO), and anti-Cyclin D1 (Zymed, South San Francisco, CA). Signal was detected by enhanced chemoluminescence techniques (Amersham Life Science, Piscataway, NJ), as described previously (Yang et al., 2005b). β-actin (Sigma, St. Louis, MO) was used as loading control.
2.5 Transfection and Luciferase Reporter Assays
To examine the effect of sulindac on β-catenin/TCF signaling, cells were plated in 24-well plate at a density of 2.5 × 105 cells/well and incubate overnight in FBS-supplemented medium. In the second day, cells were transiently transfected with the β-catenin-TCF luciferase reporter construct TOPFlash which contains multiple optimal TCF/LEF (lymphocyte enhancing factor) binding sites that induce transcription of a luciferase reporter gene when activated by β-catenin, or FOPFlash which contains mutant and inactivated TCF/LEF binding sites, respectively (Upstate Biotechnology, Lake Placid, NY). To correct the differences caused by transfection efficiency, cells were co-transfected with a Renilla reporter plasmid using LipofectAMINE 2000 according to the manufacturer's protocol. 6 h after transfection, the medium was removed and the cells were treated with fresh medium supplemented with 1.6 mM sulindac for 24 and 48 h, or treated with 0, 0.8 or 1.6 mM sulindac for 24 h. DMSO was used as control. Cells were washed with cold PBS and lysed with passive lysis buffer. Luciferase activity was measured with the Dual Luciferase Report Assay System (Promega Corporation, Madison, WI). Lysate firefly luciferase values were normalized to Renilla luciferase activity. Same experiments were triplicated independently.
3. Results
3.1 Sulindac inhibited proliferation of colorectal, breast and lung cancer cells
We and others have reported that sulindac is an effective chemopreventive agent in the prevention of colorectal cancer in vivo and in vitro (Boolbol et al., 1996; Lipkin, 1997; Rao et al., 1995; Reddy et al., 1999; Song et al., 2007; Yang et al., 2005b; Yang et al., 2001). In this study, sulindac was used to elucidate its anticancer ability to breast cancer cell line MCF7 and lung cancer cell line A549, as well as colon cancer cell line SW620. Sulindac was added to the media at a final concentration of 0, 0.8, 1.2, 1.6, 2.4 and 3.2 mM. After 24 h incubation, sulindac inhibited cell growth in a dosage-dependent manner in all three cell lines examined by MTT assay (Fig.1). Moreover, the dramatic effects were seen in all three cell lines after 72 h exposure to sulindac (Fig.1). As our previous documentation shows, colon cancer cell line SW620 was sensitive to sulindac at a concentration of 1.2 mM, but the breast cancer cell line MCF7 and lung cancer cell line A549 showed sensitive to sulindac at a little higher concentration of 1.6 mM. The proliferation rate of cells was not changed when the medium was supplemented with DMSO which was used as control. These data provides a direct evidence that sulindac could be used to prevent cancer cell proliferation from other sites, like breast and lung, as well as colon and rectum.
Figure 1.

Sulindac inhibited cancer cell proliferation in dosage-dependent and time-dependent manner: A, SW620; B, A549; C, MCF7. Cells were treated with sulindac at the final concentration from 0 to 3.2 mM for 24 h and 72 h. DMSO was used as control. Cell proliferation was determined using MTT assay. The results stood for the mean of cell proliferation (%) ± SD of triplicated determinations. (* P < 0.01, compared to the control).
3.2 Sulindac induced apoptosis in SW620 and A549 cells, but did not induce apoptosis in MCF7 cells
To determine whether sulindac has a function in inducing apoptosis in the breast and lung cancer cells like in colorectal cancer cells, flow cytometry was used to analyze apoptotic events in three cell lines with treatment of sulindac at the final concentration of 1.6 mM for 8, 24 or 48 h. As shown in figures 2A and 2B, sulindac, indeed, significantly induced apoptosis in both colorectal cancer cells and lung cancer cells and was time-dependent, but the induction of apoptosis in lung cancer cells was far less than that in colorectal cancer cells. However, the induction of apoptosis was not seen in the breast cancer cells MCF7, even with treatment of 48 h (Fig. 2C), or with longer treatment of 72 h or higher concentration (2.4 mM) of sulindac (data not shown). Thus, sulindac could be a good preventive agent in the inhibition of cell proliferation rather than the induction of apoptosis in breast and lung cancer cells, unlike in colorectal cancer cells.
Figure 2.

Sulindac induced apoptosis in colorectal (A) and lung cancer cells (B), did not induce apoptosis in breast cancer cells (C). The cells were treated with 1.6mM sulindac for 0, 8, 24 and 48 h. The DMSO was used as control. Cells were fixed with 80% ethanol for 8 h at 4 °C, then stained with propidium iodide buffer (50 μg/ml propidium iodide, 0.1% sodium citrate, and 0.1% Triton X-100) for 3 h at 4 °C. Apoptosis was analyzed using a Becton Dickinson FACScan. Columns: average from three independent experiments; bars, standard deviation. (* P < 0.01 in the comparison with control).
3.3 β-catenin expression was suppressed by sulindac in cancer cell lines
To determine whether sulindac inhibition of breast and lung cancer cell proliferation was through reduction of cyclooxygenase 2 (COX-2) since sulindac is a COX-2 selective inhibitor (Keller and Giardiello, 2003), or was through the induction of p21WAF1/cip1 similar as in colorectal cancer cells, the expression of COX-2 and p21 was analyzed by western blot. As documented previously, COX-2 was not detectable in the three cancer cell lines because COX-2 is highly expressed in stromal cells. However, in the colorectal and lung cancer cell lines, p21 was significantly induced by sulindac in colorectal cancer cell line SW620 (Fig. 3A) and lung cancer cell line A549 (Fig.3B), but the induction of p21 was not seen in breast cancer cell line MCF7 (Fig. 3C), which was consistent with the induction of apoptosis. Thus, there must be some genes other than p21, that were involved in the inhibition of cell proliferation in both breast and lung cancer cells.
Figure 3.
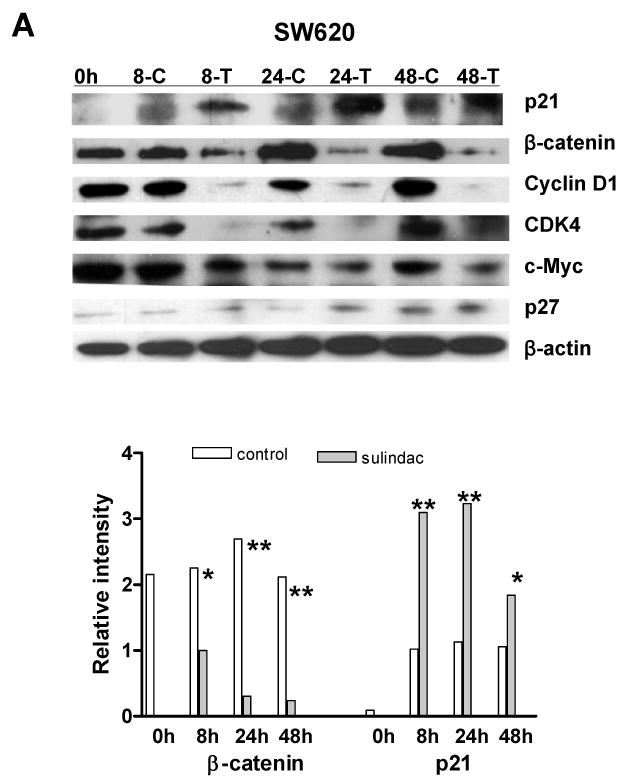
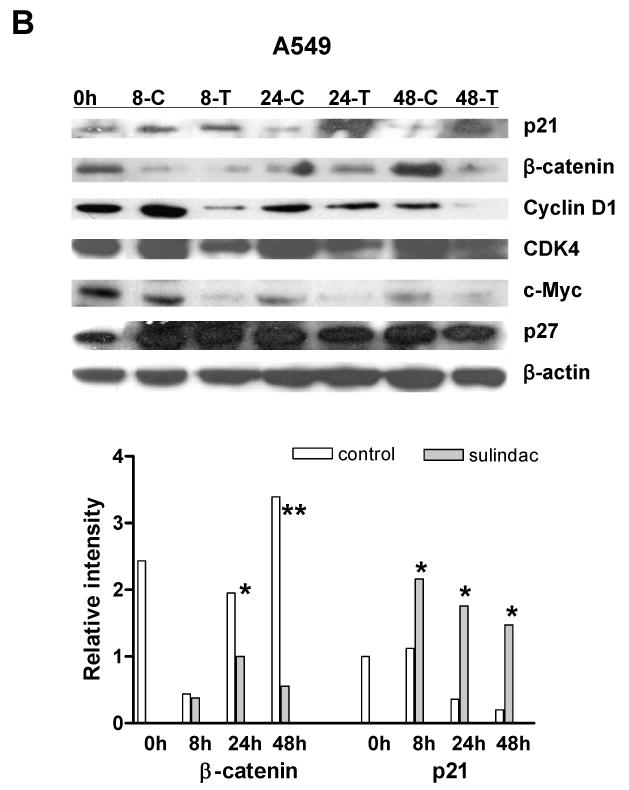
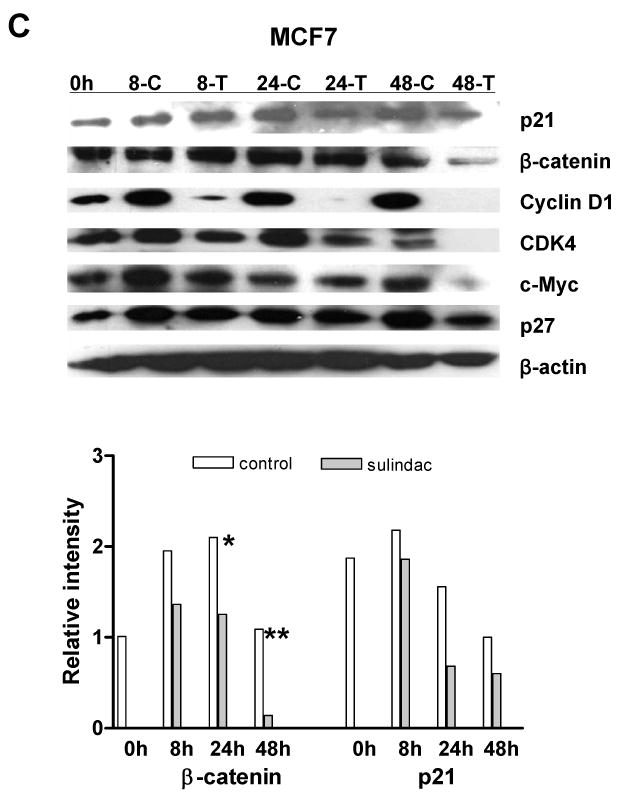
The expression of β-catenin, cyclin D1, c-myc and cdk4 were significantly suppressed by sulindac in cancer cell lines: A, SW620; B, A549; C, MCF7. The cells were treated with 1.6mM sulindac, collected at different time point and lysed for immuoblotting with antibodies including p21, β-catenin, cyclin D1, CDK4, c-Myc and p27. Lower panels indicated the quantification of signal intensity changes of β-catenin and p21. β-actin was used as sample loading control. (* P< 0.05, ** P< 0.01 in comparison with control). (C: DMSO control; T, sulindac treatment).
Previous studies have reported that β-catenin, a key player in Apc/β-catenin/TCF/c-myc pathway, might be a target of NSAIDs in cell cycle regulation (Boon et al., 2004; Chang et al., 2005; Dihlmann et al., 2001; Gardner et al., 2004), leading us to explore whether β-catenin was changed and if the alteration of β-catenin had been involved in the inhibition of cell proliferation. To our surprise, β-catenin was dramatically suppressed in the breast cancer cell line MCF7 and lung cancer cell line A549, as seen in colorectal cancer cell line SW620, by sulindac after 8 h, 24 h and 48 h, when analyzed by western blotting (Fig. 3). The transcriptional targets of β-catenin -- c-myc, cyclin D1 and cdk4, were also significantly downregulated in all three cancer cell lines (Fig. 3). But, another cyclin dependent kinase inhibitor, p27kip1 was not induced by sulindac, which was consistent with our previous report (Yang et al., 2005b).
3.4 Sulindac suppressed β-catenin-mediated transcriptional activities
To determine whether the suppression of β-catenin by sulindac corresponded to the suppression of β-catenin-mediated transcriptional activities, all three cancer cell lines were transiently co-transfected with the TCF/LEF luciferase reporter construct (TopFlash) or mutant TCF/LEF luciferase reporter construct (FopFlash) and Renilla reporter plasmid. First, sulindac significantly reduced luciferase activity (TopFlash) in all three cell lines after 24 h treatment compared to FopFlash. The reduction of transcriptional activities was in a dosage-dependent manner (Fig. 4). Second, luciferase activities were dramatically reduced in all three cell lines after 24 and 48 h of sulindac treatment compared to the control (DMSO) groups (Fig. 4). All three cell lines treated with sulindac for 24 and 48 h showed 10- to 20 – fold reductions in luciferase activity, respectively. Therefore, the reduction of β-catenin transcriptional activity was in turn to cause the decrease of its transcriptional target gene expression, in terms of the downregulated expression of c-myc, cyclin D1 and cdk4 protein (Fig. 3), leading to cancer cell growth arrest significantly (Fig.1).
Figure 4.

Sulindac suppressed β-catenin-mediated gene transcription. (A) SW620, (B) A549, and (C) MCF7 cells were transiently co-transfected with TopFlash or FopFlash and Renilla reporter plasmid with Lipofectamine 2000. 6 h following transfection, cells were treated with 0, 0.8 or 1.6mM sulindac for 24 h for dosage-dependent assay. In a time-dependent assay, cells were harvested at 24 and 48 h after 1.6 mM of sulindac treatment. DMSO was used as control. Luciferase activity was normalized for transfection efficiency with Renilla reporter activity. Up panel: relative luciferase activities of TopFlash and FopFlash. The reduction of transcriptional activities was sulindac dosage-dependent. (* P<0.01, ** P<0.05, compared to 0 mM). Low panel: ratio of TopFlash/Renilla. The transcriptional activities were reduced by sulindac at 24 and 48 h. Columns: average from three independent experiments; bars, standard deviation. (* P<0.01, compared to DMSO control).
4. Discussion
Our data provides a direct evidence that sulindac inhibits cell proliferation (rather than induces apoptosis) in breast and lung cancer cells, as well as colorectal cancer cells, which is associated with the suppression of β-catenin; and that sulindac induces apoptosis in colon and lung cancer cells, which is associated with the reduction of β-catenin and the induction of p21WAF/cip1. Thus, in addition to colorectal cancer, the Wnt/β-catenin signaling might be a target of sulindac in lung cancer and breast cancer.
The growing evidence has indicated that sulindac targets nuclear β-catenin accumulation and Wnt signaling in colorectal cancers in vivo and in vitro (Boon et al., 2004; Chang et al., 2005; Gardner et al., 2004; He et al., 1999; Koornstra et al., 2005; Rice et al., 2003; Rice et al., 2006), but that sulindac targeting β-catenin in other sites is rare. Even recent report indicated that NSAIDs inhibition of β-catenin required the high level expression of peroxisome proliferators-activated receptor γ (PPAR-γ) and its co-receptor retinoid-X-receptor α (RXR-α) (Lu et al., 2005), how they are interacting to the response to NSAIDs remains unclear and needs further investigation.
Hyperactivation of the canonical Wnt/β-catenin pathway, caused by mutations of β-catenin, APC and axin, is one of the most frequent abnormalities in human cancers, particularly colorectal cancer, but such mutation in breast cancer is rare. However, several studies have indicated the importance of Wnt signaling pathways in human breast cancer, and there is strong confirmation for an elevated level of nuclear and/or cytoplasmic β-catenin detected by immunohistochemical staining in breast carcinomas (Jonsson et al., 2000; Lin et al., 2000; Ryo et al., 2001; Turashvili et al., 2006) which correlates with the expression of β-catenin target gene cyclin D1 and poor patient prognosis (Ryo et al., 2001). However, nuclear or cytoplasmic localization of β-catenin is not detectable in normal breast tissues. In the present study, we found that sulindac targeted β-catenin in breast cancer cells, decreasing β-catenin transcriptional activities, suppressing the expression of β-catenin and its target genes cyclin D1, cdk4 and c-myc, and in turn inhibited cell proliferation. We also noted that the induction of apoptosis, unlike in colorectal and lung cancer cells, was not observed in the breast cancer cells, which might be due to the non-induction of p21 by sulindac. Thus, suppressing β-catenin expression should be a potential approach to chemoprevention and chemotherapy of breast cancer.
Mutation of the β-catenin gene is also uncommon in lung cancer (Ueda et al., 2001). However, overexpression of β-catenin has been reported in lung cancer. It has been reported that increased expression of β-catenin is associated with a high proliferative index and surprisingly with a better prognosis (Hommura et al., 2002). In fact, the real significance of β-catenin expression in lung cancer is still controversial and this might be linked to the complexity of its effects since β-catenin also functions as a cadherin-mediated cell adhesion component (Barker et al., 2000). Our present study demonstrated that both transcriptional activities and protein levels were significantly inhibited by sulindac in lung cancer, which were cooperated with the induction of p21 to inhibit proliferation and to induce apoptosis, with the effects of suppression of cyclin D1, c-myc and cdk4. Thus, similar as observed in colorectal cancer and breast cancer, β-catenin could be uses as a target for sulindac in the chemoprevention and chemotherapy of lung cancer.
In summary, our data demonstrates that sulindac suppresses β-catenin signaling pathway in lung cancer and breast cancer, in addition to colorectal cancer. The inhibition of cell proliferation and induction of apoptosis in lung cancer and colon cancer cells with sulindac administration provide important evidence for cancer chemotherapy. Therefore, targeted inhibition of Wnt/beta-catenin signaling is a rational and promising new approach for the therapy of cancers of various origins.
Acknowledgments
We would like to thank Drs. Barbara Heerdt and Tianhong Li (Albert Einstein Cancer Center, Bronx, NY) and Drs. Jonna Frasor and Zuoming Sun (University of Illinois at Chicago, Chicago, IL) for providing reagents.
Grants support: This work was supported in part by the National Cancer Institute grant R01CA112081 (to W. Yang) and the American Institute for Cancer Research grant 05A121 (to W. Yang).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barker N, Morin PJ, Clevers H. The Yin-Yang of TCF/beta-catenin signaling. Adv Cancer Res. 2000;77:1–24. doi: 10.1016/s0065-230x(08)60783-6. [DOI] [PubMed] [Google Scholar]
- Boolbol SK, Dannenberg AJ, Chadburn A, Martucci C, Guo XJ, Ramonetti JT, Abreu-Goris M, Newmark HL, Lipkin ML, DeCosse JJ, Bertagnolli MM. Cyclooxygenase-2 overexpression and tumor formation are blocked by sulindac in a murine model of familial adenomatous polyposis. Cancer Res. 1996;56:2556–2560. [PubMed] [Google Scholar]
- Boon EM, Keller JJ, Wormhoudt TA, Giardiello FM, Offerhaus GJ, van der Neut R, Pals ST. Sulindac targets nuclear beta-catenin accumulation and Wnt signalling in adenomas of patients with familial adenomatous polyposis and in human colorectal cancer cell lines. Br J Cancer. 2004;90:224–229. doi: 10.1038/sj.bjc.6601505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordonaro M, Mariadason JM, Aslam F, Heerdt BG, Augenlicht LH. Butyrate-induced apoptotic cascade in colonic carcinoma cells: modulation of the beta-catenin-Tcf pathway and concordance with effects of sulindac and trichostatin A but not curcumin. Cell Growth Differ. 1999;10:713–720. [PubMed] [Google Scholar]
- Chang WC, Everley LC, Pfeiffer GR, 2nd, Cooper HS, Barusevicius A, Clapper ML. Sulindac sulfone is most effective in modulating beta-catenin-mediated transcription in cells with mutant APC. Ann N Y Acad Sci. 2005;1059:41–55. doi: 10.1196/annals.1339.020. [DOI] [PubMed] [Google Scholar]
- Clement G, Jablons DM, Benhattar J. Targeting the Wnt signaling pathway to treat Barrett's esophagus. Expert Opin Ther Targets. 2007;11:375–389. doi: 10.1517/14728222.11.3.375. [DOI] [PubMed] [Google Scholar]
- Clevers H. Colon cancer--understanding how NSAIDs work. N Engl J Med. 2006;354:761–763. doi: 10.1056/NEJMcibr055457. [DOI] [PubMed] [Google Scholar]
- Dihlmann S, Siermann A, von Knebel Doeberitz M. The nonsteroidal anti-inflammatory drugs aspirin and indomethacin attenuate beta-catenin/TCF-4 signaling. Oncogene. 2001;20:645–653. doi: 10.1038/sj.onc.1204123. [DOI] [PubMed] [Google Scholar]
- Gardner SH, Hawcroft G, Hull MA. Effect of nonsteroidal anti-inflammatory drugs on beta-catenin protein levels and catenin-related transcription in human colorectal cancer cells. Br J Cancer. 2004;91:153–163. doi: 10.1038/sj.bjc.6601901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He TC, Chan TA, Vogelstein B, Kinzler KW. PPARdelta is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell. 1999;99:335–345. doi: 10.1016/s0092-8674(00)81664-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst A, Kolligs FT. Wnt signaling as a therapeutic target for cancer. Methods Mol Biol. 2007;361:63–91. doi: 10.1385/1-59745-208-4:63. [DOI] [PubMed] [Google Scholar]
- Hommura F, Furuuchi K, Yamazaki K, Ogura S, Kinoshita I, Shimizu M, Moriuchi T, Katoh H, Nishimura M, Dosaka-Akita H. Increased expression of beta-catenin predicts better prognosis in nonsmall cell lung carcinomas. Cancer. 2002;94:752–758. doi: 10.1002/cncr.10213. [DOI] [PubMed] [Google Scholar]
- Jonsson M, Borg A, Nilbert M, Andersson T. Involvement of adenomatous polyposis coli (APC)/beta-catenin signalling in human breast cancer. Eur J Cancer. 2000;36:242–248. doi: 10.1016/s0959-8049(99)00276-2. [DOI] [PubMed] [Google Scholar]
- Keller JJ, Giardiello FM. Chemoprevention strategies using NSAIDs and COX-2 inhibitors. Cancer Biol Ther. 2003;2:S140–149. [PubMed] [Google Scholar]
- Kelloff GJ, Lippman SM, Dannenberg AJ, Sigman CC, Pearce HL, Reid BJ, Szabo E, Jordan VC, Spitz MR, Mills GB, Papadimitrakopoulou VA, Lotan R, Aggarwal BB, Bresalier RS, Kim J, Arun B, Lu KH, Thomas ME, Rhodes HE, Brewer MA, Follen M, Shin DM, Parnes HL, Siegfried JM, Evans AA, Blot WJ, Chow WH, Blount PL, Maley CC, Wang KK, Lam S, Lee JJ, Dubinett SM, Engstrom PF, Meyskens FL, Jr, O'Shaughnessy J, Hawk ET, Levin B, Nelson WG, Hong WK. Progress in chemoprevention drug development: the promise of molecular biomarkers for prevention of intraepithelial neoplasia and cancer--a plan to move forward. Clin Cancer Res. 2006;12:3661–3697. doi: 10.1158/1078-0432.CCR-06-1104. [DOI] [PubMed] [Google Scholar]
- Koornstra JJ, Rijcken FE, Oldenhuis CN, Zwart N, van der Sluis T, Hollema H, deVries EG, Keller JJ, Offerhaus JA, Giardiello FM, Kleibeuker JH. Sulindac inhibits beta-catenin expression in normal-appearing colon of hereditary nonpolyposis colorectal cancer and familial adenomatous polyposis patients. Cancer Epidemiol Biomarkers Prev. 2005;14:1608–1612. doi: 10.1158/1055-9965.EPI-05-0112. [DOI] [PubMed] [Google Scholar]
- Lee HC, Kim M, Wands JR. Wnt/Frizzled signaling in hepatocellular carcinoma. Front Biosci. 2006;11:1901–1915. doi: 10.2741/1933. [DOI] [PubMed] [Google Scholar]
- Lin SY, Xia W, Wang JC, Kwong KY, Spohn B, Wen Y, Pestell RG, Hung MC. Beta-catenin, a novel prognostic marker for breast cancer: its roles in cyclin D1 expression and cancer progression. Proc Natl Acad Sci U S A. 2000;97:4262–4266. doi: 10.1073/pnas.060025397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipkin M. New rodent models for studies of chemopreventive agents. J Cell Biochem. 1997 28-29:144–147. [PubMed] [Google Scholar]
- Lu D, Cottam HB, Corr M, Carson DA. Repression of beta-catenin function in malignant cells by nonsteroidal antiinflammatory drugs. Proc Natl Acad Sci U S A. 2005;102:18567–18571. doi: 10.1073/pnas.0509316102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Chen J, Deng ZL, Luo X, Song WX, Sharff KA, Tang N, Haydon RC, Luu HH, He TC. Wnt signaling and human diseases: what are the therapeutic implications? Lab Invest. 2007;87:97–103. doi: 10.1038/labinvest.3700509. [DOI] [PubMed] [Google Scholar]
- Mazieres J, He B, You L, Xu Z, Jablons DM. Wnt signaling in lung cancer. Cancer Lett. 2005;222:1–10. doi: 10.1016/j.canlet.2004.08.040. [DOI] [PubMed] [Google Scholar]
- Rao CV, Rivenson A, Simi B, Zang E, Kelloff G, Steele V, Reddy BS. Chemoprevention of colon carcinogenesis by sulindac, a nonsteroidal antiinflammatory agent. Cancer Res. 1995;55:1464–1472. [PubMed] [Google Scholar]
- Reddy BS, Kawamori T, Lubet RA, Steele VE, Kelloff GJ, Rao CV. Chemopreventive efficacy of sulindac sulfone against colon cancer depends on time of administration during carcinogenic process. Cancer Res. 1999;59:3387–3391. [PubMed] [Google Scholar]
- Rice PL, Kelloff J, Sullivan H, Driggers LJ, Beard KS, Kuwada S, Piazza G, Ahnen DJ. Sulindac metabolites induce caspase- and proteasome-dependent degradation of beta-catenin protein in human colon cancer cells. Mol Cancer Ther. 2003;2:885–892. [PubMed] [Google Scholar]
- Rice PL, Peters SL, Beard KS, Ahnen DJ. Sulindac independently modulates extracellular signal-regulated kinase 1/2 and cyclic GMP-dependent protein kinase signaling pathways. Mol Cancer Ther. 2006;5:746–754. doi: 10.1158/1535-7163.MCT-05-0210. [DOI] [PubMed] [Google Scholar]
- Ryo A, Nakamura M, Wulf G, Liou YC, Lu KP. Pin1 regulates turnover and subcellular localization of beta-catenin by inhibiting its interaction with APC. Nat Cell Biol. 2001;3:793–801. doi: 10.1038/ncb0901-793. [DOI] [PubMed] [Google Scholar]
- Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- Song Z, Tong C, Liang J, Dockendorff A, Huang C, Augenlicht LH, Yang W. JNK1 is required for sulindac-mediated inhibition of cell proliferation and induction of apoptosis in vitro and in vivo. Eur J Pharmacol. 2007;560:95–100. doi: 10.1016/j.ejphar.2007.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo E. Selecting targets for cancer prevention: where do we go from here? Nat Rev Cancer. 2006;6:867–874. doi: 10.1038/nrc2008. [DOI] [PubMed] [Google Scholar]
- Terry S, Yang X, Chen MW, Vacherot F, Buttyan R. Multifaceted interaction between the androgen and Wnt signaling pathways and the implication for prostate cancer. J Cell Biochem. 2006;99:402–410. doi: 10.1002/jcb.20983. [DOI] [PubMed] [Google Scholar]
- Turashvili G, Bouchal J, Burkadze G, Kolar Z. Wnt signaling pathway in mammary gland development and carcinogenesis. Pathobiology. 2006;73:213–223. doi: 10.1159/000098207. [DOI] [PubMed] [Google Scholar]
- Ueda M, Gemmill RM, West J, Winn R, Sugita M, Tanaka N, Ueki M, Drabkin HA. Mutations of the beta- and gamma-catenin genes are uncommon in human lung, breast, kidney, cervical and ovarian carcinomas. Br J Cancer. 2001;85:64–68. doi: 10.1054/bjoc.2001.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong NA, Pignatelli M. Beta-catenin--a linchpin in colorectal carcinogenesis? Am J Pathol. 2002;160:389–401. doi: 10.1016/s0002-9440(10)64856-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Bancroft L, Augenlicht LH. Methylation in the p21(WAF1/cip1) promoter of Apc(+/-), p21(+/-) mice and lack of response to sulindac. Oncogene. 2005a;24:2104–2109. doi: 10.1038/sj.onc.1208444. [DOI] [PubMed] [Google Scholar]
- Yang W, Bancroft L, Liang J, Zhuang M, Augenlicht LH. p27kip1 in intestinal tumorigenesis and chemoprevention in the mouse. Cancer Res. 2005b;65:9363–9368. doi: 10.1158/0008-5472.CAN-05-2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Velcich A, Mariadason J, Nicholas C, Corner G, Houston M, Edelmann W, Kucherlapati R, Holt PR, Augenlicht LH. p21(WAF1/cip1) is an important determinant of intestinal cell response to sulindac in vitro and in vivo. Cancer Res. 2001;61:6297–6302. [PubMed] [Google Scholar]
- Zhang X, Morham SG, Langenbach R, Young DA. Malignant transformation and antineoplastic actions of nonsteroidal antiinflammatory drugs (NSAIDs) on cyclooxygenase-null embryo fibroblasts. J Exp Med. 1999;190:451–459. doi: 10.1084/jem.190.4.451. [DOI] [PMC free article] [PubMed] [Google Scholar]