

Abstract
Synopsis and pictogram: Gated pores in the ferritin family of protein nanocages, illustrated in the pictogram, control transfer of ferrous iron into and out of the cages by regulating contact between hydrated ferric oxide mineral inside the protein cage, and reductants such as FMNH2 on the outside. The structural and functional homology between the gated ion channel proteins in inaccessible membranes and gated ferritin pores in the stable, water soluble nanoprotein, make studies of ferritin pores models for gated pores in many ion channel proteins.
Properties of ferritin gated pores, which control rates of FMNH2 reduction of ferric iron in hydrated oxide minerals inside the protein nanocage, are discussed in terms of the conserved pore gate residues (arginine 72-apspartate 122 and leucine 110-leucine 134), of pore sensitivity to heat at temperatures 30 °C below that of the nanocage itself, and of pore sensitivity to physiological changes in urea (1–10 mM). Conditions which alter ferritin pore structure/function in solution, coupled with the high evolutionary conservation of the pore gates, suggest the presence of molecular regulators in vivo that recognize the pore gates and hold them either closed or open, depending on biological iron need. The apparent homology between ferrous ion transport through gated pores in the ferritin nanocage and ion transport through gated pores in ion channel proteins embedded in cell membranes, make studies of water soluble ferritin and the pore gating folding/unfolding a useful model for other gated pores.
1. Introduction
Rates of inorganic ion movement through organic barriers in biology are regulated by gated pores in channel proteins, usually in membranes. The movement of ferrous ions move through gated pores in channels of ferritin protein nanocages is an example of gated pores in soluble protein. Ion transfers into, out of, and within living cells through barriers that are often hydrophobic reflect the intracellular boundaries that are required to maintain varied concentrations of ions different from the surrounding environment. In the case of ferritin, the concentration of ferric iron inside the protein nanocage exceeds that in the surrounding cytoplasm by as much as 1014 fold. The protein assemblies that transfer ions across such barriers in cell membranes or subcellular organelles such as mitochondria or nuclei, are usually called ion channels or pumps. Ion channels that are particularly well understood and provide models for many other channel proteins are the potassium channels [1]. The parts of ion channel protein assemblies where ions enter or exit are called pores. Pore “gates” are regions of the pore that mediate to changes in the environment and “open” or “close” channel pores to increase or decrease rates of ion transport. Ligands that change the functional pore “size” are exemplified by serotonin which alters calcium transport in nerve cells. Mechanical stress or changes in the voltage from one side of the channel, target pore gates and alter the functional sizes of pores. The presence of gates in the pores of ferritin nanocages has been recognized only recently from structure/function studies. Physiological ligands for ferritin gated pores are still to be identified, although the effects of physiological concentrations of urea appear to be an important clue [3].
Ferritins are self-assembling, symmetrical nanocage proteins with 4 α-helix bundle, polypeptide subunits that fold and assemble through hydrophobic interactions along the helix surfaces. Two sizes of ferritin protein nanocages are known in Nature: maxi-ferritins, 12 nm in diameter with 8 nm diameter cavities, formed from 24 subunits and found in humans and other animals, plants and bacteria; mini-ferritins, 8 nm in diameter with 5 nm diameter cavities, formed from 12 subunits [2]. The ferritin gated pores form at the junctions of subunit triples. There are 8 gated pores in maxi-ferritins and 4 gated pores in mini-ferritins, symmetrically distributed around the nanocages (Fig. 1). Mini-ferritin pores, while less studied, appear to have the same amino acid pairs but are less stable, in the closed position, than maxi-ferritins (Liu and Theil, unpublished observations). Maxi-ferritins have another set of pores at the junctions of four subunits, but since the function is unknown, the presence or absence of gates cannot be tested. Ferritin pores in the context of this review refer to the gated pores at the junction of three subunits.
Fig. 1. Effects of changing ferritin protein nanocage pore gates.
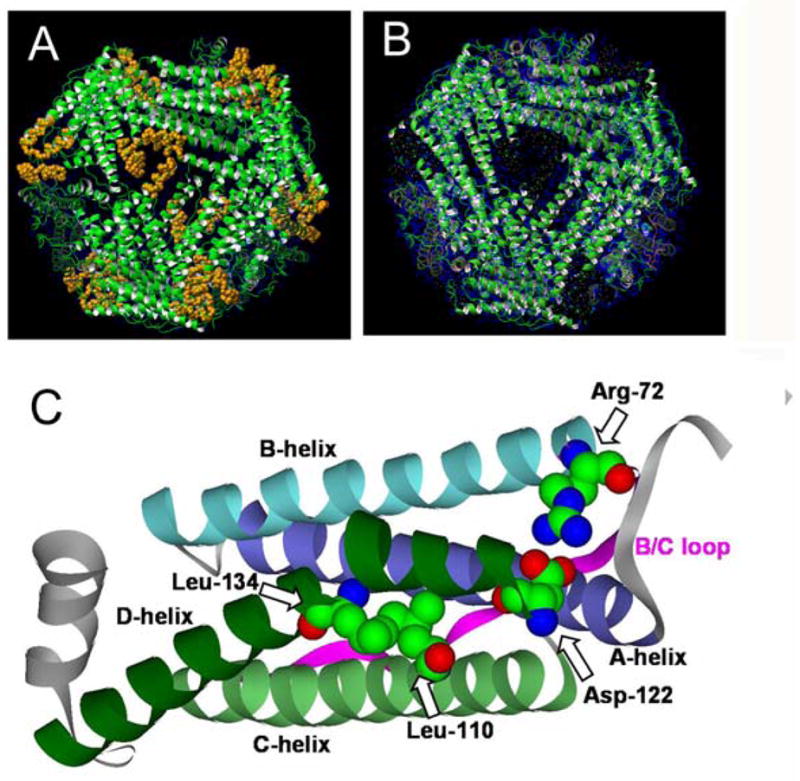
Structures of wild type ferritin with closed pores (shown in gold) and of ferritin with the L134P mutation that unfolds the pores (appearance of a “black hole” due to localized unfolding of helices C and D around the junction of three subunits. A, closed pores; reproduced with permission from ref [10] (issue cover), copyright National Academy of Sciences U.S.A, 2003; B, open pores gates and unfolded pores reproduced with permission from the American Chemical Society, 2005; C, a ferritin subunit showing the four α-helix bundle of a subunit with the A, B, C, and D helices and gates labeled; from pdb file 1MFR, using software program Viewerlite/DS Visualizer (Accelrys).
Current models of ferrous ion entry through the gated pores into the nanocage are based on competition with other metal ions [4–9], mutagenesis of non-gating residues around the pore [8,10], and co-crystal structures with metal inhibitors [11]. The model is weakened by the absence of crystal structures of the mutant proteins, by the multiple sites for metal inhibitor binding in addition to the pores, and by studies suggesting other ferrous ion entry sites besides the gated pores [12,13]. In contrast, the model of ferrous ion transfer from the reduced mineral in the cavity through the gated pores is based on parallel structural studies in solution and crystals with functional studies in solution [3,14,15].
2. Measuring the opening or closing of gated pores in proteins
The patched clamp analysis is a commonly used method for measuring gating of ion channels. Since most ion channels are parts of membranes, many studies of the properties of ion channel proteins and channel gating use a method called “patch clamp” analysis. The “Patch” is a small piece of membrane isolated in a micropipette inserted into the cell to form a tight seal around a small piece or “patch”; experimental studies of ion protein function use either patches isolated from the membrane by removing the micropipette from the living cell, or patches in situ. The “clamp” is an induced change in membrane potential which allows the change in the channel gates to be monitored as the change in electrical potential from the change in the flow of ions across the membrane. Because many novel therapeutic drugs cause changes in the transport of potassium, with risk of heart damage, the development and use of rapid throughput “patch clamp” analyses is a subject of expanding interest [16–18]. Traditional patch clamp pore gate analyses are unnecessary in studies of ferritin pore gating, since the pores can be studied in solutions of ferritin protein nanocages.
The ferritin ion channels with gated pores are part of a soluble supramolecular complex which eliminates the need to remove a “patch” for study. Since the protein pores in the ferritin nanocages control access between ferric mineral and reductants, the “patch clamp” assay for ferritins is the addition of the reductant to a solution of mineralized ferritin in the presence of a reporter for iron outside the nanocages. Formation rates of ferrous bipyridyl are the most common parameters measured for ferritin gated pore function, but since the reactions are usually carried out in air, a ferric chelator, such as desferal, can also be used as a reporter [3]. When the reaction conditions are constant, rate differences for the same protein under different conditions or for mutant proteins compared to wild type proteins, indicate changes in the protein nanocage itself (Table 1). Such a conclusion is supported by the effect of temperature on the rate, the linearity of the plot of lnVt vs. 1/T (°K) (Arrhenius plot) between 10 °C and 55 °C, and the differences in the slopes of the lines for proteins with wild type and mutated pores (X.S. Liu and E.C. Theil, unpublished observations). When the analysis for ferrous transport out of the ferritin nanocage was first developed [19], an NADH: flavin oxidoreductase was used to generate reduced flavin. The use of reduced flavin generated in the presence of NADH by the beam of a typical UV-vis spectrometer, is now more common, e.g. [20]. Electron transfer from reduced flavin to the ferric mineral is the rate limiting step in the analysis, which, based on comparisons of rates for different flavins, free and immobilized on agarose [19], appear to have limited access to the mineral. Studies with a variety of organic molecules into ferritin protein nanocage cavities shows a temperature dependence similar to that observed for pore unfolding by circular dichroism [3,21]. If contact between the mineral surface inside the ferritin nanocage and reduced flavin is increased by opening the gated pores in the protein, the rate of ferrous transfer out of the ferritin pores will increase (see Table 1 and Figs. 2, 3).
Table 1.
Fe(II) transport through ferritin pores is sensitive to mutation of conserved pore gates and physiological concentrations of urea
| Proteina | Initial rateb,c-, pH =7.0 | Initial rateb, pH=6.0 |
|---|---|---|
| Mutation | ||
| H-WT (C Helix) | 0.93 ± 0.10 | 2.48± 0.56 |
| H-L110V (C-Helix) | 3.48 ± 0.47 | NDd |
| H-L110A (C Helix) | 2.63 ± 0.41 | 5.52 ± 0.49 |
| H-L134V (D Helix) | 2.52 ± 0.64 | NDd |
| H-L134A (D Helix) | 2.93 ± 0.66 | 5.71 ± 0.76 |
| H-L134E (D Helix) | 3.03 ± 0.31 | NDd |
| H-L134G (D Helix) | 3.68 ± 0.39 | NDd |
| H-L134P (D Helix) | 4.01 ± 0.88 | 6.63 ± 1.02 |
| KVDPH insertion (C/D loop) | 3.78 ± 0.34 | 5.48 ±1.01 |
| H-R72D (B/C, C/D loops) | 3.09 ± 0.86 | NDd |
| Urea (M) | Initial rateb,c, pH =7.0 | Tm low melting domain (°C)e |
| 0 (H-WT) | 0.89 ± 0.06 | 56 |
| 0 (H-L134P) | 3.78 ± 0.41 | < 37 |
| 0.001 (H-WT) | 1.92 ± 0.09 | 43 |
| 0.010 (H-WT) | 2.61 ± 0.25 | |
| 1 (H-WT) | 4.85 ± 0.33 | |
| 2 (H-WT) | 1.27 ± 0.35 | |
Recombinant frog proteins, produced by site-directed mutagenesis, expressed in E. coli as apoproteins (≤ 2 Fe/molecule), prepared in 0.1M MOPS, 0.2M NaCl and mineralized uniformly in vitro with ferrous sulfate in 0.001 M HCl added at a 480 Fe/assembled protein. Helices and mutation sites are identified in Figure 1.
Reduction and chelation, with 2.5 mM each FMN, NADH and bipyridyl in solutions of mineralized protein (1.05 μM protein and 0.5 mM Fe). Initial rates, Δ522nm, sec−1, for mutants, and (mmoles L-1 s−1) × 103, for urea were measured for formation of the Fe(II)-bipyridyl complex that reflects transport of ferrous ions into solution outside the protein nanocage [14], after reduction of mineralized ferric and dissolving the mineral; A522 nm = 0.8 for 100% conversion of the ferric mineral to Fe(II) bipyridyl. The ferritin mineral is stable inside the proteins nanocages until the reductant is added. Initial rates were determined by a linear fit of the data points from zero to 1 min after mixing, and the data are averages of 3–5 measurements using 3 independent protein preparations for each amino substitution, with the error as the standard deviation. Data are taken from references [3,15].
The mutations has no affect on the ferrous entry/catalysts rates, except L134P which had a five-fold decrease in rate, possibly related to formation of an Fe(III)-tyrosine with Y133 which was in the disordered region induced by the L135P substitution [14,15].
ND: not determined
Measured by circular dichroism, as described in reference [3].
Fig. 2. Effects of pore gate mutation and pore unfolding.

Rates of ferrous ion transfer from the ferric mineral inside the protein were measured by adding a solution of NADH, FMN, and bipyridyl, 5 mM each, in 0.10 M MOPS, 0.1M NaCl, pH=7 to mineralized, recombinant ferritin nanocages in 0.10 M MOPS, 0.1M NaCl, pH=7. A. Rate sensitivity to conservative and nonconservative changes at residue 134 in helix D; see Fig. 1C, including the substitution of valine for leucine. B, Rate sensitivity when the C/D loop is manipulated at ion pair arginine 72 and aspartate 122, and by increasing the C/D loop size by inserting a second set of the same loop amino acids, with all the wild type gating residues present. Data from reference [9].
Fig. 3. Ferritin protein nanocage pores unfold 30°C below the nanocage structure and with low concentrations of urea.

Temperature effects on global structure were measured at 280 nm (UV absorbance) and on pore structure at 280 nm (circular dichroism), as described in reference [3], using recombinant, mineralized ferritins in 0.015M MOPS, pH = 7. Rates of ferrous ion transfer from the ferric mineral inside the protein nanocages, through the pores were measured by adding a solution of NADH, FMN, and bipyridyl, 5 mM each, in 0.10 M MOPS, 0.1oM NaCl, pH=7 and measuring the absorbance of ferrous bipyridyl (A 522nm). A. Mutations that unfolded the ferritin nanocages pores (Figure 2) had no effect on global nanocage melting. B. Pores structure unfolds at 56°C, which is 30°C below the global nanocages (> 85 °C). ■ - H-WT; □ - H-WT + 1 mM urea; ◇-H-L134P; C. Urea unfolds ferritin pores; Progress curve for formation of Fe2+-bipyridyl + urea. Chaotrope concentrations: ◆ - zero; ■-1 mM; ◊- 10 mM.; □- 1M; △- 2 M. Data are from reference [3].
The crystal structures of wild type ferritin protein and ferritin protein with a mutation in one of the four pore residues, leucine 134 (Fig. 1), showed unfolded pores that coincided with an increase in the rate of ferrous iron transport of the mineral and the ferritin nanocage. Only small differences in the speed of ferrous transfer from the reduced mineral to the chelator were observed in ferritins with differences in overall amino acids sequence as much as 15 to 33% [14]. In contrast, ferrous transfer from the reduced mineral to the chelator was thirty times faster with the substitution of a single amino acid, L134, a pore gate (Fig. 2).
In this brief review we describe studies, using the rate of formation of ferrous bipyridyl as the analysis for pore folding/unfolding, that identified: 1) the gates of the pores at the junction of three subunits in ferritin, by the coincidence between localized helix unfolding around the pores and the rate change in converting ferric mineral inside the protein nanocages to ferrous bipyridyl outside the nanocages; 2) solutes that altered pore folding, and 3) heat induced, selective melting of pore helices. Recent data show that during iron deficiency in vivo, intact ferritin protein nanocages release iron into the cytoplasm of living cells [22], but the contribution of the conserved pore gates remains to be explored directly.
3. Identifying ferritin nanocage pore gates
Identification of leucine 134, a conserved hydrophobic amino acid thought to be important in subunit stability and/or assembly, as regulator of pore unfolding, i.e. a pore gate [14], stimulated the search for other amino acids that act as gates among group of ferritin residues that had no assigned function. Early models of ion transport into and out of ferritin were based on a major role for negatively charged amino acids. The contribution of leucine to the transfer of ferrous iron out of the ferritin mineral was the first clue that helix structure itself participated in the process. Older studies of ferritin modifications that changed ferrous ion transport out of the protein, such as natural and synthetic crosslinks that locked the protein in a rigid conformation [20,23], also indicated the importance of nanocage flexibility in transfer of iron out of the nanocage cavity, but did not indicate the location of the critical sites in three dimensional space. In the search for residues that were important as ferritin pore gates, in addition to leucine 134, three criteria were used: 1) conservation in all known ferritins; 2) proximity to the triple junction of folded subunits; 3) “orphan” residues (no other known function). Only three residues, of over 170 amino acids in each sequence, met the search criteria. The putative pore gate amino acids selected by sequence searches were leucine 110, arginine 72 and aspartate 122.
Every amino acid substitution engineered in a putative pore gate indicated by the search criteria described above, significantly increased the rate of ferrous ion transfer out of the mineral and protein nanocage (Table 1). Conservative substitutions are exemplified by the substitution for leucine, valine an amino acid, aliphatic side chain of four carbons that differed from leucine by only a single CH2. Valine increased rates of ferrous transfer through the pores significantly (Table 1) but the effect was smaller than nonconservative substitution with the amino acid glycine. Glycine has a side chain consisting of a single hydrogen atom that terminates α-helices. However, any amino acid substitution of the pore gate residues, whether conservative or nonconservative, increased rates of ferrous transfer from the reduce mineral in the protein cavity through the pore by > 2.5-fold, in spite of the variations in the effects among the individual substitutions (Table 1). The selectivity of the nine pore gate residue mutations on ferrous transfer out of the protein pores is emphasized by the fact that none of the mutations affected the catalytic ferroxidase activity, except for a relatively modest effect (0.4 fold rate decrease) for L134P. In addition, none of the substations altered nanocage assembly or global heat stability [3,14,15,24]. Selectivity of the effects of amino acid substitution on pore function confirmed the accuracy of the search results using phylogenetic conservation of amino acids at sites where three subunits form a pore that had no other specific function. The results identified the ferritin pore gates as the ion pair between two loops, arginine 72 in the long B/C loop and aspartate 122 in the short C/D loop (Fig. 1), and the helix/helix interaction between leucine 110 in helix C and leucine 134 in helix D (Fig. 1), each approximately 3 helix turns from the C/D loop.
The loop of five amino acids, residues 120–125, between the C and D helices around the pores, plays a special role in gating, based on several observations. First, the loop places leucine 110 and leucine 134 in position to interact with each other and function as pore gates. In addition, the length of the C/D loop is critical since lengthening the loop by insertion of the sequence KVDPH, which duplicates the five wild type amino acids of the loop, increased the rate of ferrous iron transfer from the reduced mineral to the outside of the nanocage as much as the nonconservative substitutions of single amino acids, L134P or L134G [15]. Finally, important of C/D loop sequence is shown by the role of aspartate 122, both a loop residue and a pore gate. Disrupting the ion pair between aspartate 122 and arginine 122 changed pore function dramatically (Fig. 2). Restoring the ion pair as aspartate 72/arginine 122, still deviates from the wild type the loop sequence and did not restore normal pore function [15]. Thus both the length and the sequence in the C/D loop are important for pore function. Possibly, when the ferritin gated pore is folded or closed, aspartate 122 blocks reduction of the ferric mineral since, in crystal structures, the C/D loop is pointing into the funnel created by the pore with aspartate 122 at the base of the pore [11,25,26].
4. Selective unfolding or melting of ferritin protein nanocage pores
Ferritin protein nanocages are unusually stable to heat and to protein unfolding reagents such as urea or guanidine. No dissociation of cage subunits or helix unfolding is observed in 6 M urea or 6 M guanidine unless the pH is less than 3.5 [27]. In addition, using changes in the absorbance at 280 nm to monitor global changes in cage folding, nanocage structure was stable to 85 °C in both wild type ferritin and ferritin with pore mutations [3] (Fig. 3).
Low temperature melting subdomains in the ferritin protein nanocage can be observed by circular dichroism spectroscopy, since the α-helix content is high (85%). Approximately 15% of the ferritin nanocage helices melt with a mid-point transition of 56 °C, contrasting with the high heat stability of the nanocages itself (Fig. 3). The correlation of the low temperature helix transition with pore helix unfolding is demonstrated by several observations: 1) the melting transition initiation temperature is lower (37 °C). in the L134P ferritin mutant that has disordered peptides around the pores in protein crystals, than in the wild type protein (Fig. 3) [3,14]; 2) the % helix in L134P, determined by CD spectroscopy, is ~ 15% lower than in wild type protein (Fig. 3); and 3) low concentrations of urea in the physiological range (1 mM) increase the rate of ferrous transfer from the cavity to the solvent outside the protein nanocage (Table 1) and also shift the mid-point of the melting transition from 56 °C to 43 °C [3] (Fig. 3).
The temperature dependence of the rate of conversion of ferric iron in the mineral to ferrous-bipyridyl outside the protein nanocage, triggered by NADH/FMN, is protein dependent over the range tested, 10°C to 55°C. For example the values for the slopes in the Arrhenius plot were different for wild type and mutant L134P protein, with average values of the slope (E): 83.5 kJ/mole for wild type protein and 88.6 kJ/mole for the mutant, in two independent experiments (X.S. Liu and E.C. Theil, unpublished observations). The rate decrease in the mutant protein upon cooling indicates that the unfolding observed in crystals at room temperature (Figure 1) was reversible although a larger decrease in temperature was required to obtain the same effect in on rates for the mutant protein in than for wild type.
Under normal physiological conditions urea concentration are typically in the 2 to 7 mM range, a range where a significant fraction of ferritin pores will be unfolded at any given time making the mineral accessible to cellular reductant such as reduced flavin. The temperature sensitivity of the pores around physiological temperatures leads to the same conclusion. Evolutionary conservation of ferritin pore gates coupled with pore sensitivity to small changes in heat and urea in the physiological range suggests the presence of in vivo regulators that recognize the pore gates and hold the pores closed or open depending the amount of mineral reduction and ferrous transport needed to maintain cellular iron balance.
5. Summary and Perspective
Transport of ferrous iron out of the ferritin mineral and through gated protein pores depends upon the interaction of the mineral inside the protein nanocages and reductant outside the protein nanocage. The structure of ferritin has evolved to include 8 gated pores in the 24 subunit assembly of 4-α helix bundles (helices A,B,C,D)(Fig. 1). The pores, at the junction of three subunits, control rates of ferrous iron movement out of the mineral and protein cage into the environment outside by regulating access between mineralized ferric iron and biological reductants such as FMNH2. Ferritin gates are studied with a “patched clamp” analysis that uses NADH to trigger formation of FMNH2 and reduction of mineralized ferric iron inside the protein cage, with a chelator to monitor ferrous ion flow through the pores. The pore gates regulate ion flow by altering local folding and unfolding of the ion channel pores. Four highly conserved amino acids create the pore gates and are also present in the less studied, 12 subunit mini-ferritins (Dps proteins). In each subunit around the junction of three subunits, two amino acids form an ion pair between arginine 72 and aspartate 122, each in a loop, and a hydrophobic interaction between leucine 110 in the C helix and Leucine 134 in the D helix.
Ferritin pores melt at temperatures 30 °C below the temperature at which the nanocage itself melts. The pores are also unfolded by very low (mM) concentrations of urea or guanidine in contrast to the nanocage which is very stable to high concentrations of urea or guanidine. For example, the ferritin protein nanocage requires 104 times the concentration of protons plus 103 times the concentration of urea or guanidine before unfolding, emphasizing the selectivity of pore folding/unfolding controlled by the pore gates. Given the similarity of physiological concentrations of urea that unfold ferritin pores, it is likely that biological regulators recognize the pore gates and/or the folded/unfolded states to protect the ferric iron mineral from cellular reductants until the iron is needed for synthesis of iron cofactors. The apparent functional and structural (helix bundles with hydrophobic interactions) homology between ferritin gated pores and gated ion channel proteins in membranes and the water solubility of ferritin nanocages, make ferritin an attractive model for the study of gated pore structure, folding and unfolding.
Acknowledgments
The work of the authors described has been supported by DK 20251 (ECT, XSL, and TT), The Cooley’s Anemia Foundations (XSL) and the JSPS Fellowship for Research Abroad (TT). The authors are also grateful for the contributions of all the members of the Theil Lab, especially Drs. Weili Jin and Hidenori Takagi, as well as our collaborators.
Biographies
 ELIZABETH C. THEIL, educated at Cornell (B.S.) and Columbia (Ph.D.) Universities, is currently Senior Scientist at CHORI (Children’s Hospital Oakland Research Institute), where she directs CeBIC (Council on BioIron at CHORI) and is Professor of Nutritional and Molecular Toxicology at the University of California, Berkeley (adj.) and Professor of Molecular and Structural Biochemistry at North Carolina State University (NCSU) (adj.). Previously, she was the University Professor of Biochemistry and Physics at NCSU.
ELIZABETH C. THEIL, educated at Cornell (B.S.) and Columbia (Ph.D.) Universities, is currently Senior Scientist at CHORI (Children’s Hospital Oakland Research Institute), where she directs CeBIC (Council on BioIron at CHORI) and is Professor of Nutritional and Molecular Toxicology at the University of California, Berkeley (adj.) and Professor of Molecular and Structural Biochemistry at North Carolina State University (NCSU) (adj.). Previously, she was the University Professor of Biochemistry and Physics at NCSU.
 XIAOFENG S. LIU is currently Professor of Life Sciences at Nanjing University. He was educated in Physiology (B.S.) at Changchun College of Traditional Chinese Medicine, and in Biochemistry and Biophysics (Ph.D.) at Iowa State University. From 2001 to 2007 he was a Postdoctoral Fellow, Cooley’s Anemia Foundation Fellow and Assistant Staff Scientist in the Molecular BioIron Group at CHORI.
XIAOFENG S. LIU is currently Professor of Life Sciences at Nanjing University. He was educated in Physiology (B.S.) at Changchun College of Traditional Chinese Medicine, and in Biochemistry and Biophysics (Ph.D.) at Iowa State University. From 2001 to 2007 he was a Postdoctoral Fellow, Cooley’s Anemia Foundation Fellow and Assistant Staff Scientist in the Molecular BioIron Group at CHORI.
 TAKEHIKO TOSHA currently holds a Japan Society for the Promotion of Science (JSPS) Fellowship for Research Abroad and is a Postdoctoral Fellow in the Molecular BioIron Group at CHORI. He was educated in Chemistry (B.S) and Engineering (Ph.D.) at Kyoto University, and was a JSPS Postdoctoral Fellow at the Okazaki Institute for Integrative Bioscience, National Institutes of Natural Sciences, before beginning his research at CHORI in 2006.
TAKEHIKO TOSHA currently holds a Japan Society for the Promotion of Science (JSPS) Fellowship for Research Abroad and is a Postdoctoral Fellow in the Molecular BioIron Group at CHORI. He was educated in Chemistry (B.S) and Engineering (Ph.D.) at Kyoto University, and was a JSPS Postdoctoral Fellow at the Okazaki Institute for Integrative Bioscience, National Institutes of Natural Sciences, before beginning his research at CHORI in 2006.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Elizabeth C. Theil, Council for BioIron at CHORI, Children’s Hospital Oakland Research Institute, Oakland, CA, 94609 and The Department of Nutritional Sciences and Toxicology, University Of California, Berkeley, CA 94720
Xiaofeng S. Liu, State Laboratory of Pharmaceutical Biotechnology and College of Life Sciences, Nanjing University, Nanjing, China, 210093
Takehiko Tosha, Council for BioIron at CHORI, Children’s Hospital Oakland Research Institute, Oakland, CA, 94609 and The Department of Nutritional Sciences and Toxicology, University Of California, Berkeley, CA 94720.
References
- 1.Gouaux E, Mackinnon R. Science. 2005;310:1461. doi: 10.1126/science.1113666. [DOI] [PubMed] [Google Scholar]
- 2.Liu X, Theil EC. Ann NY Acad Sci. 2005;1054:136. doi: 10.1196/annals.1345.016. [DOI] [PubMed] [Google Scholar]
- 3.Liu X, Jin W, Theil EC. Proc Natl Acad Sci USA. 2003;100:3653. doi: 10.1073/pnas.0636928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Treffry A, Harrison PM. J Inorg Biochem. 1984;21:9. doi: 10.1016/0162-0134(84)85035-7. [DOI] [PubMed] [Google Scholar]
- 5.Wardeska JG, Viglione B, Chasteen ND. J Biol Chem. 1986;261:6677. [PubMed] [Google Scholar]
- 6.Stefanini S, Desideri A, Vecchini P, Drakenberg T, Chiancone E. Biochemistry. 1989;28:378. doi: 10.1021/bi00427a052. [DOI] [PubMed] [Google Scholar]
- 7.Yablonski MJ, Theil EC. Biochemistry. 1992;31:9680. doi: 10.1021/bi00155a022. [DOI] [PubMed] [Google Scholar]
- 8.Treffry A, Bauminger ER, Hechel D, Hodson NW, Nowik I, Yewdall SJ, Harrison PM. Biochem J. 1993;296:721. doi: 10.1042/bj2960721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barnes CM, Theil EC, Raymond KN. Proc Natl Acad Sci USA. 2002;99:5195. doi: 10.1073/pnas.032089399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levi S, Luzzago A, Franceschinelli F, Santambrogio P, Cesareni G, Arosio P. Biochem J. 1989;264:381. doi: 10.1042/bj2640381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hempstead PD, Yewdall SJ, Fernie AR, Lawson DM, Artymiuk PJ, Rice DW, Ford GC, Harrison PM. J Mol Biol. 1997;268:424. doi: 10.1006/jmbi.1997.0970. [DOI] [PubMed] [Google Scholar]
- 12.Macedo S, Romao CV, Mitchell E, Matias PM, Liu MY, Xavier AV, LeGall J, Teixeira M, Lindley P, Carrondo MA. Nat Struct Biol. 2003;10:285. doi: 10.1038/nsb909. [DOI] [PubMed] [Google Scholar]
- 13.Carrondo MA, Macedo S, Romao CV, Mitchell E, Matias PM, Liu MY, Xavier AV, LeGall J, Teixeira M, Lindley P, Carrondo MA. Embo J. 2003;22:1959. doi: 10.1038/nsb909. [DOI] [PubMed] [Google Scholar]
- 14.Takagi H, Shi D, Ha Y, Allewell NM, Theil EC. J Biol Chem. 1998;273:18685. doi: 10.1074/jbc.273.30.18685. [DOI] [PubMed] [Google Scholar]
- 15.Jin W, Takagi H, Pancorbo NM, Theil EC. Biochemistry. 2001;40:7525. doi: 10.1021/bi002509c. [DOI] [PubMed] [Google Scholar]
- 16.Liu H, Gao ZB, Yao Z, Zheng S, Li Y, Zhu W, Tan X, Luo X, Shen J, Chen K, Hu GY, Jiang H. J Med Chem. 2007;50:83. doi: 10.1021/jm060414o. [DOI] [PubMed] [Google Scholar]
- 17.Vasilyev D, Merrill T, Iwanow A, Dunlop J, Bowlby M. Pflugers Arch. 2006;452:240. doi: 10.1007/s00424-005-0029-2. [DOI] [PubMed] [Google Scholar]
- 18.Ionescu-Zanetti C, Shaw RM, Seo J, Jan YN, Jan LY, Lee LP. Proc Natl Acad Sci USA. 2005;102:9112. doi: 10.1073/pnas.0503418102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones T, Spencer R, Walsh C. Biochemistry. 1978;17:4011. doi: 10.1021/bi00612a021. [DOI] [PubMed] [Google Scholar]
- 20.Mertz JR, Theil EC. J Biol Chem. 1983;258:11719. [PubMed] [Google Scholar]
- 21.Yang X, Arosio P, Chasteen ND. Biophys J. 2000;78:2049. doi: 10.1016/S0006-3495(00)76752-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Domenico I, Vaughn MB, Li L, Bagley D, Musci G, Ward DM, Kaplan J. EMBO J. 2006;25:5396. doi: 10.1038/sj.emboj.7601409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McKenzie RA, Yablonski MJ, Gillespie GY, Theil EC. Arch Biochem Biophys. 1989;272:88. doi: 10.1016/0003-9861(89)90198-7. [DOI] [PubMed] [Google Scholar]
- 24.Santambrogio P, Levi S, Arosio P, Palagi L, Vecchio G, Lawson DM, Yewdall SJ, Artymiuk PJ, Harrison PM, Jappelli R, et al. J Biol Chem. 1992;267:14077. [PubMed] [Google Scholar]
- 25.Trikha J, Theil EC, Allewell NM. J Mol Biol. 1995;248:949. doi: 10.1006/jmbi.1995.0274. [DOI] [PubMed] [Google Scholar]
- 26.Ha Y, Shi D, Small GW, Theil EC, Allewell NM. J Biol Inorg Chem. 1999;4:243. doi: 10.1007/s007750050310. [DOI] [PubMed] [Google Scholar]
- 27.Listowsky I, Blauer G, Enlard S, Betheil JJ. Biochemistry. 1972;11:2176. doi: 10.1021/bi00761a026. [DOI] [PubMed] [Google Scholar]