

Abstract
Inteins are protein-splicing elements, most of which contain conserved sequence blocks that define a family of homing endonucleases. Like group I introns that encode such endonucleases, inteins are mobile genetic elements. Recent crystallography and computer modeling studies suggest that inteins consist of two structural domains that correspond to the endonuclease and the protein-splicing elements. To determine whether the bipartite structure of inteins is mirrored by the functional independence of the protein-splicing domain, the entire endonuclease component was deleted from the Mycobacterium tuberculosis recA intein. Guided by computer modeling studies, and taking advantage of genetic systems designed to monitor intein function, the 440-aa Mtu recA intein was reduced to a functional mini-intein of 137 aa. The accuracy of splicing of several mini-inteins was verified. This work not only substantiates structure predictions for intein function but also supports the hypothesis that, like group I introns, mobile inteins arose by an endonuclease gene invading a sequence encoding a small, functional splicing element.
Inteins are protein-splicing elements that exist as in-frame fusions with flanking protein sequences called exteins. Inteins are self-splicing at the protein level, with their excision being coupled to extein ligation (1–3). Most of the inteins that have been described are in the 400- to 500-aa range with little absolute sequence conservation among the elements (4, 5). However, Cys or Ser residues are required at the amino termini of both the intein and the second extein, and a His and Asn are present at the carboxy terminus of the intein (Fig. 1A Top). Most inteins contain eight conserved sequence blocks (A–H), two of these being the LAGLIDADG motifs (blocks C and E) that define a family of intron-homing endonucleases (refs. 4 and 5; Fig. 1A). Consistent with the occurrence of these motifs, several inteins have been shown to have site-specific endonuclease activity (6), and PI-SceI, the VMA1 intein of Saccharomyces cerevisiae, is capable of homing into a cognate inteinless allele (7). The sporadic distribution of inteins in all three biological kingdoms is consistent with their being mobile elements.
Figure 1.
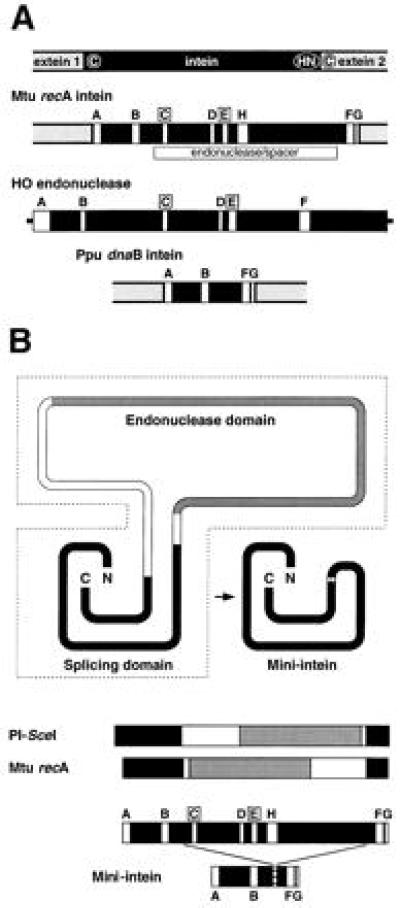
Structural features of inteins. (A) Intein and endonuclease maps. The top map shows the generic relationship of an intein to its flanking exteins. Conserved amino acids are circled (see text). In the intein and endonuclease maps below, conserved sequence blocks are labeled A–H, with the LAGLIDADG endonuclease motifs, C and E, boxed (4, 5). (B) Intein structures. The top schematic depicts the two domains and spacer regions of the 440-aa Mtu recA intein according to modeling predictions (ref. 13; J.Z.D., et al., unpublished data) and the structure of PI-SceI (12), with corresponding linear maps below. Solid areas, protein-splicing domain; shaded, endonuclease domain; open, spacer regions. The smallest functional mini-inteins from this study (137 aa; Fig. 2B) are shown in both representations, with the linear form below a map of the Mtu recA intein with conserved sequence blocks.
Endonuclease genes have been assumed to be invasive genetic elements that colonized group I introns, converting them into mobile genetic elements (8–11). Similarly, mobile inteins appear to be derived from invasive endonuclease genes. Recent structural studies indeed suggest that the protein-splicing and endonuclease domains are separate and that their two activities may have evolved independently. First, the crystal structure of PI-SceI has recently been solved (12). This 454-aa protein is folded into two distinct structural domains. Second, hidden Markov models have been used to define two conserved functional domains of inteins, corresponding to independent endonuclease and splicing modules, separated by nonconserved spacer regions of variable lengths (ref. 13; J.Z.D., A. Klar, M. J. Moser, W. R. Holley, A. Chatterjee, and I. S. Mian, unpublished results; Fig. 1B). Third, three putative inteins have recently been reported that are in the 150-aa size range and lack endonuclease motifs, although it is not clear whether these smaller elements retain splicing function (4, 5, 13). Finally, a newly identified Synechocystis intein does not contain a LAGLIDADG endonuclease but instead contains a member of the H-N-H family of group I intron endonucleases (13, 28).
Site-directed mutagenesis experiments have shown that endonuclease activity is not required for protein-splicing function (14, 15), and deletion of a region encompassing the LAGLIDADG motifs of PI-SceI has confirmed this conclusion (16). However, despite the apparent structural autonomy of the protein-splicing and endonuclease domains of PI-SceI, they do appear to collaborate in interacting with the homing site DNA (12). Therefore, it is important to determine whether the bipartite structure of inteins is mirrored by the functional independence of their two components. We tested the prediction that the entire endonuclease domain and spacer sequences between the domains can be deleted from a protein-splicing element to generate a mini-intein that is splicing proficient. To this end, we used the 440-aa intein from the Mycobacterium tuberculosis recA gene expressed in Escherichia coli. The Mtu recA intein contains a conventional LAGLIDADG endonuclease domain (4, 5), although endonuclease activity has not yet been demonstrated. Guided by junctions inferred from structure models (ref. 13; Fig. 1B), a series of mini-intein derivatives was tested in two genetic systems developed to screen for splicing activity in vivo and in vitro. A number of mini-inteins deleted for the entire endonuclease domain were shown to be capable of protein splicing in both contexts, consistent with structure predictions. These results support the model that homing inteins evolved through an endonuclease gene invading a DNA sequence encoding a functional mini-intein.
MATERIALS AND METHODS
Construction of td:Mini-Intein Fusions.
Plasmid pKKtdC238-I containing the Mtu recA intein is derived from pKK223 (Pharmacia). The plasmid contains an in-frame fusion of the intein with the intronless T4 td gene, which encodes thymidylate synthase (TS), such that Cys-238 of TS is the N-terminal residue of the second extein (circled in Fig. 2A; D.W.W., V.D., W.W., Georges Belfort, and M.B., unpublished results). pKKtdC238-I was used as a template for inverse PCRs to generate a series of plasmids for expression of TS:mini-intein in-frame fusion proteins. The coding sequences for the fusion proteins were sequenced in their entirety.
Figure 2.

Summary of phenotypes of mini-intein constructs. (A) The td-based genetic system. Schematic of the TS:intein in-frame fusion system shows precursors and splice products (Left). TS phenotype of td:intein in-frame fusion derivatives (Right). Patches of TS− cells (D1210ΔthyA::KanR) containing plasmids expressing td:intein fusions were tested for growth on minimal medium plates at different temperatures (27). td, no intein; td Mtu, full-length Mtu recA intein; td MtuAA, full-length Mtu recA intein with C-terminal His and Asn residues mutated to Ala; remainder, td:mini-intein constructs. Numbering in brackets is as in B. (B) Summary of protein-splicing activity. Full-length and mini-intein constructs are numbered 1–21. The constructs are defined by the deletion junctions, with Ala residues inserted in the cloning listed in parentheses [e.g., 94Δ383 has all residues between 94 and 383 deleted, whereas 101Δ405(A7) has residues between 101 and 405 deleted and replaced with 7 Ala residues]. Constructs 4, 9, 12, and 18 have Ala–Arg inserted between the junction residues as indicated by a prime. TS phenotype: +5, growth at 23°C, 30°C, and 37°C; +4, growth at 23°C, 30°C, and weak growth at 37°C; +3, growth at 23°C and at 30°C; +2, growth at 23°C; +1, weak growth at 23°C; —, no growth on minimal media. MIC splicing is quantitated as the percentage of precursor that was converted to ligated exteins after 3 h of induction: +5, 80–100%; +4, 60–80%; +3, 40–60%; +2, 20–40%; +1, ≤20%. Those constructs indicated by an asterisk splice further upon incubation at 4°C. Intein size represents Mtu recA residues only, excluding Ala and Arg residues incorporated as part of the cloning process. Arrowheads mark largest deletion on each side, consistent with splicing function.
The primers used in the PCRs carried a terminal BssHII restriction site, such that digestion of the products with this enzyme and religation generated in-frame td:mini-intein fusions with central deletions marked by a BssHII site and, hence, an Ala–Arg dipeptide. Where possible, these residues replaced the same or similar amino acids in the final fusion proteins. In some cases this required the addition of an Ala residue between the junctions (Fig. 2B), and in constructs 101Δ395(A5′), 123Δ372′, 129Δ372′, and 129Δ400′ (Fig. 2B, constructs 4, 9, 12, and 18), deleted residues are replaced with Ala–Arg.
Construction of Tripartite Fusion Derivatives, Detection of Splicing, and Protein Purification.
Mini-intein derivatives were transferred to a tripartite fusion system (MIC) (D.W.W. et al., unpublished results) for in vitro characterization. This encodes maltose binding protein (M), intein (I), and the C-terminal domain of I-TevI (C) (ref. 17; Fig. 3). Fragments encoding the pKKtdC238-mini-intein derivatives were PCR-amplified using primers encoding EcoRI (5′) and BsrGI (3′) restriction sites for cloning into the parent MIC expression plasmid (pMIC), which contains silent EcoRI and BsrGI sites close to the intein–extein junctions. pMIC is derived from pMalC2 (New England Biolabs). Again, the mini-intein portion of each pMIC derivative was sequenced. pMIC25 is a slow-splicing derivative of full-length pMIC (D.W.W. et al., unpublished results). The tripartite MIT fusion, which has the I-TevI domain replaced with TS (D.W.W. et al., unpublished results), was used as a control in Western blots.
Figure 3.
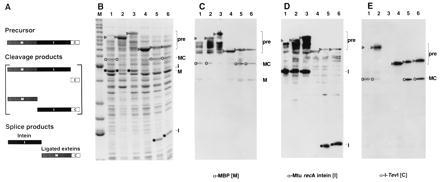
Splicing in vivo. (A) Schematic of the MIC in-frame fusion. Precursor, cleavage, and splice products are shown. (B) Twelve percent Coomassie-stained SDS polyacrylamide gel of cell lysates of MIC constructs induced at 37°C for 3 h. Lane M, molecular mass marker bands (Benchmark, GIBCO/BRL) from the bottom up correspond to 15, 20, 25, 30, 40, 50, 60, 70, 80, 90, 100, 120, 160, and 220 kDa (bold numbers correspond to the two high-intensity bands). Lanes: 1, full-length MIC; 2, MIC25, a slow-splicing mutant derivative of full-length MIC (D.W.W. et al., unpublished results); 3, MIT with TS as C-terminal extein; 4, 101Δ405(A7) (Fig. 2, construct 3); 5, 110Δ383 (Fig. 2, construct 6); 6, 114Δ372 (Fig. 2, construct 8). ▹, precursor (MIC or MIT); ○, ligated exteins (MC); •, intein. Arrowheads in lane 1 mark position of precursor. (C) Western blot of 12% SDS polyacrylamide gel with maltose binding protein antiserum. (D) Western blot of 12% SDS polyacrylamide gel with Mtu recA intein antiserum. (E) Western blot of 10% SDS polyacrylamide gel with I-TevI antiserum. The assignments were verified on a 12% gel (data not shown). Background bands from cross-reactivity to the polyclonal antisera are also evident in C–E. Lanes and symbols are as in B.
pMIC derivatives were maintained in E. coli D1210ΔthyA::KanR [F−Δ(gpt-proA)62 leuB6 supE44 ara-14 galK2 lacY1 Δ(mcrC-mrr) rpsL20 (Strr) xyl-5 mtl-1 recA13 lacIq]. Cells were grown to mid-log phase at 37°C in L Broth with 100 μg/ml ampicillin, and MIC expression was induced for 3 h by the addition of 1 mM isopropyl β-d-thiogalactoside (IPTG). Splicing products were visualized by running cell lysates on SDS polyacrylamide gels (18). Western blot analyses were carried out using a Bio-Rad semi-dry blot apparatus and the Amersham ECL detection kit according to the manufacturers’ instructions. Polyclonal antisera to I-TevI and the Mtu recA intein were raised in rabbits and that to maltose binding protein was purchased from New England Biolabs.
MIC and the mini-intein in-frame fusion proteins were purified on amylose columns (New England Biolabs) according to the manufacturer’s instructions. Quantitation of MIC splicing was achieved by densitometry of samples fractionated on 12% Coomassie-stained SDS/PAGE gels to determine the percentage of precursor converted to ligated exteins.
RESULTS
Construction of Mini-Inteins and Examining Boundaries of Splicing Function.
Guided by sequence alignments of all known inteins (13), a series of deletion derivatives of the Mtu recA intein was constructed. We aimed to remove all of the endonuclease domain, leaving behind a minimal splicing domain (Fig. 1). Initially intein function was assayed in a genetic system, in which the intein is expressed from a plasmid as an in-frame fusion with the TS gene (td) of bacteriophage T4 (D.W.W. et al., unpublished results; Fig. 2A). Because three in-frame deletion derivatives of this intein had been shown previously to be inactive by Western blot analysis (19), the increased sensitivity of the in vivo system would likely prove useful for detecting low-level splicing activity. Furthermore, we reasoned that even if the endonuclease exists as an independent domain, it might be difficult to generate a functional mini-intein, because crudely snipping out a large region of protein could well leave the remainder of the protein unable to fold properly. Therefore, we designed a series of derivatives with more or less of the central portion of the intein deleted. The derivatives were generated by inverse PCR using the td expression plasmid containing the full-length Mtu recA intein as template.
Fig. 2 shows the TS phenotype of a number of mini-intein derivatives. Whereas the full-length td:intein fusion was TS+ at all temperatures up to 37°C (Fig. 2B, construct 1), and the splicing-defective MtuAA mutant was TS− at all temperatures (Fig. 2B, construct 2), most of the mini-intein derivatives were TS+ only at lower temperatures (Fig. 2B, constructs 3–12, 14, 15, 17, 18, 20, and 21). These phenotypes suggest that many of the mini-inteins are capable of splicing, although less efficiently than the wild-type Mtu recA intein. Although there is apparently no direct correlation between deletion size and phenotype, it is interesting to note that the shorter mini-inteins gave the strongest TS+ phenotypes (Fig. 2B, constructs 7, 14, and 15), whereas the longest mini-intein was TS− (Fig. 2B, construct 13). Perhaps the most surprising result is the frequency with which TS+ mini-inteins were generated. Derivatives with deletions of up to 303 aa [101Δ405(A7) and 96Δ400′ (Fig. 2B, constructs 3 and 21)] still retain splicing function. Construct 101Δ405(A7) corresponds precisely to the predicted minimal splicing domain as defined by computer modeling (13).
Once it was apparent that a number of mini-inteins were active and that TS phenotype correlated with protein-splicing activity (see below), it became of interest to approximate the boundaries of the splicing domains at the N and C termini of the intein (Fig. 2B, constructs 14–21). Mini-inteins 94Δ383 and 114Δ406 (Fig. 2B, constructs 15 and 17, see arrowhead) that are TS+ indicate that splicing function is contained within the first 94 and last 35 aa of the intein.
Characterization of Mini-Intein Splicing Products.
To further assess splicing activity, the mini-intein derivatives were transferred to a tripartite fusion system (MIC) for in vitro characterization. The tripartite system comprises maltose binding protein (M) as the first extein and as an affinity tag for purification, fused in-frame to the intein (I) and then to the second extein, the C-terminal domain of the homing endonuclease I-TevI (C) (ref. 17; Fig. 3A). This C-terminal domain was chosen because of the solubility of this protein module and the availability of antibody to I-TevI. Indeed, antibody to each component of MIC allowed accurate identification of all precursors, intermediates, and products of the splicing reaction by Western blot analysis (Fig. 3 C–E).
Overexpression of the tripartite fusion proteins was induced by the addition of IPTG, and the precursors and products of the splicing reactions were separated and identified (representative data shown in Fig. 3 B–E). For most of the derivatives the products of the splicing reaction, ligated exteins and free intein, were readily detected on Coomassie gels and their identity was verified by Western blot analysis. In addition, the precursor and intermediates or side-products of the reaction corresponding to N-terminal or C-terminal intein cleavage without ligation were also seen.
A Western blot using maltose binding protein antiserum (Fig. 3C) shows the presence of precursors in MIC25, a full-length mutant derivative (lane 2), in MIC mini-intein constructs (lanes 4–6), and in MIT (lane 3), a control tripartite fusion of M, I, and intact TS (D.W.W. et al., unpublished work), although full-length wild-type MIC splices so efficiently that no precursor was visible (lane 1). In addition to full-length protein, several products containing the maltose binding domain were detected, including ligated exteins (MC) and free maltose binding protein (M). Cleavage products containing the intein (MI) and other unidentified species carrying the maltose binding protein were also observed between the precursor and MC bands. Mtu recA intein antiserum (Fig. 3D) identified inteins from splicing-proficient derivatives. Full-length intein was seen as a product of splicing of full-length MIC (wild type and the MIC25 mutant, lanes 1 and 2) and MIT (lane 3), whereas mini-inteins of the appropriate size were visible for constructs 110Δ383 and 114Δ372 (lanes 5 and 6, corresponding to constructs 6 and 8 in Fig. 2B). For construct 101Δ405(A7) the intein band was detectable only on longer exposures (lane 4, corresponding to construct 3 in Fig. 2B), because this construct splices slowly (see below; Fig. 4B). Finally, I-TevI antiserum (Fig. 3E) clearly identified precursors, as well as ligated exteins (MC), in splicing-proficient MIC derivatives (lanes 1, 2, 5, and 6) but, as expected, not in the MIT construct (lane 3). Importantly, there is agreement between predicted and observed sizes of the different inteins (Fig. 3D), and the size of ligated exteins was constant for all MIC derivatives, regardless of intein size (Fig. 3 C and E; see also below; Fig. 4A).
Figure 4.
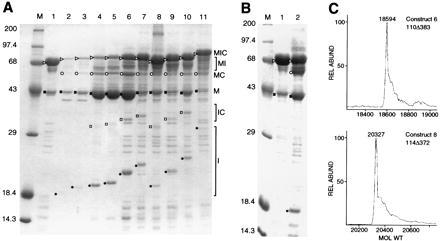
Purification and characterization of mini-intein constructs. (A) Purified MIC mini-intein derivatives. 12% SDS polyacrylamide gel is shown. M, markers (kDa), and lanes: 1, 101Δ405(A7); 2, 101Δ395(A5′); 3, 101Δ383(A7); 4, 110Δ383; 5, 114Δ384; 6, 114Δ372; 7, 123Δ372′; 8, 129Δ405(A1); 9, 129Δ383(A1); 10, 129Δ372′; 11, 129Δ271(A1), corresponding to constructs 3–13, respectively, in Fig. 2B. (B) In vitro splicing. 12% SDS polyacrylamide gel of MIC101Δ405(A7) (Fig. 2B, construct 3) immediately after purification on amylose resin (lane 1) and after 18 days at 4°C (lane 2). M, markers (kDa). In both A and B, ▹, precursor (MIC); ○, ligated exteins (MC); •, intein (I); □, N-terminal cleavage product (IC); ▪, maltose binding protein (M). (C) Precise molecular masses of selected mini-inteins. HPLC-purified mini-inteins were analyzed by infusion on a Finnigan-MAT TSQ-700 triple quadrupole mass spectrometer equipped with a Finnigan ESI source (San Jose, CA).
Purification and Properties of Mini-Intein Derivatives.
Overexpressed proteins were affinity-purified on amylose resin for most of the MIC derivatives (Fig. 4). In each case, as expected, all fusion proteins containing maltose binding protein were purified (Fig. 4A): tripartite precursor (MIC); ligated exteins (MC); side-products (M, which was highly enriched in these preparations) and (MI). Free intein (I) as well as C and IC were also visible in some preparations, either because of association of these species with proteins bound to the column and/or because splicing occurs during or after purification.
TS phenotype as a function of temperature provides a semiquantitative measure of splicing efficiency. In the MIC context, splicing was judged by quantitating the appearance of ligated exteins upon overexpression of the tripartite fusion. In most cases there was agreement between the activity in the two contexts, although 129Δ271(A1) (Fig. 2B, construct 13) was absolutely TS− but sometimes exhibited very low level MIC splicing (Fig. 4A). In addition, 101Δ405(A7) and 129Δ405(A1) (Fig. 2B, constructs 3 and 10), which were splicing proficient as judged by TS phenotype, exhibited very low level MIC splicing on initial induction (Figs. 3 and 4A). However, upon storage in elution buffer at 4°C, these fusion proteins continued to splice in vitro (Fig. 4B; data not shown). Despite these subtle context effects, there is a general correlation between TS phenotype and splicing proficiency (Fig. 2B).
Constructs that tentatively delineate the N- and C-terminal boundaries of the protein-splicing domains in the TS context (94Δ383 and 114Δ406; Fig. 2B, constructs 15 and 17, arrowheads) also splice as MIC derivatives (Fig. 2B; data not shown). The splicing properties of these derivatives confirm that the functional domains of the Mtu recA intein are contained within the first 94 and the last 35 aa.
Assessment of Splicing Fidelity.
Ligated exteins (MC) resulting from MIC splicing were of the same apparent molecular mass (56.1 kDa) for all constructs including the full-length intein (Figs. 3 and 4). To verify that the splicing reaction was proceeding accurately, the molecular masses of two mini-intein products from MIC splicing reactions were determined by mass spectrometry. Mini-inteins from MIC constructs 110Δ383 and 114Δ372 (Fig. 2B, constructs 6 and 8) with predicted molecular masses of 18,594 and 20,326 Da, respectively, were determined to be 18,594 and 20,327 Da by electrospray ionization mass spectroscopy (Fig. 4C). Together, these data indicate not only that the mini-inteins are capable of protein splicing but also that the accuracy of the reaction remains uncompromised.
DISCUSSION
Mini-Inteins Deleted for the Entire Endonuclease Domain of the Mtu recA Intein Are Splicing Proficient.
Derivatives of the Mtu recA intein have been constructed that retain splicing activity despite removal of the entire endonuclease domain, which constitutes more than two-thirds of the wild-type protein. These results are in accord with deletion of ca. 80% of the PI-SceI endonuclease domain while maintaining splicing function (16). The Mtu recA mini-inteins were analyzed using two in-frame fusion systems, for which a general correlation between TS phenotype and MIC splicing was found. These results are consistent with the expectation that all information necessary for splicing is carried within the intein itself and that splicing activity is not an artifact of a particular fusion context. It was thereby shown that all information required for splicing function is carried within the first 94 and final 35 aa of the 440-residue intein. This size and configuration of the mini-intein corresponds well with both the naturally occurring inteins without endonuclease domains (5, 13) and with the computer alignments that designated the first 101 and last 35 aa as constituting the protein-splicing element (13).
It is clear from the analysis of MIC mini-intein fusions that, in addition to bona fide splicing, as judged by the presence of ligated exteins, there is a significant amount of substrate cleavage in the absence of ligation (Fig. 4A), as has been seen in other intein expression constructs (20). First, the artificial nature of tripartite fusion systems, wherein the individual components are, by design, stable structural domains rather than the intein interrupting a protein of defined structure, likely increases accumulation of side-products. Second, the precise nature of the deletion appears to affect the build-up of cleavage products as reflected by variability in amount of side-products among the different constructs (Fig. 4A). Third, removal of the endonuclease domain may compromise splicing—it is not unlikely that the activity of the protein-splicing domain is affected by the endonuclease domain, just as endonuclease interaction with its homing site seems to be influenced by the intein domain (12). Nevertheless, the appearance of ligated exteins validates the splicing proficiency of the mini-inteins.
Structural and Functional Domains of Mobile Inteins.
The localization of protein-splicing activity in the Mtu recA intein as defined by the mini-inteins described in this work is entirely consistent with the two-domain structure of PI-SceI (12) and the statistical modeling that predicts that all endonuclease-containing inteins are folded into two domains (13). The central endonuclease domain is predicted to be separated from the minimal protein-splicing domains by variable spacer sequences (ref. 13; J.Z.D. et al., unpublished results; Fig. 1B), which may serve to enable the protein to accommodate the dual functions of the endonuclease-containing inteins. The tolerance of Mtu recA intein function to different central deletions is likely a consequence of the flexibility of these spacers.
It is clear, however, that inteins are not tolerant of all internal deletions. For example, the 166Δ201 deletion of the Mtu recA intein (19) does not retain activity, despite the presence of all residues required for splicing function. Similarly, the 129Δ271(A1) derivative (Fig. 2B, construct 13) exhibits very low activity and only in the MIC context. Additionally, a derivative of PI-SceI with a 184-aa deletion spanning the endonuclease motifs had no protein-splicing activity unless modified by the addition of peptide linkers at the site of the deletion (16). The lack of splicing activity in these constructs containing an intact protein-splicing domain presumably reflects protein-folding problems. These constraints on intein function have undoubtedly played an important role in guiding their evolution.
Evolution of Mobile Inteins.
The invasiveness of homing endonuclease genes is likely to form the basis of the maintenance and spread of both mobile introns and inteins (refs. 8–12; Fig. 5). According to one model, an endonuclease gene invaded a protein-coding sequence and evolved protein-splicing activity to preserve the functional integrity of the host protein (Fig. 5A, model 1). According to a second model, the endonuclease gene invaded the DNA sequence of a primitive protein-splicing element (Fig. 5A, model 2). Model 1 was supported by the observation that HO endonuclease, a free-standing LAGLIDADG endonuclease, has six intein motifs (ref. 4; Fig. 1A) but is unable to splice as an in-frame protein fusion (J. Platko and F. Perler, cited in ref. 5; V.D. and M.B., unpublished results), suggesting that HO is part-way along the path to evolving intein function. However, the combination of crystallographic studies (12), molecular modeling (13), and our genetic analysis support a structurally and functionally independent protein-splicing domain, in favor of model 2, as argued further below. In this case, HO endonuclease could be a defunct intein.
Figure 5.
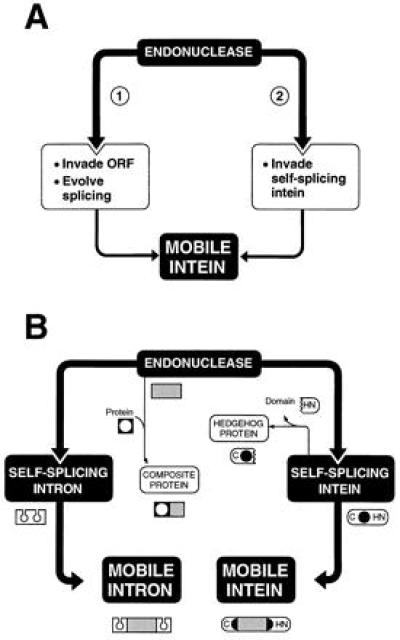
Intein evolution. (A) Models for the coexistence of endonuclease and protein-splicing functions in mobile inteins. (B) Mobile intron and intein evolution. An endonuclease gene is shown invading DNA encoding a self-splicing intron or intein to generate a mobile intron or intein, respectively. The sharing of endonuclease motifs with other proteins to form composite proteins is shown. The formation of a hedgehog protein from an intein is depicted by loss of the C-terminal domain responsible for protein ligation. Symbols are depictions of DNA, with the endonuclease ORF represented by a gray rectangle.
Predicted endonuclease target sites flanking the endonuclease gene that serve as markers of the initial invasion event have been found in mobile group I introns (11). These recognition sites have not, however, been observed in inteins (ref. 12; W.W. and J.Z.D., unpublished results). This work, demonstrating the functional independence of the protein-splicing and endonuclease domains, is therefore of particular importance in supporting their separate origin. Furthermore, the ability of the intein to function in the absence of the entire endonuclease domain favors the scenario in which the endonuclease gene invaded a preexisting intein (Fig. 5A, model 2). The clear, functional independence of the protein-splicing domain is counter to model 1, in which some splicing function would be predicted to reside in the endonuclease moiety itself.
Endonuclease-containing inteins are far more common in modern genomes than the endonuclease-free inteins. The endonuclease seems to provide the means for inteins to be maintained and, indeed, to spread among different genes, organisms, and perhaps even kingdoms. Through their ability to splice, autocatalytic inteins, like self-splicing introns, in turn provide a genetically silent haven for the invasive endonuclease ORFs and the vehicles for their propagation. Accordingly, endonucleases can be viewed as a substantial driving force in molecular evolution. Through their capacity to make nicks and breaks in DNA, endonuclease genes can invade sequences to form molecular associations that not only mobilize introns and inteins but can also provide catalytic function to other proteins (Fig. 5B). An example of the latter is the H-N-H endonuclease cassette, which is present in mobile group I intron- and intein-encoded proteins, as well as in the colicin family of bacterial toxins and in the tripartite reverse transcriptase-maturase-endonuclease proteins of mobile group II introns (13, 21, 22, 28). The propensity of endonuclease genes to colonize genomes therefore can influence genome stability and configuration by promoting lesions in DNA and subsequent intron- or intein-based rearrangements. Furthermore, intron endonucleases can provide selective advantage in both phage and archaeal systems (29, 39), whereas colicins promote host defense, thereby influencing the stability of entire microbial populations.
Although the antiquity of the original self-splicing introns has been argued (23), nothing is known of the evolutionary history of “ancestral” endonuclease-free inteins. Although there are examples of endonuclease-free inteins in all three biological kingdoms (5, 13), it would be premature to assume that these elements existed in the last common ancestor. It is, however, interesting to note that mechanistic parallels have been drawn between inteins and the self-activating amidohydrolases (24). Furthermore, the endonuclease-free inteins have been hypothesized to be evolutionarily related to the self-cleaving hedgehog proteins, which are involved in eukaryotic developmental pathways (13, 25, 26). It has been suggested that the hedgehog family, which exists in arthropods and all vertebrates from amphibians to mammals, arose from an intein that lost ligation activity (ref. 13; Fig. 5). Regardless, finding such nonmobile, autocatalytic, intein-like molecules with related functions in deeply branching organisms will ultimately help address issues relating to intein ancestry and evolutionary age.
Acknowledgments
We are grateful to Benoit Cousineau, Mathias Holpert, Joseph Kowalski, Richard Lease, Monica Parker, and George Silva for critical reading of the manuscript; Georges Belfort and Fred Gimble for useful discussions; Maryellen Carl for expert secretarial assistance; Robert F. Stack and Charles R. Hauer III for the mass spectrometry; and Maureen Belisle for making the figures. The Molecular Genetics Core Facility at the Wadsworth Center provided DNA oligonucleotides and DNA sequencing service. This work was supported by National Institutes of Health Grants GM39422 and GM44844 to M.B. J.Z.D. is supported by a grant from the Danish Natural Science Research Council and by the National Cancer Institute, Department of Health and Human Services.
ABBREVIATION
- TS
thymidylate synthase
References
- 1.Colston M J, Davis E O. Mol Microbiol. 1994;12:359–363. doi: 10.1111/j.1365-2958.1994.tb01025.x. [DOI] [PubMed] [Google Scholar]
- 2.Perler F B, Davis E O, Dean G E, Gimble F S, Jack W E, Neff N, Noren C J, Thorner J, Belfort M. Nucleic Acids Res. 1994;22:1125–1127. doi: 10.1093/nar/22.7.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cooper A A, Stevens T H. TIBS. 1995;20:351–356. doi: 10.1016/s0968-0004(00)89075-1. [DOI] [PubMed] [Google Scholar]
- 4.Pietrokovski S. Protein Sci. 1994;3:2340–2350. doi: 10.1002/pro.5560031218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perler F B, Olsen G J, Adam E. Nucleic Acids Res. 1997;25:1087–1093. doi: 10.1093/nar/25.6.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belfort M, Roberts R J. Nucleic Acids Res. 1997;25:3379–3388. doi: 10.1093/nar/25.17.3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gimble F S, Thorner J. Nature (London) 1992;357:301–306. doi: 10.1038/357301a0. [DOI] [PubMed] [Google Scholar]
- 8.Perlman P S, Butow R A. Science. 1989;246:1106–1109. doi: 10.1126/science.2479980. [DOI] [PubMed] [Google Scholar]
- 9.Belfort M. Trends Genet. 1989;5:209–213. doi: 10.1016/0168-9525(89)90083-8. [DOI] [PubMed] [Google Scholar]
- 10.Lambowitz A M. Cell. 1989;56:323–326. doi: 10.1016/0092-8674(89)90232-8. [DOI] [PubMed] [Google Scholar]
- 11.Loizos N, Tillier E R M, Belfort M. Proc Natl Acad Sci USA. 1994;91:11983–11987. doi: 10.1073/pnas.91.25.11983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duan X, Gimble F S, Quiocho F A. Cell. 1997;89:555–564. doi: 10.1016/s0092-8674(00)80237-8. [DOI] [PubMed] [Google Scholar]
- 13.Dalgaard J Z, Moser M J, Hughey R, Mian I S. J Comput Biol. 1997;4:193–214. doi: 10.1089/cmb.1997.4.193. [DOI] [PubMed] [Google Scholar]
- 14.Hodges R A, Perler F B, Noren C J, Jack W E. Nucleic Acids Res. 1992;20:6153–6157. doi: 10.1093/nar/20.23.6153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gimble F S, Stephens B W. J Biol Chem. 1995;270:5849–5856. doi: 10.1074/jbc.270.11.5849. [DOI] [PubMed] [Google Scholar]
- 16.Chong S, Xu M-Q. J Biol Chem. 1997;272:15587–15590. doi: 10.1074/jbc.272.25.15587. [DOI] [PubMed] [Google Scholar]
- 17.Derbyshire V, Kowalski J C, Dansereau J T, Hauer C R, Belfort M. J Mol Biol. 1997;265:494–506. doi: 10.1006/jmbi.1996.0754. [DOI] [PubMed] [Google Scholar]
- 18.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 19.Davis E O, Jenner P J, Brooks P C, Colston M J, Sedgwick S G. Cell. 1992;71:201–210. doi: 10.1016/0092-8674(92)90349-h. [DOI] [PubMed] [Google Scholar]
- 20.Xu M-Q, Southworth M W, Mersha F B, Hornstra L J, Perler F B. Cell. 1993;75:1371–1377. doi: 10.1016/0092-8674(93)90623-x. [DOI] [PubMed] [Google Scholar]
- 21.Gorbalenya A E. Protein Sci. 1994;3:1117–1120. doi: 10.1002/pro.5560030716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shub D A, Goodrich-Blair H, Eddy S R. Trends Biochem. 1994;19:402–404. doi: 10.1016/0968-0004(94)90086-8. [DOI] [PubMed] [Google Scholar]
- 23.Cech T R. Int Rev Cytol. 1985;93:3–22. doi: 10.1016/s0074-7696(08)61370-4. [DOI] [PubMed] [Google Scholar]
- 24.Brannigan J A, Dodson G, Duggleby H J, Moody P C E, Smith J L, Tomchick D R, Murzin A G. Nature (London) 1995;378:416–419. doi: 10.1038/378416a0. [DOI] [PubMed] [Google Scholar]
- 25.Koonin E V. Trends Biochem Sci. 1995;20:141–142. doi: 10.1016/s0968-0004(00)88989-6. [DOI] [PubMed] [Google Scholar]
- 26.Lee J J, Ekker S C, Von Kessler D P, Porter J A, Sun B I, Beachy P A. Science. 1994;266:1528–1537. doi: 10.1126/science.7985023. [DOI] [PubMed] [Google Scholar]
- 27.Belfort M, Ehrenman K, Chandry P S. Methods Enzymol. 1990;181:521–539. doi: 10.1016/0076-6879(90)81149-o. [DOI] [PubMed] [Google Scholar]
- 28.Pietrokovski, S. (1997) Protein Sci., in press.
- 29.Goodrich-Blair H, Shub D A. Cell. 1996;84:211–221. doi: 10.1016/s0092-8674(00)80976-9. [DOI] [PubMed] [Google Scholar]
- 30.Aagaard C, Dalgaard J Z, Garrett R A. Proc Natl Acad Sci USA. 1995;92:12285–12289. doi: 10.1073/pnas.92.26.12285. [DOI] [PMC free article] [PubMed] [Google Scholar]