

Abstract
STAT1 is a cytoplasmic transcription factor that is phosphorylated by Janus kinases (Jak) in response to interferon-γ (IFNγ). Phosphorylated STAT1 translocates to the nucleus, where it turns on specific sets of IFNγ-inducible genes. Here, we show that UV light interferes with tyrosine phosphorylation of STAT1, thereby hindering IFNγ from exerting its biological effects. This effect is not due to a down-regulation of the IFNγ receptor because phosphorylation of upstream-located Jak1 and Jak2 was not suppressed by UV light. In contrast, UV light had no effect on the phosphorylation of STAT3, which is activated by the proinflammatory cytokine interleukin 6. The UV light effect on STAT1 phosphorylation could be antagonized by vanadate, indicating at least partial involvement of a protein tyrosine phosphatase. Therefore, this study indicates a mechanism by which UV light can inhibit gene activation and suggests STAT1 as a new extranuclear UV target closely located to the membrane.
Keywords: signal transduction, immunosuppression, cytokines, phosphorylation
Interferon γ (IFNγ) is a pleiotropic cytokine with antiproliferative and immunomodulatory activities that are crucial for the regulation of immune responses (1). IFNγ signals through a multimeric receptor complex consisting of a ligand binding chain and a transmembrane accessory factor (2, 3). IFNγ signaling is initiated when IFNγ binds to its receptor, thereby inducing its dimerization. Upon this event, the receptor-associated Janus kinases Jak1 and Jak2 start to transphosphorylate each other, resulting in kinase activation. The cytoplasmic domains of the receptors then become tyrosine phosphorylated, enabling interaction with the signal transducer and activator of transcription protein STAT1 (4–6). Finally, STAT1 is activated by tyrosine phosphorylation, leading to its dimerization, and translocates to the nucleus, where it activates transcription of IFNγ-responsive genes by binding to the IFNγ-activated sequences (GAS) (7, 8).
UV light is known for its immunosuppressive properties, which are best demonstrated by the inhibition of cellular immune reactions and by the exacerbation of infectious diseases (9, 10). UV light induces the release of immunosuppressive cytokines (11) but obviously also can interfere with the biological effects of cytokines; we recently found that UV light counteracts biological activities of IFNγ (12). Specifically, we observed that stimulation of the murine keratinocyte line Pam212 with IFNγ induced mRNA expression of the cytokine interleukin 7 (IL-7) whereas IL-7 transcripts were suppressed in cells that were UV light-exposed before IFNγ treatment. The inhibitory effect of UV light does not, as initially anticipated, target the IL-7 promoter in a negative regulatory way but hinders IFNγ from exerting its biological activity. IFNγ induces IL-7 mRNA expression via activation of an IFN-stimulated response element located in the 5′ upstream region of the IL-7 gene (13) by inducing the transcription factor IFN regulatory factor 1 (IRF-1), which binds to the IFN-stimulated response element (14). Electrophoretic mobility-shift assays (EMSA) revealed that IFNγ significantly induced IRF-1 binding whereas UV treatment remarkably decreased IFNγ-induced IRF-1 binding. Northern blot analysis indicated that UV light reduced IFNγ-induced IRF-1 mRNA (12).
Because IFNγ is an important immunomodulatory cytokine, inhibition of its activity by UV light may contribute to its immunosuppressive properties. Therefore and based on the fact that inhibition of cytokine activities may represent a new biological activity of UV light, we were interested to clarify the molecular mechanisms underlying the inhibition of IRF-1 mRNA expression by UV light. Here we show that UV light interferes with tyrosine phosphorylation of STAT1, thereby preventing binding of STAT1 to the GAS element in the IRF-1 promoter. This study both indicates a mechanism by which UV light inhibits gene activation and suggests STAT1 as a further extranuclear, closely membrane-located UV light target.
MATERIALS AND METHODS
Cell Culture and Treatment of Cells.
The murine keratinocyte cell line Pam212 was cultured in supplemented RPMI 1640 medium. UV irradiation was performed as described (15) using FS-40 bulbs (Westinghouse Electric, Pittsburgh), which emit most of the energy in the UVB range (290–320 nm). A dose of 250 J/m2 was used throughout the study because this dose yielded the optimal effect without significantly affecting cell viability. Thereafter, cells were medium supplemented with or without recombinant murine IFNγ (Genzyme) added. Unless otherwise stated, experiments were performed at least three times, and data shown are representative of one of those.
Chloramphenicol Acetyltransferase (CAT) Assay.
Using a calcium/phosphate coprecipitation method, cells were transfected with the GAS oligonucleotide (5′-AGCCTGATTTCCCCGAAATGATGAGGCCGAGTGC-3′) and cloned just upstream of the thymidine kinase promoter; CAT coding sequencing (pGAScat) followed. Thirty six hours after transfection, cells were treated with IFNγ and UV light, respectively; 18 h later protein fractions were extracted. Enzymatic activity of CAT was determined using [3H]-chloramphenicol, and CAT activities obtained were normalized by β-galactosidase activity derived from cotransfection of a cytomegalovirus promoter-driven β-galactosidase expression vector.
EMSA.
Fifteen minutes after stimulation, cells were swelled in hypotonic buffer as described (12). After addition of Nonidet P-40, cell suspensions were minifuged, and supernatants were kept for Western blot analysis as cytosolic protein fractions. To extract nuclear proteins, pellets were resuspended in hypertonic buffer (hypotonic buffer supplemented with 0.4 M NaCl), incubated for 15 min on ice, and minifuged. EMSA was performed as described (12), and binding reactions were carried out by addition of 2 μg of poly(dI⋅dC) (Boehringer Mannheim) and 104 cpm of 32P-labeled, double-stranded oligonucleotide for 20 min at 22°C. To characterize binding, antibodies (Ab) were added to the reactions and incubated at 4°C for 30 min. Reaction mixture was applied onto native high ionic gel [1× Tris/glycine buffer/4% acrylamide (acrylamide/bis ratio, 30:1)/2.5% glycerol), electrophoresis was done at 120 V for 1.5 h, and blots were autoradiographed. Oligonucleotides derived from the IRF-1 gene (GAS-IRF-1) had the following sequence: 5′-AGCCTGATTTCCCCGAAATGATGAGGCCGAGTGC-3′ (IFN response element responsible for GAS activation by STAT1 is underlined).
Competition analysis was performed by adding the following competitors: IFN-stimulated response element–IL7: 5′-AGTAGAAACTGAAAGTACC-3′; mutant (mu) GAS: 5′-AGCCTGATTTCCCCGTTTTGATGAGGCCGAGTGC-3′ (the substitution from AAA to TTT to abolish the binding region of the GAS–IRF1 probe is underlined).
Immunoprecipitation and Western Blot Analysis.
Fifteen minutes after treatment, cells were washed with PBS containing 1 mM sodium orthovanadate (Na3VO4) and were lysed in radioimmunoprecipitation assay buffer (10 mM Tris, pH 8/150 mM sodium chloride/1% Nonidet P-40/0.5% sodium deoxycholate/0.1% SDS/1 mM Na3VO4/1 mM phenylmethylsulfonyl fluoride/4 μg/ml aprotinin). Proteins from lysates were immunoprecipitated with Ab against STAT1 or STAT3 (Upstate Biotechnology, Lake Placid, NY) and protein A at 4°C overnight and washed four times in radioimmunoprecipitation assay buffer by centrifugation at 2500 rpm, 4°C for 5 min. Precipitated proteins were released from protein A beads by boiling in the presence of 5% 2-mercaptoethanol. Samples were fractionated in 7.5% SDS/PAGE and blotted onto nitrocellulose membranes (Hybond enhanced chemiluminescence, Amersham), and Western blot analysis was performed using an Ab against phosphotyrosine (4G10; Upstate Biotechnology). The same membranes were reprobed with Ab directed against STAT1 or STAT3 (Santa Cruz Biotechnology). Signals were detected using an enhanced chemiluminescence kit. For detection of IκB, cytosolic protein fractions were analyzed in 12% SDS/PAGE by Western blot using an Ab against IκB (Santa Cruz Biotechnology).
Autophosphorylation Assay.
Five minutes after stimulation, cells were washed twice with ice-cold PBS containing 1 mM Na3VO4 and detached with a rubberpoliceman. Cells were resuspended in 1 ml of lysis buffer (30 mM Tris, pH 7.5/150 mM NaCl/1 mM phenylmethylsulfonyl fluoride/60 μg/ml aprotinin/1 mM Na3VO4/1% Nonidet P-40/10% glycerol), incubated on ice for 15 min, vortexed, and minifuged at 14,000 rpm, 4°C for 15 min. Protein A bead-coupled Ab directed against Jak1 and Jak2 (Upstate Biotechnology) were added to supernatants, and immunoprecipitation was performed at 4°C for 3 h. Complexes were washed twice in lysis buffer by centrifugation at 2500 rpm, 4°C for 5 min and twice in kinase buffer [20 mM Hepes, pH 7.2/10 mM MnCl2/0.1% Brij 35 (Pierce)/30 μM Na3VO4]. Pellets were resuspended in 30 μl of kinase buffer containing 5 μCi [32P]ATPγ, and kinase reaction was carried out at 25°C for 15 min. Reaction was stopped by SDS sample buffer containing 2.5% 2-mercaptoethanol and boiling for 5 min, and labeled samples were separated from protein A beads by centrifugation. Samples were run on 7.5% SDS/PAGE at 120 V for 2 h. After blotting, membranes were exposed to x-ray films at room temperature. Western blots were performed using Ab against Jak1 and Jak2.
RESULTS
UV Light Inhibits Activation of the GAS Element by IFNγ.
Induction of IRF-1 mRNA by IFNγ is controlled by the GAS element (16), so we addressed whether UV light inhibits activation of the GAS element. Thus, Pam212 cells were transfected with a construct containing the GAS oligonucleotide just upstream of the thymidine kinase promoter followed by CAT coding sequence. Whereas addition of IFNγ significantly induced CAT activity, exposure of cells to 250 J/m2 UV light immediately before IFNγ addition almost completely abolished CAT induction (Fig. 1A). Because activated STAT1 interacts with the GAS sequence (17), we performed EMSA with nuclear protein extracts from Pam212 cells that were exposed to IFNγ alone or to IFNγ plus UV light. UV exposure suppressed IFNγ-induced STAT1 binding (Fig. 1B). In most of the EMSA, nonspecific bands appeared whose nature was not identified, so specificity was proven by incubating the extracts with a STAT1-specific Ab, resulting in loss of the corresponding band (Fig. 1B, lane 5). Sequence specificity of STAT1 binding to the GAS–IRF-1 element was demonstrated by competition analysis shown in Fig. 1C.
Figure 1.

UV light suppresses IFNγ-induced GAS activation and binding of STAT1 to GAS. (A) pGAScat-transfected Pam212 cells were exposed to 20 ng/ml IFNγ, 250 J/m2 UV light followed by IFNγ, or UV light alone or were left untreated (control); 18 h thereafter protein fractions were extracted. CAT activities obtained were normalized by β-galactosidase activity derived from cotransfection of a cytomegalovirus promoter-driven β-galactosidase expression vector. (B) Pam212 cells were treated with 20 ng/ml IFNγ (lanes 2, 5 and 6), UV light (250 J/m2) plus 20 ng/ml IFNγ added immediately after UV exposure (lane 4), or UV light plus IFNγ added 15 min after UV exposure (lane 3) or were left untreated (lane 1). EMSA was performed by incubating nuclear protein extracts with GAS–IRF1 oligonucleotides. In lanes 5 and 6, proteins extracted from cells stimulated with IFNγ were incubated with an Ab directed against STAT1 or with nonimmune rabbit serum (NRS). (C) Competition analysis was performed with nuclear protein fractions prepared from Pam212 cells that were stimulated with IFNγ (20 ng/ml) or left untreated by adding a 50-molar excess of GAS, IFN-stimulated response element–IL7, or mutant (mu) GAS oligonucleotides as competitors.
UV Light Interferes with Phosphorylation of STAT1.
To address whether UV light interferes with the phosphorylation of STAT1, proteins from lysates of Pam212 cells were immunoprecipitated with an Ab against STAT1, and Western blot analysis was performed using Ab against phosphotyrosine and STAT1. Whereas stimulation of Pam212 cells with IFNγ resulted in phosphorylation of STAT1, STAT1 precipitated from IFNγ plus UV light-treated cells was not phosphorylated (Fig. 2A). This suggests that UV light interferes with IFNγ-induced tyrosine phosphorylation of STAT1, thereby preventing activation of the GAS element in the IRF-1 promoter by IFNγ.
Figure 2.

UV light interferes with phosphorylation of STAT1 but not of STAT3. (A) Pam212 cells were either irradiated with 250 J/m2 UV light, treated with 20 ng/ml IFNγ, treated with UV light plus IFNγ, or left untreated. Proteins were immunoprecipitated with an Ab directed against STAT1, resolved by SDS/PAGE, and analyzed by anti-phosphotyrosine and anti-STAT1 immunoblotting. (B) Pam212 cells were irradiated with 250 J/m2 UV light, treated with 100 ng/ml recombinant murine IL-6 or with UV light plus IL-6, or left untreated. Proteins were immunoprecipitated with an Ab directed against mouse STAT3 and analyzed by anti-phosphotyrosine and anti-STAT3 immunoblotting.
UV light is known to activate several transcription factors including NF-κB or AP-1 (18–21), thus the present findings indicate that UV light can affect transcription activators in a negative regulatory way and may do so by interfering with phosphorylation. At present, the sequences of six mammalian STAT family members have been reported (8). STAT proteins exhibit Src homology region 2 domains that bind to receptor phosphotyrosines, after which the STAT proteins are phosphorylated. To address whether UV light interferes with STAT protein phosphorylation in general, we studied the effect of UV light on STAT3 phosphorylation. STAT3 is involved in IL-6 signaling (22, 23). Binding of IL-6 to its receptor leads to phosphorylation of the kinases Jak1, Jak2, and Tyk2, finally inducing tyrosine phosphorylation of STAT3. Pam212 cells were stimulated with IL-6, UV light alone, or UV light followed by IL-6 stimulation. Fifteen minutes thereafter, proteins were immunoprecipitated with an anti-STAT3 Ab, and Western blot analysis was performed using Ab against phosphotyrosine and STAT3. Whereas stimulation with IL-6 resulted in STAT3 phosphorylation, treatment of cells with UV light alone had no effect on STAT3 (Fig. 2B). In contrast to the findings with STAT1, STAT3 proteins precipitated from cells treated with UV light plus IL-6 turned out to be phosphorylated as in cells stimulated with IL-6 alone.
UV Light Does Not Inhibit Jak1 and Jak2 Phosphorylation.
Phosphorylation of STAT1 is a consequence of activation of the IFNγ receptor after ligand binding, so UV light could indirectly inhibit STAT1 phosphorylation simply by down-regulating the IFNγ receptor. To determine whether UV light interferes with IFNγ-activated phosphorylation of STAT1 or rather indirectly via modulation of the IFNγ receptor, the effect of UV light on the IFNγ receptor-associated tyrosine kinases Jak1 and Jak2 was addressed. Jak1 and Jak2 are both required for the IFNγ response and become tyrosine-phosphorylated upon ligand binding (24, 25). Lysates from IFNγ-treated or IFNγ plus UV light-treated cells were immunoprecipitated with an Ab against Jak1 (Fig. 3A) and analyzed by anti-phosphotyrosine immunoblotting. IFNγ-induced Jak1 phosphorylation was not affected when cells were preexposed to UV light. Similar findings were observed after Jak2 immunoprecipitation (Fig. 3B).
Figure 3.

UV light does not suppress tyrosine phosphorylation of Jak1 and Jak2. Pam212 cells were either irradiated with 250 J/m2 UV light, treated with 20 ng/ml IFNγ, treated with UV light plus IFNγ, or left untreated; 5 min thereafter immunoprecipitations were performed using Ab directed against Jak1 (A) or Jak2 (B). Western blot analysis was performed using an Ab directed against phosphotyrosine. The same membrane was reprobed with an Ab directed against Jak1 (A) and Jak2 (B). (C) Pam212 cells were treated with 20 ng/ml IFNγ (lane 2), UV light (250 J/m2) plus IFNγ (lane 3), or UV light (lane 4) or were left untreated (lane 1). Cell lysates were precipitated with Ab directed against Jak1 and Jak2. For autophosphorylation (AP), precipitated proteins were reacted with [32P]ATPγ. To monitor equal loading of samples, the same membranes were subjected to Western blot analysis (WB) using Ab directed against Jak1 and Jak2.
To determine whether Jak1 and Jak2 precipitated from UV light-exposed cells are functionally active, we addressed the autophosphorylation capacity of both proteins by in vitro kinase assays. Cells were exposed to IFNγ, UV light, or IFNγ plus UV light or were left untreated; 5 min later immunoprecipitation was performed, and precipitated Jaks were reacted with [32P]ATPγ. Although weak autophosphorylation of Jak1 was observed in precipitates from untreated controls, autophosphorylation was significantly induced in IFNγ-treated cells, which, however, was not suppressed but even enhanced in precipitates from cells that were UV light-exposed before IFNγ stimulation (Fig. 3C). Similar findings were observed with Jak2 extracts. These data suggest that UV light-mediated inhibition of STAT1 phosphorylation takes place downstream of active sites of Jak1 and Jak2 and that the inhibitory effect of UV light may directly affect the STAT1 protein or, even if indirectly, should be very near this phosphorylation event.
Vanadate but Not Lactacystin Counteracts the UV Light Effect.
UV light could down-regulate phosphorylated STAT1 by directly inhibiting IFNγ-induced phosphorylation of STAT1. However, it recently was recognized that the amount of phosphorylated STAT1 does not only depend on the rate of active phosphorylation but also can be influenced by dephosphorylation. The amount of phosphorylated STAT1 is maximal between 15 and 30 min after IFNγ treatment and then rapidly decreases to undetectable levels within 2 h (17, 26). Recently, it was shown that the amounts of phosphorylated STAT1 in IFNγ-treated cells are, at least in part, controlled by ubiquitin-dependent proteolysis of STAT1 because STAT1 activated by IFNγ was stabilized by the proteasome inhibitor MG132 (26). To determine whether this pathway was involved in our system, Pam 212 cells were incubated with the proteasome inhibitor lactacystin and stimulated with IFNγ or IFNγ plus UV light or were left unstimulated. Fifteen minutes later, EMSA was performed using the synthetic GAS oligonucleotide. Although IFNγ induced STAT1 binding, binding was significantly reduced in extracts obtained from cells exposed to UV light before IFNγ treatment (Fig. 4A). The inhibitory effect of UV light, however, was not affected by lactacystin, suggesting that UV light-induced down-regulation of STAT1 phosphorylation may not be mediated via the proteasome pathway. To prove whether lactacystin acts as a proteasome inhibitor in our hands, the effect of IκB degradation was addressed. It has been reported that proteasome inhibitors block inducible degradation of IκB in mammalian cells (27). UV light is known to activate NF-κB (18, 20), so the effect of lactacystin on UV light-induced IκB degradation was addressed. Therefore, from the same cell preparations, cytosolic protein fractions were subjected to Western blot analysis with an Ab against IκB. The efficacy of lactacystin to inhibit the proteasome pathway is demonstrated in the lower panel of Fig. 4A, showing that lactacystin inhibited UV light-induced IκB degradation.
Figure 4.
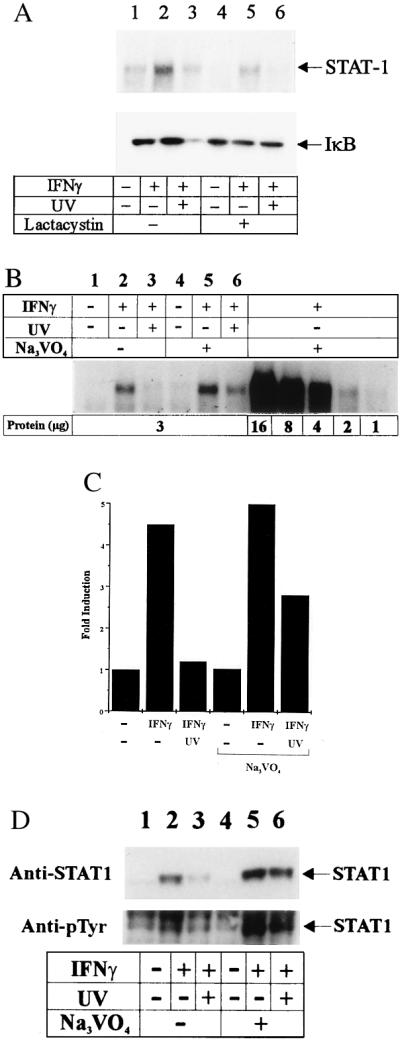
Vanadate but not lactacystin counteracts UV light-mediated suppression of IFNγ-activated STAT1. (A) Cells were first incubated in the presence (lanes 4–6) or absence (lanes 1–3) of 50 μM lactacystin for 30 min, washed, and stimulated with IFNγ (lanes 2 and 5) or IFNγ plus UV light (lanes 3 and 6) or left unstimulated (lanes 1 and 4); 15 min later, nuclear protein fractions were extracted, and EMSA was performed using the GAS oligonucleotide. From the same cell preparations, cytosolic protein fractions were obtained and fractionated on SDS/PAGE, and Western blot was analysis performed using an Ab directed against IκB. (B) Cells were first incubated in the presence (lanes 4–6) or absence (lanes 1–3) of 1 mM sodium orthovanadate for 30 min, washed extensively twice with PBS, and stimulated with IFNγ (lanes 2 and 5) or IFNγ plus UV light (lanes 3 and 6) or left unstimulated (lanes 1 and 4). EMSA was performed using the GAS oligonucleotide. To avoid overloading of the gels, 3 μg of protein was loaded according to protein titration (Right). (C) Lanes 1–6 were quantitated by densitometry. (D) Cells were treated in the same way as in B; 15 min thereafter nuclear protein fractions were obtained, fractionated by 7.5% SDS/PAGE, and subjected to Western blot analysis using an Ab directed against STAT1 (Upper) and phosphotyrosine (Lower).
There is also evidence that removal of activated STAT1 molecules from the nucleus appears not to be proteolytic but depends on a protein tyrosine phosphatase(s) (28). To determine whether phosphatase(s) may be involved in down-regulating phosphorylation of STAT1 by UV light, EMSA was performed in the presence of the phosphatase inhibitor sodium orthovanadate. Cells were incubated with 1 mM sodium orthovanadate for 30 min, washed extensively, and stimulated with IFNγ or IFNγ plus UV light or left unstimulated. Fifteen minutes later, EMSA was performed using the synthetic GAS oligonucleotide. Vanadate counteracted UV light-mediated inhibition of STAT1 binding (Fig. 4B). To confirm these data, nuclear extracts were obtained 15 min after stimulation, and Western blot analysis was performed with Ab directed against STAT1 and phosphotyrosine, respectively. In accordance with the EMSA, phosphorylated STAT1 was significantly reduced after UV exposure, and this inhibition was reduced again by vanadate (Fig. 4D). Activation of a phosphatase by UV light should induce an increased rate of dephosphorylation, and thus UV light should be active even when applied after IFNγ. Thus, Pam212 cells were first treated with IFNγ and UV light exposed immediately, 30 or 45 min thereafter. EMSA revealed reduced binding activity of the samples treated with UV light after IFNγ (Fig. 5); however, the suppressive effect became weaker with increasing time lag between IFNγ and UV light treatment. Together, these data suggest that activation of a phosphatase by UV light may partially contribute to this inhibitory effect; however, direct inhibition of the phosphorylation of the STAT1 protein by UV light has to be considered as a further possibility because vanadate did not completely counteract the UV light effect and furthermore both pathways are not mutually exclusive.
Figure 5.

Effect of UV light when applied after IFNγ. Cells were stimulated with IFNγ (20 ng/ml) (lanes 2–5) or left untreated (lane 1) and exposed to UV light immediately (lane 3), 30 min (lane 4), or 45 min (lane 5) thereafter; 60 min after IFNγ stimulation, nuclear protein fractions were obtained, and EMSA was performed using the GAS oligonucleotide.
DISCUSSION
UV light, in particular the middle wave length range between 290 and 320 nm (UVB), has one of the most relevant environmental impacts because of its health-hazardous effects, including skin aging, induction of skin cancer, and exacerbation of infectious diseases (10, 29, 30). Like other adverse agents (alkylating chemicals, oxidants), UV light induces changes in mammalian gene expression (31–33). Induction of gene expression depends on the activation of specific transcription factors, including activation of NF-κB and AP-1 (18–21). Among a variety of others, genes encoding for cytokines are induced by UV light, ultimately resulting in the release of the majority of these mediators. Consequently, UV light has been recognized as a potent inducer of cytokine secretion (11). Because cytokines are important inflammatory and immunologic mediators, UV light, by exerting this effect, significantly influences immunologic and inflammatory reactions. Besides its inflammatory activities, UV light exhibits potent immunosuppressive properties. In contrast to the proinflammatory activities of UV light, the immunosuppressive properties are less well understood. Recently, we observed that UV light, in addition to its influence on the release of cytokines, also may interfere with their biological activities (12). UV light was found to counteract particular activities of IFNγ by inhibiting the expression of the transcription factor IRF-1. This may represent a new biological effect of UV light and may contribute to its immunosuppressive properties, so we were interested in clarifying the underlying molecular mechanism.
IFNγ induces IRF-1 expression by activating the GAS element located in the IRF-1 promoter, so first we addressed whether UV light inhibits activation of GAS. Usage of a minimal promoter construct containing the GAS oligonucleotide indicated that UV light prevents IFNγ-induced GAS activation. This was confirmed by EMSA, revealing that UV light almost completely inhibited binding of the GAS oligonucleotides to protein extracts from cells that were exposed to UV light immediately before stimulation with IFNγ. Because STAT1 needs to be tyrosine-phosphorylated to bind to the GAS element (17), we asked whether UV light interferes with the phosphorylation of STAT1. Immunoprecipitates from cells that were UV light-exposed before stimulation with IFNγ revealed STAT1 not to be tyrosine-phosphorylated, suggesting that UV light inhibits IFNγ-induced IRF-1 expression by preventing phosphorylation of STAT1. Because all STAT proteins bind to receptor phosphotyrosine via Src homology region domains [which results in the phosphorylation of STAT and ultimately enables DNA binding (8)], the question emerged whether UV light interferes with phosphorylation of STAT proteins in general. This, however, does not appear to be the case, because UV light did not affect phosphorylation of STAT3, which is involved in IL-6 signaling (22, 23). IL-6 is a proinflammatory cytokine whose release by keratinocytes is significantly induced by UV light and that contributes to mediation of UV light-induced local and systemic inflammation (34, 35). The inability of UV light to inhibit IL-6 signaling is not surprising because it would not make sense that UV light induces the release of IL-6 on the one hand, but would prevent this cytokine from mediating its effects by interfering with its signal transduction pathway on the other. Because IL-6 is a proinflammatory cytokine and IFNγ is an immunomodulatory one, the present findings of differential effects on cytokine signaling by UV light may help to explain the diverse biological effects of UV light, which causes inflammation (via inducing the release of inflammatory cytokines) on the one hand but inhibits immune reactions (presumably by interrupting the signal transduction of immunomodulatory cytokines) on the other. However, we have not yet addressed whether the findings obtained with IFNγ and IL-6 are representative for other inflammatory and immunomodulatory cytokines.
STAT1 is tyrosine-phosphorylated as an ultimate consequence of binding of IFNγ to its receptor, so one has to take into account that failing phosphorylation of STAT1 after UV light exposure is just a consequence of down-regulation of the IFNγ receptor by UV light. After dimerization of the IFNγ receptor after IFNγ binding, Jak1 and Jak2, which are associated with the cytoplasmic domains of the receptor, are brought into apposition and transphosphorylate each other. Therefore, if UV light influences the IFNγ receptor in a negative way, Jak1 and Jak2 should not be phosphorylated after UV light exposure. However, immunoprecipitations showed that UV light has no inhibitory effect on IFNγ-induced Jak1 and Jak2 phosphorylation, and, based on the autophosphorylation data, UV light even appeared to increase IFNγ-mediated activation of Jak1 and Jak2. This event is located upstream of the phosphorylation of STAT1, thus suggesting that inhibition of STAT1 phosphorylation is not a consequence of IFNγ receptor modulation by UV light.
The data indicate that UV light may interfere with IFNγ signaling by interfering with STAT1 phosphorylation, suggesting STAT1 protein as a new UV light target. However, we have no evidence yet whether phosphorylation of STAT1 is directly inhibited by UV light. Data are accumulating that show that the amount of phosphorylated STAT1 does not only depend on the rate of phosphorylation but also on the rate of dephosphorylation. Kim and Maniatis (26) recently proposed that the amounts of phosphorylated STAT1 in IFNγ-treated cells are controlled by ubiquitin-dependent proteolysis because STAT1 activated by IFNγ treatment was shown to be stabilized by the proteasome inhibitor MG132. Thus, we had to encounter the possibility that UV light activates the proteasome pathway, thereby down-regulating the amounts of phosphorylated STAT1. However, EMSA performed with nuclear protein extracts from the same cell preparations revealed that, in the presence of the proteasome inhibitor lactacystin, IFNγ-activated STAT1 binding was still reduced by UV light, suggesting that dephosphorylation by the proteasome pathway may not explain the inhibitory UV light effect.
Furthermore, it recently was reported that the amount of activated STAT1 in the nucleus is controlled by dephosphorylation upon a protein tyrosine phosphatase, which can be blocked by vanadate (28). Therefore, Pam212 cells were exposed to IFNγ or to IFNγ plus UV light in the presence of vanadate. Preincubation with vanadate antagonized UV light-mediated down-regulation of STAT1 binding, suggesting that activation of a vanadate-inhibitable phosphatase may contribute to the inhibitory effect of UV light. However, these data have to be interpreted cautiously; because vanadate stabilizes IFNγ-activated STAT1, it is difficult to differentiate in this experimental design whether the binding-active STAT1 detected in the UV light sample was caused by inhibition of the UV light effect or simply was caused by stabilization of residual STAT1. However, in the case of the latter possibility, the ratio of binding-active STAT1 between the unirradiated and the UV light-exposed groups should not be changed. Therefore, EMSA was performed with protein concentrations far below the saturation level, which indicated that the binding signal response was linear from the low signal detected in the absence of vanadate to the higher signal seen in its presence. Because, after addition of vanadate, the difference in the amounts of binding-active STAT1 between the IFNγ- and the UV light-treated samples was significantly reduced as determined by densitometry (Fig. 4D), this favors at least partial involvement of a phosphatase upon UV light exposure. This also was confirmed by anti-STAT1 Western blotting showing significantly increased amounts of phosphorylated STAT1 in the UV light samples after vanadate treatment. Moreover, involvement of a phosphatase also was supported by the observation that the inhibitory effect of UV light is still present when cells are UV light-exposed up to 45 min after IFNγ treatment, suggesting dephosphorylation as one mechanism. Together, these data indicate at least partial involvement of a phosphatase; however, this does not exclude the possibility that, in addition, UV light could directly inhibit phosphorylation of STAT1 because both mechanisms are not mutually exclusive and because vanadate did not completely antagonize the UV light effect.
UV light induces ligand-independent activation of various receptors, receptor tyrosine kinases, and protein tyrosine kinases at the inner side of the plasma membrane (36–40). Recently, UV light was found to cause epidermal and platelet-derived growth factor receptor autophosphorylation by inhibiting dephosphorylation of these receptors, presumably by targeting membrane-associated protein tyrosine phosphatases (41). Although these data suggest inhibition of phosphatase activity by UV light, our data imply activation of a phosphatase(s) by UV light. The reasons for this discrepancy remain to be determined; however, because the phosphatases involved in both systems have not been characterized, it is not clear whether the same phosphatase(s) are involved. Selective influences on the multiple phosphatases by UV light also are supported by the fact that autophosphorylation of Jak1 and Jak2 was not inhibited by UV light but even enhanced, suggesting that different phosphatases are involved and modulated by UV light in a selective way. Furthermore, the UV doses applied in the study by Knebel et al. (41) differ significantly from those applied in our study. In the study by Knebel et al., UVB and UVC doses up to 3000 J/m2 were applied, which are lethal, whereas in our study only UVB doses up to 250 J/m2 were used, which do not kill the cells at significant levels and therefore imply physiological relevance.
Acknowledgments
We thank J. Neumann for discussions, S. Grabbe and H. Riemann for reading the manuscript, B. Pöppelmann and K. Grosse-Heitmeyer for technical assistance, and J. Bückmann and P. Wissel for preparing the graphs. Supported by grants from the German Research Foundation (Schw 625/1-2) and the European Community (EV5V-CT94-0564).
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: IL, interleukin; Ab, antibody; EMSA, electrophoretic mobility-shift assay; IFNγ, interferon γ; IRF-1, IFN regulatory factor-1; CAT, chloramphenicol acetyltransferase.
References
- 1.Farrar M A, Schreiber R D. Annu Rev Immunol. 1993;11:571–611. doi: 10.1146/annurev.iy.11.040193.003035. [DOI] [PubMed] [Google Scholar]
- 2.Marsters S A, Pennica D, Bach E, Schreiber R D, Ashkenazi A. Proc Natl Acad Sci USA. 1995;92:5401–5495. doi: 10.1073/pnas.92.12.5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kotenko S V, Izotova L S, Pollack B P, Mariano T M, Donnelly R J, et al. J Biol Chem. 1995;270:20915–20921. doi: 10.1074/jbc.270.36.20915. [DOI] [PubMed] [Google Scholar]
- 4.Sakatsume M, Igarashi K, Winestock K D, Garotta G, Larner A C, Finbloom D S. J Biol Chem. 1995;270:17528–17534. doi: 10.1074/jbc.270.29.17528. [DOI] [PubMed] [Google Scholar]
- 5.Greenlund A C, Farrar M A, Viviano B L, Schreiber R D. EMBO J. 1994;13:1591–1600. doi: 10.1002/j.1460-2075.1994.tb06422.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greenlund A C, Morales M O, Viviano B L, Yan H, Krolewski J, Schreiber R D. Immunity. 1995;2:677–687. doi: 10.1016/1074-7613(95)90012-8. [DOI] [PubMed] [Google Scholar]
- 7.Darnell J E, Jr, Kerr I M, Stark G R. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 8.Leaman D W, Leung S, Li X, Stark G R. FASEB J. 1996;10:1578–1588. [PubMed] [Google Scholar]
- 9.Kripke M L. Photochem Photobiol. 1990;52:919–924. doi: 10.1111/j.1751-1097.1990.tb08703.x. [DOI] [PubMed] [Google Scholar]
- 10.Chapman R S, Cooper K D, DeFabo E C, Frederich J E, Gelatt K N, et al. Photochem Photobiol. 1995;61:223–247. doi: 10.1111/j.1751-1097.1995.tb03966.x. [DOI] [PubMed] [Google Scholar]
- 11.Schwarz T, Urbanski A, Luger T A. In: Epidermal Growth Factors and Cytokines. Luger T A, Schwarz T, editors; Luger T A, Schwarz T, editors. New York: Dekker; 1993. pp. 453–473. [Google Scholar]
- 12.Aragane Y, Schwarz A, Luger T A, Ariizumi K, Takashima A, Schwarz T. J Immunol. 1997;158:5393–5399. [PubMed] [Google Scholar]
- 13.Ariizumi K, Meng Y, Bergstresser P R, Takashima A. J Immunol. 1995;154:6031–6039. [PubMed] [Google Scholar]
- 14.Harada H, Fujita T, Miyamoto M, Kumura Y, Maruyama M, Furia A, Miyata T, Taniguchi T. Cell. 1989;58:729–739. doi: 10.1016/0092-8674(89)90107-4. [DOI] [PubMed] [Google Scholar]
- 15.Köck A, Schwarz T, Kirnbauer R, Urbanski A, Perry O, Ansel J C, Luger T A. J Exp Med. 1990;172:1609–1614. doi: 10.1084/jem.172.6.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pine R, Canova A, Schindler C. EMBO J. 1994;13:158–167. doi: 10.1002/j.1460-2075.1994.tb06245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shuai K, Schindler C, Prezioso V R, Darnell J E., Jr Science. 1992;258:1808–1812. doi: 10.1126/science.1281555. [DOI] [PubMed] [Google Scholar]
- 18.Devary Y, Rosette C, DiDonato J A, Karin M. Science. 1993;261:1442–1445. doi: 10.1126/science.8367725. [DOI] [PubMed] [Google Scholar]
- 19.Engelberg D, Klein C, Martinetto H, Struhl K, Karin M. Cell. 1994;77:381–390. doi: 10.1016/0092-8674(94)90153-8. [DOI] [PubMed] [Google Scholar]
- 20.Simon M M, Aragane Y, Schwarz A, Luger T A, Schwarz T. J Invest Dermatol. 1994;102:422–427. doi: 10.1111/1523-1747.ep12372194. [DOI] [PubMed] [Google Scholar]
- 21.Stein B, Angel P, Dam van H, Ponta H, Herrlich P, Eb van der A, Rahmsdorf H J. Photochem Photobiol. 1992;55:409–415. doi: 10.1111/j.1751-1097.1992.tb04255.x. [DOI] [PubMed] [Google Scholar]
- 22.Zhong Z, Wen J E, Darnell J E., Jr Science. 1994;264:95–98. doi: 10.1126/science.8140422. [DOI] [PubMed] [Google Scholar]
- 23.Akira S, Nishio Y, Inoue M, Wang X-J, Wei S, Matsusaka T, Yoshida K, Sudo T, Naruto M, Kishimoto T. Cell. 1994;77:63–71. doi: 10.1016/0092-8674(94)90235-6. [DOI] [PubMed] [Google Scholar]
- 24.Müller M, Briscoe J, Laxton C, Guschin D, Ziemiecki A, Silvennoinen O, Harpur A G, Barbieri G, Witthuhn B A, Schindler C, Pellegrini S, Wilks A F, Ihle J, Stark G R, Kerr I M. Nature (London) 1993;366:129–135. doi: 10.1038/366129a0. [DOI] [PubMed] [Google Scholar]
- 25.Silvennoinen G, Ihle J N, Schlessinger J, Levy D E. Nature (London) 1993;366:583–585. doi: 10.1038/366583a0. [DOI] [PubMed] [Google Scholar]
- 26.Kim T K, Maniatis T. Science. 1996;273:1717–1719. doi: 10.1126/science.273.5282.1717. [DOI] [PubMed] [Google Scholar]
- 27.Palombella V J, Rando O J, Goldberg A L, Maniatis T. Cell. 1994;78:773–785. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- 28.Haspel R L, Salditt-Georgieff M, Darnell J E., Jr EMBO J. 1996;15:6262–6268. [PMC free article] [PubMed] [Google Scholar]
- 29.Fisher G J, Datta S C, Talwar H S, Wang Z Q, Varani J, Kang S, Voorhees J J. Nature (London) 1996;379:335–339. doi: 10.1038/379335a0. [DOI] [PubMed] [Google Scholar]
- 30.Katsambas A, Nicolaidou E. Arch Dermatol. 1996;132:444–450. [PubMed] [Google Scholar]
- 31.Herrlich P, Ponta H, Rahmsdorf H J. Rev Physiol Biochem Pharmacol. 1992;119:187–223. doi: 10.1007/3540551921_7. [DOI] [PubMed] [Google Scholar]
- 32.Herrlich P, Rahmsdorf H J. Curr Opin Cell Biol. 1994;6:425–431. doi: 10.1016/0955-0674(94)90036-1. [DOI] [PubMed] [Google Scholar]
- 33.Schenk H, Klein M, Erdbrügger W, Dröge W, Schulze-Osthoff K. Proc Natl Acad Sci USA. 1994;91:1672–1676. doi: 10.1073/pnas.91.5.1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kirnbauer R, Köck A, Neuner P, Förster R, Krutmann J, Urbanski A, Ansel J C, Schwarz T, Luger T A. J Invest Dermatol. 1991;96:484–489. doi: 10.1111/1523-1747.ep12470181. [DOI] [PubMed] [Google Scholar]
- 35.Urbanski A, Schwarz T, Neuner P, Krutmann J, Kirnbauer R, Köck A, Luger T A. J Invest Dermatol. 1990;94:808–811. doi: 10.1111/1523-1747.ep12874666. [DOI] [PubMed] [Google Scholar]
- 36.Rosette C, Karin M. Science. 1996;274:1194–1197. doi: 10.1126/science.274.5290.1194. [DOI] [PubMed] [Google Scholar]
- 37.Devary Y, Gottlieb R A, Smeal T, Karin M. Cell. 1992;71:1081–1091. doi: 10.1016/s0092-8674(05)80058-3. [DOI] [PubMed] [Google Scholar]
- 38.Sachsenmaier C, Radler-Pohl A, Zinch R, Nordheim A, Herrlich P, Rahmsdorf H J. Cell. 1994;78:963–972. doi: 10.1016/0092-8674(94)90272-0. [DOI] [PubMed] [Google Scholar]
- 39.Schieven G L, Mittler R S, Nadler S G, Kirihara J M, Bolen J B, Kanner S B, Ledbetter J A. J Biol Chem. 1994;269:20718–20726. [PubMed] [Google Scholar]
- 40.Coffer P J, Burgering B M, Peppelenbosch M P, Bos J L, Kruijer W. Oncogene. 1995;11:561–569. [PubMed] [Google Scholar]
- 41.Knebel A, Rahmsdorf H J, Ullrich A, Herrlich P. EMBO J. 1996;15:5314–5325. [PMC free article] [PubMed] [Google Scholar]