

Abstract
The anesthetic propofol is thought to induce rapid hypnotic sedation by facilitating a GABAergic tonic current in forebrain neurons. The depression of cardiovascular and respiratory regulation often observed during propofol suggests potential additional actions within the brainstem. Here we determined the impacts of propofol on both GABAergic and glutamatergic synaptic mechanisms in a class of solitary tract nucleus (NTS) neurons common to brainstem reflex pathways. In horizontal brainstem slices, we recorded from NTS neurons directly activated by solitary tract (ST) axons. We identified these second-order NTS neurons by time-invariant (“jitter” <200 µs), “all-or-none” glutamatergic excitatory postsynaptic currents (EPSCs) in response to shocks to the ST. In order to assess propofol actions, we measured ST-evoked, spontaneous and miniature EPSCs and inhibitory postsynaptic currents (IPSCs) during propofol exposure. Propofol prolonged miniature IPSC decay time constants by 50% above control at 1.8 µM. Low concentrations of gabazine (SR-95531) blocked phasic GABA currents. At higher concentrations, propofol (30 µM) induced a gabazine-insensitive tonic current that was blocked by picrotoxin or bicuculline. In contrast, total propofol concentrations up to 30 µM had no effect on EPSCs. Thus, propofol enhanced phasic GABA events in NTS at lower concentrations than tonic current induction, opposite to the relative sensitivity observed in forebrain regions. These data suggest that therapeutic levels of propofol facilitate phasic (synaptic) inhibitory transmission in second order NTS neurons which likely inhibits autonomic reflex pathways during anesthesia.
Keywords: brainstem, propofol, glutamate, EPSC, IPSC, anesthetic
1. Introduction
Propofol (2,6-di-isopropylphenol) is widely favored as an intravenous general anesthetic (Rudolph & Antkowiak, 2004). Strong mechanistic links have been established, mostly in forebrain regions, between several of its clinical features and underlying cellular-molecular actions (Rudolph & Antkowiak, 2004; Franks, 2006). The hypnotic effects of propofol are primarily attributed to the enhancement of A-type γ-aminobutyric acid (GABAA) receptor function by inducing extended GABAA channel open times (Kitamura et al, 2004) and slowing desensitization (Bai et al, 1999; Bai et al, 2001), both attributes of phasic GABAergic transmission. Increasing interest has focused on propofol actions to induce an inhibitory tonic current that is pharmacologically and biophysically separable from phasic GABAergic currents (Hales & Lambert, 1991; Bai et al, 2001; Yeung et al, 2003). In hippocampal neurons, this tonic, “extrasynaptic” GABAA component is more sensitive to anesthetics than actions on phasic currents in the same neurons (Hemmings, Jr. et al, 2005) and seemingly plays a substantial role in suppressing neuronal excitability (Bieda & MacIver, 2004). In addition, ion channels and in particular voltage-gated sodium channels are reported to be inhibited by low micromolar concentrations of propofol (Frenkel & Urban, 1991; Ouyang et al, 2003; Jones et al, 2007). Given the diversity in composition and expression of native GABAA receptors as well as potential ion channel targets, accurate predictions of general anesthetic actions within any specific brain region is problematic.
Hypotension, bradycardia, desaturation and apnea are noted side effects of propofol administration and are consistent with depression of cardiorespiratory control mechanisms (Trapani et al, 2000; Nieuwenhuijs et al, 2000). Neurons responsible for these homeostatic regulatory mechanisms are concentrated in the brainstem (Loewy, 1990; Andresen & Kunze, 1994; Guyenet, 2006) and actions in the brainstem are consistent with the substantial autonomic impact of propofol in humans (Ebert & Muzi, 1994; Ebert, 2005). Common to most of these reflex pathways is the nucleus of the solitary tract (NTS), the site of central terminations of visceral cranial afferent nerves (including the vagus) from the cardiovascular, respiratory and gastrointestinal systems (Saper, 2002; Andresen et al, 2004; Travagli et al, 2006; Guyenet, 2006). Arterial baroreceptors and respiratory afferents terminate within medial portions of the caudal NTS and are a source of excitatory glutamatergic input onto second-order NTS neurons (Mendelowitz et al, 1992; Doyle & Andresen, 2001; Kubin et al, 2006). GABAergic transmission in NTS is also critical to normal cardiorespiratory reflex performance (Andresen & Kunze, 1994; Kubin et al, 2006) as well as pathophysiological conditions (Urbanski & Sapru, 1988; Callera et al, 2000; Mei et al, 2003; Tolstykh et al, 2004).
To assess the cellular mechanisms of propofol in the NTS, we recorded GABAergic and glutamatergic synaptic currents from synaptically identified, second-order neurons in horizontal brainstem slices. Propofol facilitated spontaneous and miniature inhibitory postsynaptic currents (IPSCs) and evoked a tonic, GABA-mediated current without altering glutamatergic, excitatory postsynaptic currents (EPSCs). Interestingly and unlike that observed in some forebrain neurons, inhibitory phasic currents were more sensitive to propofol than the evoked GABAA mediated tonic conductance. Taken together, these results suggest that the facilitation of inhibitory phasic neurotransmission dominates propofol actions within NTS.
2. Materials & Methods
All animal procedures were performed with the approval of the Institutional Animal Care and Use Committee at Oregon Health & Science University (Portland, Oregon) and conform to the guidelines laid out in the National Institutes of Health publication “Guide for the Care and Use of Laboratory Animals”.
2.1 Brain Stem Slice Preparation
Brain stem slices were prepared from adult (>160 g) Sprague Dawley rats (Charles River Laboratories, Inc., Wilmington, MA) as described in detail previously (Doyle & Andresen, 2001). Briefly, rats were deeply anesthetized with isoflurane and killed by cervical dislocation. The medulla was rapidly removed, cooled and trimmed rostrally and caudally to yield a brainstem block centered on obex. The ventral surface of brain tissue was cut to yield a single horizontal slice 250 µm thick which contained the ST together with the neural cell bodies in the medial NTS region. Slices were cut with a sapphire knife (Delaware Diamond Knives, Wilmington, DE) mounted in a vibrating microtome (VT1000S; Leica Microsystems Inc., Bannockburn, IL). The external solution was artificial cerebrospinal fluid (ACSF) containing the following (mM): 125 NaCl, 3 KCl, 1.2 KH2PO4, 1.2 MgSO4, 25 NaHCO3, 10 dextrose, and 2 CaCl2, bubbled with 95% O2-5% CO2. Slices were secured with a nylon mesh in a perfusion chamber and perfused with ACSF at 32–35°C, 300 mOsm, bubbled with 95% O2-5% CO2.
2.2 Whole Cell Recordings
The medial sub-nucleus of caudal NTS was targeted for recording using anatomical landmarks visualized within horizontal slices (Doyle et al, 2004). Recorded neurons were located medial to the ST and within 200 µm rostral or caudal from obex – an area containing the densest synaptic terminals from aortic baroreceptors and substantial pulmonary afferent terminals (Mendelowitz et al, 1992; Doyle & Andresen, 2001; Kubin et al, 2006). Electrodes (3.5–4.5 MΩ) were visually guided to neurons using infrared illumination and differential interference contrast optics (40X water immersion lens) on an Axioskop-2 FS plus fixed stage microscope (Zeiss, Thornwood, NJ) with camera (Hamamatsu Photonic Systems, Bridgewater, NJ).
The Cl− content of the recording electrode solutions and therefore the intracellular concentration were varied depending upon the experimental protocol. For spontaneous and miniature IPSC measurements (sIPSC and mIPSC, respectively), a high Cl− intracellular solution (ECl = −25mV) was used and contained (mM): 10 NaCl, 40 KCl, 70 K -gluconate, 11 EGTA, 1 CaCl2, 1 MgCl2, 10 N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), 2 Na2ATP, and 0.2 Na2GTP. For spontaneous and miniature EPSC measurements (sEPSC and mEPSC, respectively), a low Cl− intracellular solution (ECl = −69mV) was used and contained (mM): 6 NaCl, 4 NaOH, 130 K-gluconate, 11 EGTA, 1 CaCl2, 1 MgCl2, 10 HEPES, 2 Na2ATP, and 0.2 Na2GTP. For ST-evoked EPSCs (ST-EPSCs), a medium Cl− content intracellular solution (ECl = −60mV) was used and contained (mM): 10 NaCl, 130 K-gluconate, 11 EGTA, 1 CaCl2, 1 MgCl2, 10 HEPES, 2 Na2ATP, and 0.2 Na2GTP. All intracellular solutions were pH 7.3 and 296 mOsm. All recordings were made in open, whole cell patch configuration under voltage clamp using an Axopatch 200A or Multiclamp 700B amplifier (Axon Instruments, Foster City, CA). Signals were filtered at 10 kHz and sampled at 30 kHz using p-Clamp software (versions 8.2 or 9.2, Axon Instruments, MDS Analytical Technologies, Sunnyvale, CA).
2.3 Functional identification of second- order NTS neurons
2.3.1 Remote activation of ST primary afferents
A fine-tipped, concentric bipolar stimulating electrode (200 µm outer diameter; Frederick Haer Co., Bowdoinham, ME) was placed on a rostral site of the visible ST axons 2–3 mm from recorded neuron cell bodies. This remote placement minimized the likelihood that electrical shocks would activate non-ST axons or local neurons (Doyle & Andresen, 2001; Doyle et al, 2004). A Master-8 isolated programmable stimulator (A.M.P.I., Jerusalem, Israel) generated bursts of five ST shocks at 50 Hz every 3 s (shock duration 0.1 msec). From the analysis of responses to such ST burst protocols, we calculated synaptic latency, frequency-dependent amplitude depression and failure rates for ST-EPSCs. Latency was defined as the time between the ST shock and onset of the resulting EPSC (Doyle & Andresen, 2001). Synaptic jitter was calculated as the standard deviation of 30 – 40 ST-EPSC latencies within each neuron. Jitter reliably distinguishes between direct monosynaptic and indirect polysynaptic afferent contacts onto NTS neurons (Doyle & Andresen, 2001; Bailey et al, 2006a). In addition, multi-neuronal pathways (>200 µs jitter) (Bailey et al, 2006a) are particularly prone to synaptic failure. Suprathreshold ST shocks that failed to evoke an identifiable EPSC were counted as synaptic failures and failure rates were calculated as a percent of total ST shocks delivered at a constant but supra-threshold intensity over 30 – 40 trials (5 shocks @ 50 Hz per sweep). Monosynaptic ST responses typically exhibited failure rates of less then 0.1%. For the purposes of this study, neurons that exhibited a ST-EPSC jitter >200 µs and/or >0.1% failure rates were excluded from the study.
2.3.2 ST stimulus intensity-recruitment curves
Action potentials are triggered and conducted in an all-or-none fashion. Thus, a given shock at the ST may result in two outcomes: ineffectual (sub-threshold) or successful (supra-threshold synaptic response). Therefore synaptic event amplitudes were independent of shock intensity once threshold is exceeded. The all-or-none profile of shock intensity-recruitment curves indicates that such synaptic responses in a given NTS neuron rely on the activation of a single axon. (Andresen & Yang, 1995; Bailey et al, 2006b). For determining synaptic thresholds, shock intensities were finely graded and the amplitude of each evoked synaptic event was measured. The appearance of multiple ST- synced EPSCs or changes in the kinetic profile of synaptic responses over the range of shock intensities tested indicated the recruitment of additional afferent fibers with different, discrete shock thresholds and latency characteristics (Bailey et al, 2006a).
2.4 Drugs
All drugs were administered via the bath ACSF perfusate. In order to study GABAergic neurotransmission, ionotropic glutamate receptors were blocked with 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (NBQX; 20 µM) and D-2-amino-5-phosphonovalerate (AP-5; 100 µM). Glutamatergic EPSCs and tonic GABA currents were studied in the presence of gabazine (SR95531; 3 µM) to block phasic GABAA synaptic events. Picrotoxin (50 or 100 µM; Sigma RBI, St. Louis MO) and bicuculline (100 µM) blocked all GABAergic receptor activity. Miniature events (mEPSCs and mIPSCs) were recorded in the presence of tetrodotoxin (TTX; 3 µM). A concentrated stock solution of propofol (Aldrich, Milwaukee, WI) was created by combination with DMSO (1:3000 v/v) and this propofol stock was then diluted to final concentrations using ACSF. Propofol was perfused for 7 min (0.3, 1, 3, or 10 µM) or 8 min (30 µM) at a flow rate of 1.5 to 2 ml/min. Analysis of synaptic responses utilized the final 5 min period of each concentration exposure. All drugs were purchased from Tocris Bioscience (Ellisville, MO) unless otherwise noted.
2.5 Data and Statistics Analysis
All spontaneous and miniature synaptic events were detected and analyzed from digitized waveforms using MiniAnalysis (Synaptosoft, Decatur, GA). Except for determination of frequency rates, events <15 pA and those with multiple peaks were excluded from waveform analyses. The number of events that were part of multi-peak clusters was expressed as a percentage of all events detected. Baseline currents were measured over a 2 msec section of the recorded traces prior to every detected event. In cases in which phasic currents were blocked, 30 seconds of the digitized current traces was plotted as an all-points histogram (e.g. (Bieda & MacIver, 2004)) in 0.5 pA bins and all values were then averaged to represent the mean current for that time period. The all-points histograms express variation in current during the time period. Decay-time constants represent decay kinetics independent of amplitude and were calculated by least squares fitting of a single exponential between the 10% and 90% peak amplitude portion of the current relaxation. For statistical comparison of spontaneous and miniature synaptic events, waveform characteristic values (decay-time constant and amplitude), event frequency and baseline values across each group were averaged over the last 5 minutes of each cumulative concentration step. Results were compared with the Friedman repeated measures ANOVA on ranks (frequency, decay-time constant and amplitude) or a one-way, repeated measures ANOVA (baseline values, ST-EPSC1 amplitude and latency) each with Bonferroni post hoc testing (SigmaStat, San Jose, CA). All data are represented as mean ± SEM and p<0.05 was considered statistically significant.
3. Results
3.1 Second-order NTS neurons
We studied propofol actions on a total of 45 neurons meeting all ST-synaptic criteria for second-order neurons (Doyle et al, 2004; Bailey et al, 2006b). The ST-evoked EPSCs had aggregate means for latency and jitter for these neurons of 4.9±0.2 msec (range 1.7 to 8.3 msec) and 87±5 µsec (range 33 to 187 µsec), respectively. Such ST-EPSCs were blocked completely by non-NMDA (NBQX, 20 µM) and NMDA receptor (AP-5, 100 µM) antagonists leaving spontaneous IPSCs (Figure 1A).
Figure 1.

Isolation and event analysis of GABAergic inhibitory postsynaptic currents (IPSCs) for original traces derived from a single representative neuron. (A) ST shocks (five shocks, 50 Hz) evoked short, consistent latency ST-EPSCs (left panel, 10 sweeps overlaid, arrows indicate ST shocks) that depressed with repeated shocks and the jitter of the first EPSC identified the neuron as directly innervated by an ST afferent (latency = 3.4 ms, jitter = 91 µs). Blockade of the ST-EPSC with non-NMDA (20 µM NBQX) and NMDA (100 µM AP-5) antagonists (right panel). (B) Glutamatergic receptor blockade (NBQX and AP-5) reduced the number of spontaneous events and isolated GABA-mediated IPSCs. Traces display 2 sec epochs sampled from the complete dataset (see Figure 2 for this neuron). Introduction of propofol increased the decay time and incidence of large multi-peaked events from 1 to 30 µM. (C) Individual events were detected and then analyzed to determine amplitude, decay-time constant (tau of single exponential fit between 10% and 90% of total amplitude, circles and curved broken line) and baseline values (sampled before each detected event) depicted by the broken horizontal line. This analysis resulted in single neuron datasets and aggregate summaries as in the following figures.
3.2 Propofol enhances GABAergic spontaneous IPSCs
Following ST synaptic characterization and block of glutamatergic transmission (Figure 1A), spontaneous EPSCs (sEPSCs) were also eliminated and all second-order NTS neurons displayed spontaneous IPSCs (sIPSCs) (Figure 1A right panel and top two panels of 1B). Under conditions of elevated intracellular Cl− (ECl = −25 mV), GABAergic currents were large and inward when voltage-clamped near the normal resting potential (VH= −60 mV; Figure 2B).
Figure 2.

Analyses of spontaneous IPSCs (sIPSC) during cumulative perfusion of propofol. (A) Diary plots of sIPSC event characteristics display events of one representative second-order NTS neuron (original traces, Figure 1) during increasing propofol followed by a wash out period (left to right). NBQX (20 µM) and AP-5 (100 µM) isolated sIPSCs for study. The decay-time constant (top panel), amplitude (second panel) and baseline values (bottom panel) for each detected spontaneous IPSC are displayed (excluding multi-peaked events). All events were counted to establish frequency values (third panel). Note the ~3-fold increase in decay-time constant and the shift in baseline holding current to more inward values as propofol concentration increased. Wash resulted in a return toward control levels (top & bottom panels). Corresponding cumulative fractions of event measurements for the last 5 minutes of control (0 µM), 1, 3, 10 and 30 µM propofol perfusion periods as shown in individual values in 2A. Propofol increased sIPSC decay-time constant and baseline in a dose dependent manner (top and bottom panels, P > 0.05, KS test), in contrast to sIPSC amplitudes and event interval which remained at control levels throughout. (B) Summary aggregate data (n = 6 neurons) for spontaneous IPSC (sIPSC) events. Measurements were normalized to fractions of control levels for decay-time constant, amplitude and frequency within neurons before compiling averages. Changes in the baseline holding currents were calculated as changes from control in absolute pA. Dashed lines indicate control levels. Overall decay-time constant of sIPSCs increased significantly from control at 10 µM and 30 µM propofol (*P > 0.05, vs. control, RM ANOVA). Propofol significantly altered the basal holding current from control at 30 µM propofol (*P < 0.05, vs. control, RM ANOVA). Propofol did not alter either sIPSC amplitudes or the frequency of events.
Introduction of propofol rapidly broadened sIPSCs in a concentration related fashion beginning at 1 µM (Figure 1B). At high concentrations of propofol, the prolonged events began to overlap in time with successive sIPSCs and produced clusters of events with multiple peaks (10 – 30 µM propofol, Figure 1B). Detailed analysis of these records consisted of an examination of all phasic events plus measurements of the baseline preceding each event. Once an event was detected a sample of the preceding baseline generated a baseline current level that was also used as an index of the tonic current level (Figure 1C). The decay phase of each sIPSC was fitted with a single exponential to estimate a decay-time constant (tau; Figure 1C). Analysis of the waveform characteristics of the full record of events for the representative neuron (short epochs displayed in Figure 1) showed that the sIPSC decay-time constant values increased with increases in propofol concentration while low decay-time constant values became less common (Figures 2A). However, neither the amplitudes nor the frequency of sIPSC events were altered by increasing concentrations of propofol (Figure 2A). Note that propofol induced an inward shift in the baseline current in a concentration dependent manner despite constant VH (Figure 2A). Propofol actions reversed only gradually with washing in propofol free solution (Figure 2A).
On average (n= 6, Figure 2B), propofol consistently increased sIPSC time constant and the holding current at 10 µM and 30 µM, respectively. Such calculated means obscure the large ranges in responsiveness across individual neurons in which significant within experiment changes were identified at concentrations as low as 1 µM (Figure 2A). The magnitude of this heterogeneity in the propofol effects varied across neurons such that the range of change in relative decay-time constants for sIPSCs was 1.7 to 3.8 fold increases (n=6) at 10 µM propofol. The propofol evoked tonic current ranged from little or no net current up to −35 pA (n=6) at 30 µM. Propofol increased the incidence of multi-peaked events from 8±3 % of all events in control to 8±3 %, 11±4 %, 12±4 %, 16±6 % and 22±9 % at 0.3 µM, 1 µM, 3 µM, 10 µM and 30 µM propofol, respectively. Neither the spontaneous IPSC amplitudes nor frequencies were unaltered by propofol, both within individual experiments (distribution shifts) and in aggregate means (Figure 2).
3.3 Propofol enhances miniature GABAergic IPSCs
Spontaneous, action potential dependent processes can contribute to release from nerve terminals. To focus on nerve terminal release alone, we tested propofol actions on miniature GABAergic events (mIPSCs) in the presence of TTX as well as NBQX and AP-5. Miniature IPSCs occurred at a rate of 0.98±0.27 Hz, a mean amplitude of 41±7 pA and a mean decay time constant (τ) of 18±2 ms under control conditions (n = 10). Beginning between 1 and 3 µM, propofol significantly increased the decay-time constant of mIPSCs (Figure 3A & C; 3 µM p < 0.01, KS test), such that the decay phase of the average mIPSC waveforms (Figure 3C) extended with increasing concentrations yet peak amplitude remained constant. Note that propofol shifted the distribution of decay time constant values to longer values in a concentration dependent manner in this neuron and that, at the highest levels, the shortest tau events almost disappeared (Figure 3C, right panel). Evidence of a tonic inward current appeared only when propofol reached 10 µM in this neuron (Figure 3A, bottom panel; p < 0.01, KS test). Note that adding gabazine (3 µM) rapidly blocked all phasic currents (mIPSCs) but did not alter the propofol induced tonic current (Figures 3A & B).
Figure 3.

Analysis of propofol actions on miniature IPSCs (mIPSCs). (A) Diary plots of mIPSC event characteristics as recorded from one representative second-order NTS neuron exposed to increasing concentrations of propofol followed by gabazine (GBZ, 3 µM). Tetrodotoxin (TTX; 3 µM) together with NBQX (20 µM) and AP-5 (100 µM) isolated mIPSCs for study. The decay-time constant (top panel), amplitude (second panel) and baseline values (bottom panel) for each detected spontaneous IPSC are displayed (excluding multi-peaked events). All events were counted to establish frequency values (third panel). With increasing propofol exposure, mIPSC duration increased as reflected in the increased decay-time constant and basal holding current shifted to more negative values (inward current). (B) Original experimental trace corresponding to the time of onset of GBZ showing the blockade of phasic mIPSCs but no reversal of baseline holding current to control levels. Broken horizontal lines display the ΔIH from control to 30 µM propofol (−20 pA) in this neuron. (C) An average waveform was generated from mIPSCs for the last 5 minutes for control (0 µM), 1, 3, and 10 µM propofol exposure periods and then fitted with a single exponential at each concentration (left panel). Fits measured the elongation of the decay phase of the mIPSC events with increasing propofol (left panel, 0.3 and 30 µM not shown). Similarly, distributions of decay-time constant values for all individual mIPSCs shifted right with increasing propofol (right panel, 0.3 and 30 µM not shown). (D) Summary aggregate data (n = 10 neurons) for mIPSC events. Measurements were normalized to fractions of the control level within each neuron for decay-time constant, amplitude and frequency before compiling averages. Changes in the baseline holding currents were calculated as changes from control in absolute pA. Dashed lines represent control levels. Aggregate decay-time constant of mIPSCs significantly increased at 3, 10 and 30 µM propofol (*p > 0.05, vs. control, RM ANOVA). Propofol significantly shifted the basal holding current inward compared to control, on average, only at 30 µM propofol (*p > 0.05, vs. control, RM ANOVA). Propofol did not alter either mIPSC amplitudes or their frequency.
On average (n = 10), 3 µM propofol increased the decay-time constant for mIPSCs significantly above control (p=0.006, Figure 3D). The decay-time constant for mIPSCs averaged 18±2 ms at control and increased to 22±2 ms, 34±5 ms and 60±7 ms at 1 µM, 3 µM, and 10 µM propofol respectively. Note that even at 30 µM, as is commonly observed, the propofol induced increase in decay-time constant showed no indication of saturating and therefore EC50 values could not be determined. Expressed as functional endpoints, the mIPSC decay-time constant increased 50% increases over control on average at 1.8 µM and doubled at 3.6 µM propofol (Figure 3D). Similar to sIPSCs, propofol increased the prevalence of multi-peak mIPSCs from control levels of 8±2% of all events to 7±2 %, 10±2 %, 11±2 %, 16±4 % and 21±4 % in 0.3 µM, 1 µM, 3 µM, 10 µM and 30 µM propofol, respectively. Given the varied magnitude of tonic currents across neurons, only 30 µM propofol significantly induced a significantly larger average tonic current compared to control (Figure 3D, range: +21 to −71 pA). Propofol did not alter mIPSC event amplitudes or frequencies (Figure 3D). Such results are consistent with selective propofol action at postsynaptic GABAA receptors to enhance phasic events at therapeutic (low micromolar) concentrations, whereas tonic currents were induced at substantially higher concentrations.
3.4 Propofol induces an inward, tonic Cl− current in the absence of phasic events
The presence of long lasting and multi-peaked phasic currents during high levels of propofol complicated the intermittent measurements shifts in the holding current, i.e. the tonic GABA current. To better discern the propofol evoked tonic current, we blocked all phasic events (NBQX, AP-5 and gabazine) and examined the propofol evoked current in isolation (Figure 4). Under these conditions, 30 µM propofol induced an inward current (range: −6 to −48 pA, n=5, Figure 4A, B, C) that was eliminated by either 100 µM picrotoxin (n=3) or bicuculline (100 µM, not shown, n=2). Propofol also increased the variation or ‘noise’ of the tonic current as evidenced by a broader distribution of baseline values as the mean current shifted (Figure 4B, all points graph). Subtractions of the ionic currents evoked by command ramps of voltage (VH from −90 to −40 mV over 750 ms) during control (NBQX and gabazine) and during 10 µM propofol revealed a net (subtracted) propofol-induced, tonic current that reversed at −61.6±1.0 mV (n=3, Figure 4D). The reversal potential for this propofol induced current was close to the calculated ECl (−60 mV) for these conditions. Picrotoxin (50 µM) subsequently blocked these propofol induced Cl− currents (data not shown). Therefore, the tonic current induced by propofol is pharmacologically distinct from phasic GABAA currents and relies on a Cl− selective conductance.
Figure 4.

Propofol actions on isolated tonic holding current. All phasic synaptic events were blocked by NBQX (20 µM), AP-5 (100 µM) and gabazine (3 µM). (A) Full record of holding current during application of 30 µM propofol to evoke an inward tonic current. Note that propofol substantially increased the baseline excursions (i.e. ‘noise’). Both the increased noise and tonic current were eliminated by picrotoxin (100 µM). (B) Thirty second, raw record epochs were extracted from the full recorded and all points distributions of holding current values (right) show that propofol shifted the mean current (dashed lines) to more negative values and broadened the distribution – in this case beyond control levels. (C) The average change in tonic current for group data (n = 5 neurons) for propofol was reversed by addition of GABAA antagonists, either 100 µM picrotoxin or biccuculine (*p > 0.05, vs. control, RM ANOVA; #p > 0.05, vs. blocker, RM ANOVA). (D) The voltage dependence of ΔIH (difference current subtracting the control from 10 µM propofol) was measured using command voltage ramps (66 mV/s). The net (subtracted) propofol-induced tonic current reversed at −62 mV in this neuron, close to the calculated ECl (−60 mV) for these conditions and was subsequently blocked by picrotoxin (50 µM, not shown).
3.5 ST afferent and glutamatergic synaptic transmission is propofol resistant
The consistent timing and waveforms of ST-evoked EPSCs makes them sensitive assays of afferent glutamatergic transmission (Bailey et al, 2006b). Even high concentrations (30 µM), of propofol had no effect on ST-EPSCs (Figure 5A). Thus, on average, the mean amplitude of the first evoked ST-EPSC (ST-EPSC1) remained unchanged from control up to the highest concentration tested propofol (30 µM, Figure 5B). Although in cortical neurons sodium channels, spiking and nerve conduction are inhibited by low micromolar propofol concentrations (Rehberg & Duch, 1999; Bieda & MacIver, 2004; Martella et al, 2005; Jones et al, 2007), ST-EPSC latency remained constant during propofol (p=0.958, n=5, Figure 5C), a finding that suggests that sensory axon conduction and glutamate release are unimpeded by the anesthetic. Likewise, following pharmacological isolation of sEPSCs with gabazine, propofol did not alter decay-time constants, amplitudes, frequencies or the holding current at any concentration tested (n=6, Figure 5D). Miniature EPSCs isolated by TTX (and gabazine) occurred at a rate of 2.3±0.5 Hz, had a mean amplitude of 38±5 pA and a mean decay time constant (τ) of 3.4±0.4 ms under control conditions (n = 10) and were unaffected by propofol (Figure 5E). A tonic current was not detected during propofol in this subset of experiments but this was likely due to recording conditions that held the neurons close to the chloride reversal potential.
Figure 5.
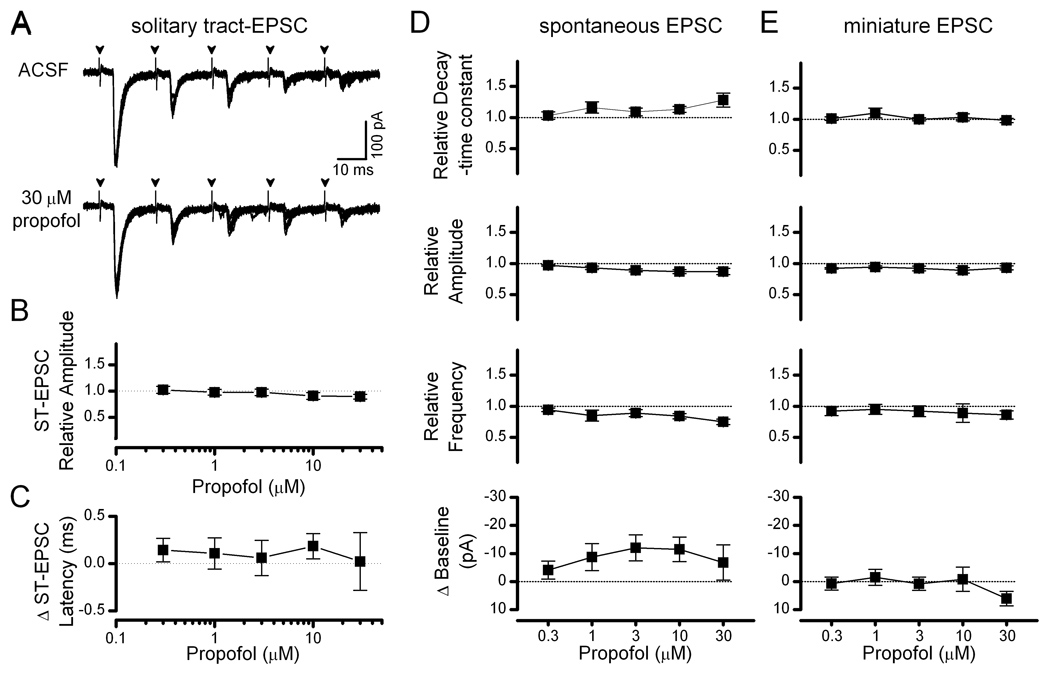
Evoked solitary tract glutamatergic transmission in second-order NTS neurons was propofol resistant. (A) Solitary tract-evoked excitatory postsynaptic currents (ST-EPSC) were not altered by propofol (traces of 8 replicate trials overlapped from a single representative neuron). (B) On average for group data (n = 5 neurons), the amplitude of ST-EPSC1 (the first evoked EPSC in the series of 5) was not altered by propofol. (C) Mean latency of ST-EPSC1 was not altered by propofol in second order NTS neurons (n = 5). (D & E) Summary aggregate data (n = 6–10) for spontaneous and miniature EPSCs showed no changes with propofol. Measurements were normalized to fractions of control levels for decay-time constant, amplitude and frequency. Changes in the baseline holding currents were calculated as changes from control in absolute pA. Dashed lines represent control levels.
4. Discussion
The brainstem neurons studied in the present series of experiments are positioned at the first CNS transmission point for a broad array of homeostatic reflexes. Our studies demonstrated that propofol selectively enhanced inhibitory mechanisms by facilitating phasic GABAA currents onto second order NTS neurons with no discernable effect on glutamatergic transmission. Thus, propofol at low micromolar concentrations appears to act only on postsynaptic GABAA receptors to facilitate phasic inhibitory currents. Approximately 10-fold higher concentrations were required for propofol to induce a tonic GABAergic current. Our NTS data suggest that the sensitivity of phasic GABAA receptors to propofol in this adult tissue corresponds to estimated therapeutic concentrations (Rudolph & Antkowiak, 2004; Urban et al, 2006; Franks, 2006) and these mechanisms may well contribute to the autonomic dysregulation observed in patients that includes baroreflex depression and sympathoinhibition (Ebert et al, 1992; Ebert, 2005) as in animal models (Seagard et al, 1983; Lee et al, 2002).
A wide range of membrane proteins have been suggested as targets for general anesthetic action and most studies have focused on forebrain and spinal cord neurons closely related to hypnotic, amnesiac, immobilizing and analgesic outcomes (Rudolph & Antkowiak, 2004; Franks, 2006). Although GABAA receptors are the most prominent target, ion channels and other neurotransmitter receptors including glutamate have been identified as potential sites of anesthetic action in various neurons. Certainly the strongest evidence links general anesthetic actions to GABAA ionotropic receptors and this is true for propofol in particular (Rudolph & Antkowiak, 2004; Franks, 2006). Propofol acts at GABAA receptors where it increases both decay times and the open channel probability (Kitamura et al, 2004) and slows desensitization (Bai et al, 1999; Bai et al, 2001). Diverse expression and the multitude of naturally occurring sub-unit combinations of GABAA receptors make it difficult to predict anesthetic sensitivity and mechanisms (Yeung et al, 2003). This receptor diversity differs prominently across brain regions and is thought to be responsible for pharmacological differences between phasic and tonic GABA receptor mediated currents within and across neurons (Yeung et al, 2003). In this context, our goal in the present studies was to try to identify the potential targets for anesthetic actions in adult NTS neurons with fully mature development.
4.1 Use of second-order NTS neurons
The second-order neuron phenotype adds the specific context of known pathway structure and their relationship to peripheral afferents including the vagus nerve (Andresen & Kunze, 1994). The use of adult rats avoided the developmentally transient expression of potential target proteins including GABA mechanisms observed in the brainstem (Awatramani et al, 2004) as well as cortical neurons (Brickley et al, 1996). The high level of experimental precision offered by ST-evoked EPSC measures in second order NTS neurons makes the negative result of propofol effects on ST responses particularly remarkable since multiple presynaptic properties are assessed that include conduction sites along the afferent axon as well as the terminal release site itself (Bailey et al, 2006b).
4.2 Propofol enhances phasic GABAergic currents in NTS
In NTS neurons, propofol enhanced both phasic and tonic GABA-mediated currents albeit with different sensitivities. Propofol increased the duration (decay-time constant) of IPSCs in a concentration dependent manner beginning at 1 µM. Similar response patterns for propofol actions (from 1 µM) on GABAergic miniature and/or spontaneous IPSCs occur in brainstem cardiac projection neurons (Wang et al, 2004). Propofol likewise affected GABAA IPSCs in supramedullary neurons from embryonic to early postnatal brain slices from the hippocampus (from 0.2 µM) (Bai et al, 2001), paraventricular nucleus (from 10 µM, decay time (Shirasaka et al, 2004)) and cortex (primary culture, from 1 µM (Kitamura et al, 2003)). Note that experimental design factors such as Cl− gradient and driving force influence discrimination of such threshold values particularly regarding GABAA mediated currents.
4.3 Propofol induces a tonic inhibitory current
Propofol-induced tonic currents shifted the holding current in NTS neurons and developed even in the presence of gabazine, which blocked all phasic IPSCs. Total propofol concentrations of 10 – 30 µM were required in NTS neurons to induce a persistent, inhibitory tonic current. Tonic GABAA currents increase during development in cerebellar neurons (Brickley et al, 1996) and anesthetic sensitivity of tonic currents can be much greater than for phasic events, for example Belelli et al. (2005). The relative insensitivity of tonic currents in NTS neurons for propofol contrasts with the high sensitivity for tonic GABAA currents in hippocampal and neocortical neurons to propofol and isoflurane (Bai et al, 2001; Caraiscos et al, 2004a; Caraiscos et al, 2004b; Drasbek et al, 2007). This contrasting pattern of NTS compared to forebrain regions may represent differences in GABAA receptor subunit composition (Feng & Macdonald, 2004; Caraiscos et al, 2004a) and/or extrasynaptic cellular loci (Yeung et al, 2003). Indeed, a unique possibility might include actions of propofol on GABAA receptors containing ρ1 subunits which are expressed in NTS (Milligan et al, 2004). In intact animals, ongoing activity of GABA pathways was suggested by observations that GABA antagonists increased spontaneous firing (Bennett et al, 1987). The molecular basis of tonic GABAA currents is controversial. Propofol can induce inhibitory currents in the absence of GABA in cultured hypothalamic neurons at concentrations above 10 µM (Adodra & Hales, 1995). The potential for induction of sub-conductance states of GABAA receptors (Smith & McBurney, 1989) offers multiple possible mechanisms by which propofol may be inducing tonic currents in NTS neurons.
The absolute sensitivity of phasic compared to tonic GABAA targets to propofol in NTS suggests that propofol may dampen NTS neuron excitability dynamically. Targeting of phasic events means that the functional consequences of propofol’s actions will occur in synchrony with GABA synaptic volleys and this pattern is similar to effects on cardiac vagal preganglionic neurons within the nucleus ambiguus (Wang et al, 2004; Bouairi et al, 2006). This phasic enhancement contrasts with the more continuous silencing of the neurons that might result from an overt hyperpolarization (Bai et al, 2001; Orser et al, 2002). Therefore, timing of enhanced GABAergic synaptic events in NTS neurons may be particularly important in autonomic reflex performance during propofol administration. Clearly, propofol has the potential to induce an additional inhibitory tonic current and silence NTS neurons. Such actions should fully block such reflexes, but our in vitro results suggest that these effects may manifest only at concentrations corresponding to clinical doses possibly achieved via bolus injections.
4.4 Propofol does not affect glutamatergic transmission in second-order NTS neurons
Cranial visceral afferent excitation of second-order NTS neurons relies on non-NMDA receptor mediated glutamatergic transmission (Jin et al, 2004a; Jin et al, 2004b). Propofol did not alter the shape of EPSC events whether evoked or spontaneous, a result consistent with cortical neurons (Kitamura et al, 2003). The latency of ST triggered events is a sensitive, integrated index of axon conduction and terminal excitation that includes voltage dependent channel function such as sodium channels (Jin et al, 2004b; Bailey et al, 2006b). Latency of ST-EPSCs was unaffected by 30 µM propofol. In contrast, in cortical neurons, sodium channels, spiking and nerve conduction are inhibited by low micromolar propofol concentrations (Rehberg & Duch, 1999; Bieda & MacIver, 2004; Martella et al, 2005; Jones et al, 2007). We conclude that the afferent axon and terminal excitation process is remarkably insensitive to propofol and this may reflect the expression of a particular complement of ion channels within peripheral afferent neuron axons (Schild et al, 1994; Schild & Kunze, 1995).
4.5 Propofol actions in NTS in vitro and autonomic control
In healthy subjects, sedative doses of propofol substantially inhibit sympathetic nerve activity and reflex responses to hypotension and bring significant decreases in mean blood pressure (Reves et al, 2000; Ebert, 2005). This decrease in mean arterial pressure is not accompanied by an appropriate reflex tachycardia and, collectively, these observations are consistent with propofol acting centrally to depress baroreflexes. The neurons responsible for propofol depression of cardiorespiratory control are unknown, but most likely homeostatic depression involves autonomic pathways within the brainstem that are compromised by general anesthetics (Lee et al, 2002). Indeed modulation of GABA neurotransmission within the NTS may alter multiple homeostatic processes within the gastrointestinal (Travagli et al, 2006), respiratory (Wasserman et al, 2002) and cardiovascular systems. Propofol likely enhances solitary tract driven GABAergic inputs as well as other major sources of inhibitory input to NTS from the forebrain (Saha, 2005). Thus propofol enhancement of IPSCs may greatly disrupt the widespread EPSC-IPSC temporal sequence triggered by afferent activation (Andresen & Yang, 1995; Mifflin & Felder, 1988; Smith et al, 1998).
In conclusion, propofol enhances both phasic and tonic GABAergic currents in second-order NTS neurons. The concentration dependence suggests that GABAA receptors mediating phasic IPSCs are more sensitive than the receptors responsible for the inhibitory tonic current. The differential pharmacological profiles for these two effects of propofol are consistent with at least two functionally distinct groups of GABAA receptors in NTS.
Acknowledgments
Supported by grants from the National Institutes of Health (HL-058760) and the National Health and Medical Research Council of Australia for which SJM is a C.J. Martin Fellow.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Stuart J. McDougall, Department of Physiology & Pharmacology, Oregon Health & Science University, Portland OR.
Timothy W. Bailey, Department of Physiology & Pharmacology, Oregon Health & Science University, Portland OR.
David Mendelowitz, Department of Pharmacology & Physiology, George Washington University, Washington DC..
Michael C. Andresen, Department of Physiology & Pharmacology, Oregon Health & Science University, Portland OR.
References
- Adodra S, Hales TG. Potentiation, activation and blockade of GABAA receptors of clonal murine hypothalamic GT1-7 neurones by propofol. British Journal of Pharmacology. 1995;115:953–960. doi: 10.1111/j.1476-5381.1995.tb15903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andresen MC, Doyle MW, Bailey TW, Jin Y-H. Differentiation of autonomic reflex control begins with cellular mechanisms at the first synapse within the nucleus tractus solitarius. Brazilian Journal of Medical and Biological Research. 2004;37:549–558. doi: 10.1590/s0100-879x2004000400012. [DOI] [PubMed] [Google Scholar]
- Andresen MC, Kunze DL. Nucleus tractus solitarius: gateway to neural circulatory control. Annual Review of Physiology. 1994;56:93–116. doi: 10.1146/annurev.ph.56.030194.000521. [DOI] [PubMed] [Google Scholar]
- Andresen MC, Yang M. Dynamics of sensory afferent synaptic transmission in aortic baroreceptor regions of nucleus tractus solitarius. Journal of Neurophysiology. 1995;74:1518–1528. doi: 10.1152/jn.1995.74.4.1518. [DOI] [PubMed] [Google Scholar]
- Awatramani GB, Turecek R, Trussell LO. Staggered development of GABAergic and glycinergic transmission in the MNTB. Journal of Neurophysiology. 2005;93:819–828. doi: 10.1152/jn.00798.2004. [DOI] [PubMed] [Google Scholar]
- Bai D, Pennefather PS, MacDonald JF, Orser BA. The general anesthetic propofol slows deactivation and desensitization of GABA(A) receptors. Journal of Neuroscience. 1999;19:10635–10646. doi: 10.1523/JNEUROSCI.19-24-10635.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai D, Zhu G, Pennefather P, Jackson MF, MacDonald JF, Orser BA. Distinct functional and pharmacological properties of tonic and quantal inhibitory postsynaptic currents mediated by gamma-aminobutyric acid(A) receptors in hippocampal neurons. Molecular Pharmacology. 2001;59:814–824. doi: 10.1124/mol.59.4.814. [DOI] [PubMed] [Google Scholar]
- Bailey TW, Hermes SM, Andresen MC, Aicher SA. Cranial visceral afferent pathways through the nucleus of the solitary tract to caudal ventrolateral medulla or paraventricular hypothalamus: Target-specific synaptic reliability and convergence patterns. Journal of Neuroscience. 2006a;26:11893–11902. doi: 10.1523/JNEUROSCI.2044-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TW, Jin Y-H, Doyle MW, Smith SM, Andresen MC. Vasopressin inhibits glutamate release via two distinct modes in the brainstem. Journal of Neuroscience. 2006b;26:6131–6142. doi: 10.1523/JNEUROSCI.5176-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belelli D, Peden DR, Rosahl TW, Wafford KA, Lambert JJ. Extrasynaptic GABAA receptors of thalamocortical neurons: a molecular target for hypnotics. J Neuroscience. 2005;25:11513–11520. doi: 10.1523/JNEUROSCI.2679-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett JA, McWilliam PN, Shepherd SL. A gamma-aminobutyric-acid-mediated inhibition of neurones in the in nucleus tractus solitarius of the cat. Journal of Physiology. 1987;392:417. doi: 10.1113/jphysiol.1987.sp016788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieda MC, MacIver MB. Major role for tonic GABAA conductances in anesthetic suppression of intrinsic neuronal excitability. Journal of Neurophysiology. 2004;92:1658–1667. doi: 10.1152/jn.00223.2004. [DOI] [PubMed] [Google Scholar]
- Bouairi E, Kamendi H, Gorini C, Mendelowitz D. Multiple types of GABAA receptors mediate inhibition in brainstem parasympathetic cardiac neurons in the nucleus ambiguus. Journal of Neurophysiology. 2006;96:3266–3272. doi: 10.1152/jn.00590.2006. [DOI] [PubMed] [Google Scholar]
- Brickley SG, Cull-Candy SG, Farrant M. Development of a tonic form of synaptic inhibition in rat cerebellar granule cells resulting from persistent activation of GABAA receptors. Journal of Physiology. 1996;497:753–759. doi: 10.1113/jphysiol.1996.sp021806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callera JC, Bonagamba LG, Nosjean A, Laguzzi R, Machado BH. Activation of GABA receptors in the NTS of awake rats reduces the gain of baroreflex bradycardia. Autonomic Neuroscience, Basic and Clinical. 2000;84:58–67. doi: 10.1016/S1566-0702(00)00184-3. [DOI] [PubMed] [Google Scholar]
- Caraiscos VB, Elliott EM, You T, Cheng VY, Belelli D, Newell JG, Jackson MF, Lambert JJ, Rosahl TW, Wafford KA, MacDonald JF, Orser BA. Tonic inhibition in mouse hippocampal CA1 pyramidal neurons is mediated by alpha5 subunit-containing gamma-aminobutyric acid type A receptors. Proceedings of the National Academy of Sciences of the United States of America. 2004a;101:3662–3667. doi: 10.1073/pnas.0307231101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caraiscos VB, Newell JG, You T, Elliott EM, Rosahl TW, Wafford KA, MacDonald JF, Orser BA. Selective enhancement of tonic GABAergic inhibition in murine hippocampal neurons by low concentrations of the volatile anesthetic isoflurane. Journal of Neuroscience. 2004b;24:8454–8458. doi: 10.1523/JNEUROSCI.2063-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle MW, Andresen MC. Reliability of monosynaptic transmission in brain stem neurons in vitro. Journal of Neurophysiology. 2001;85:2213–2223. doi: 10.1152/jn.2001.85.5.2213. [DOI] [PubMed] [Google Scholar]
- Doyle MW, Bailey TW, Jin Y-H, Appleyard SM, Low MJ, Andresen MC. Strategies for cellular identification in nucleus tractus solitarius slices. Journal of Neuroscience Methods. 2004;37:37–48. doi: 10.1016/j.jneumeth.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Drasbek KR, Hoestgaard-Jensen K, Jensen K. Modulation of extrasynaptic THIP conductances by GABAA-receptor modulators in mouse neocortex. Journal of Neurophysiology. 2007;97:2293–2300. doi: 10.1152/jn.00651.2006. [DOI] [PubMed] [Google Scholar]
- Ebert TJ. Sympathetic and hemodynamic effects of moderate and deep sedation with propofol in humans. Anesthesiology. 2005;103:20–24. doi: 10.1097/00000542-200507000-00007. [DOI] [PubMed] [Google Scholar]
- Ebert TJ, Muzi M. Propofol and autonomic reflex function in humans. Anesthesia and Analgesia. 1994;78:369–375. doi: 10.1213/00000539-199402000-00029. [DOI] [PubMed] [Google Scholar]
- Ebert TJ, Muzi M, Berens R, Goff D, Kampine JP. Sympathetic responses to induction of anesthesia in humans with propofol or etomidate. Anesthesiology. 1992;76:725–733. doi: 10.1097/00000542-199205000-00010. [DOI] [PubMed] [Google Scholar]
- Feng HJ, Macdonald RL. Multiple actions of propofol on alphabetagamma and alphabetadelta GABAA receptors. Molecular Pharmacology. 2004;66:1517–1524. doi: 10.1124/mol.104.003426. [DOI] [PubMed] [Google Scholar]
- Franks NP. Molecular targets underlying general anaesthesia. British Journal of Pharmacology. 2006;147:S72–S81. doi: 10.1038/sj.bjp.0706441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkel C, Urban BW. Human brain sodium channels as one of the molecular target sites for the new intravenous anaesthetic propofol (2,6-diisopropylphenol) European Journal of Pharmacology. 1991;208:75–79. doi: 10.1016/0922-4106(91)90054-l. [DOI] [PubMed] [Google Scholar]
- Guyenet PG. The sympathetic control of blood pressure. Nature Reviews Neuroscience. 2006;7:335–346. doi: 10.1038/nrn1902. [DOI] [PubMed] [Google Scholar]
- Hales TG, Lambert JJ. The actions of propofol on inhibitory amino acid receptors of bovine adrenomedullary chromaffin cells and rodent central neurones. British Journal of Pharmacology. 1991;104:619–628. doi: 10.1111/j.1476-5381.1991.tb12479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmings HC, Jr, Akabas MH, Goldstein PA, Trudell JR, Orser BA, Harrison NL. Emerging molecular mechanisms of general anesthetic action. Trends in Pharmacological Sciences. 2005;26:503–510. doi: 10.1016/j.tips.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Jin Y-H, Bailey TW, Andresen MC. Cranial afferent glutamate heterosynaptically modulates GABA release onto second order neurons via distinctly segregated mGluRs. Journal of Neuroscience. 2004a;24:9332–9340. doi: 10.1523/JNEUROSCI.1991-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y-H, Bailey TW, Li BY, Schild JH, Andresen MC. Purinergic and vanilloid receptor activation releases glutamate from separate cranial afferent terminals. Journal of Neuroscience. 2004b;24:4709–4717. doi: 10.1523/JNEUROSCI.0753-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PJ, Wang Y, Smith MD, Hargus NJ, Eidam HS, White HS, Kapur J, Brown ML, Patel MK. Hydroxyamide analogs of propofol exhibit state-dependent block of sodium channels in hippocampal neurons: implications for anticonvulsant activity. Journal of Pharmacological and Experimental Therapeutics. 2007;320:828–836. doi: 10.1124/jpet.106.111542. [DOI] [PubMed] [Google Scholar]
- Kitamura A, Marszalec W, Yeh JZ, Narahashi T. Effects of halothane and propofol on excitatory and inhibitory synaptic transmission in rat cortical neurons. Journal of Pharmacological and Experimental Therapeutics. 2003;304:162–171. doi: 10.1124/jpet.102.043273. [DOI] [PubMed] [Google Scholar]
- Kitamura A, Sato Sato, Marszalec W, Yeh JZ, Ogawa R, Narahashi T. Halothane and propofol modulation of gamma-aminobutyric acidA receptor single-channel currents. Anesthesia and Analgesia. 2004;99:409–415. doi: 10.1213/01.ANE.0000131969.46439.71. [DOI] [PubMed] [Google Scholar]
- Kubin L, Alheid GF, Zuperku EJ, McCrimmon DR. Central pathways of pulmonary and lower airway vagal afferents. Journal of Applied Physiology. 2006;101:618–627. doi: 10.1152/japplphysiol.00252.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Andresen MC, Morrow D, Chang KSK. Isoflurane depresses baroreflex control of heart rate in decerebrate rats. Anesthesiology. 2002;96:1214–1222. doi: 10.1097/00000542-200205000-00026. [DOI] [PubMed] [Google Scholar]
- Loewy AD. Central autonomic pathways. In: Loewy AD, Spyer KM, editors. Central regulation of autonomic functions. New York: Oxford; 1990. pp. 88–103. [Google Scholar]
- Martella G, De Persis C, Bonsi P, Natoli S, Cuomo D, Bernardi G, Calabresi P, Pisani A. Inhibition of persistent sodium current fraction and voltage-gated L-type calcium current by propofol in cortical neurons: implications for its antiepileptic activity. Epilepsia. 2005;46:624–635. doi: 10.1111/j.1528-1167.2005.34904.x. [DOI] [PubMed] [Google Scholar]
- Mei L, Zhang J, Mifflin SW. Hypertension alters GABA receptor-mediated inhibition of neurons in the nucleus of the solitary tract. American Journal of Physiology, Regulatory Integrative and Comparative Physiology. 2003;285:R1276–R1286. doi: 10.1152/ajpregu.00255.2003. [DOI] [PubMed] [Google Scholar]
- Mendelowitz D, Yang M, Andresen MC, Kunze DL. Localization and retention in vitro of fluorescently labeled aortic baroreceptor terminals on neurons from the nucleus tractus solitarius. Brain Research. 1992;581:339–343. doi: 10.1016/0006-8993(92)90729-s. [DOI] [PubMed] [Google Scholar]
- Mifflin SW, Felder RB. An intracellular study of time-dependent cardiovascular afferent interactions in nucleus tractus solitarius. Journal of Neurophysiology. 1988;59:1798–1813. doi: 10.1152/jn.1988.59.6.1798. [DOI] [PubMed] [Google Scholar]
- Milligan CJ, Buckley NJ, Garret M, Deuchars J, Deuchars SA. Evidence for inhibition mediated by coassembly of GABAA and GABAC receptor subunits in native central neurons. Journal of Neuroscience. 2004;24:9241–9250. doi: 10.1523/JNEUROSCI.1979-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwenhuijs D, Sarton E, Teppema L, Dahan A. Propofol for monitored anesthesia care: implications on hypoxic control of cardiorespiratory responses. Anesthesiology. 2000;92:46–54. doi: 10.1097/00000542-200001000-00013. [DOI] [PubMed] [Google Scholar]
- Orser BA, Canning KJ, MacDonald JF. Mechanisms of general anesthesia. Current Opinion in Anaesthesiology. 2002;15:427–433. doi: 10.1097/00001503-200208000-00004. [DOI] [PubMed] [Google Scholar]
- Ouyang W, Wang G, Hemmings HC., Jr Isoflurane and propofol inhibit voltage-gated sodium channels in isolated rat neurohypophysial nerve terminals. Molecular Pharmacology. 2003;64:373–381. doi: 10.1124/mol.64.2.373. [DOI] [PubMed] [Google Scholar]
- Rehberg B, Duch DS. Suppression of central nervous system sodium channels by propofol. Anesthesiology. 1999;91:512–520. doi: 10.1097/00000542-199908000-00026. [DOI] [PubMed] [Google Scholar]
- Reves JG, Glass PSA, Lubarsky DA. Nonbarbiturate intravenous anesthetics. In: Miller RD, editor. Anesthesia. New York: Churchill Livingstone; 2000. pp. 228–272. [Google Scholar]
- Rudolph U, Antkowiak B. Molecular and neuronal substrates for general anaesthetics. Nature Reviews Neuroscience. 2004;5:709–720. doi: 10.1038/nrn1496. [DOI] [PubMed] [Google Scholar]
- Saha S. Role of the central nucleus of the amygdala in the control of blood pressure: descending pathways to medullary cardiovascular nuclei. Clinical and Experimental Pharmacology and Physiology. 2005;32:450–456. doi: 10.1111/j.1440-1681.2005.04210.x. [DOI] [PubMed] [Google Scholar]
- Saper CB. The central autonomic nervous system: Conscious visceral perception and autonomic pattern generation. Annual Review of Neuroscience. 2002;25:433–469. doi: 10.1146/annurev.neuro.25.032502.111311. [DOI] [PubMed] [Google Scholar]
- Schild JH, Clark JW, Hay M, Mendelowitz D, Andresen MC, Kunze DL. A- and C-type nodose sensory neurons: Model interpretations of dynamic discharge characteristics. Journal of Neurophysiology. 1994;71:2338–2358. doi: 10.1152/jn.1994.71.6.2338. [DOI] [PubMed] [Google Scholar]
- Schild JH, Kunze DL. The dynamic properties of a TTX-resistant Na+ current may underlie the heterogenous discharge of rat nodose sensory neurons. Society for Neuroscience Abstracts. 1995;21:61. [Google Scholar]
- Seagard JL, Elegbe E, Hopp FA, Bosnjak ZJ, von Colditz J, Kalbfleisch J, Kampine J. Effects of isoflurane on the baroreceptor reflex. Anesthesiology. 1983;59:511–520. doi: 10.1097/00000542-198312000-00005. [DOI] [PubMed] [Google Scholar]
- Shirasaka T, Yoshimura Y, Qiu DL, Takasaki M. The effects of propofol on hypothalamic paraventricular nucleus neurons in the rat. Anesthesia and Analgesia. 2004;98:1017–1023. doi: 10.1213/01.ANE.0000107960.89818.35. [DOI] [PubMed] [Google Scholar]
- Smith BN, Dou P, Barber WD, Dudek FE. Vagally evoked synaptic currents in the immature rat nucleus tractus solitarii in an intact in vitro preparation. Journal of Physiology. 1998;512:149–162. doi: 10.1111/j.1469-7793.1998.149bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM, McBurney RN. Caesium ions: a glycine-activated channel agonist in rat spinal cord neurones grown in cell culture. British Journal of Pharmacology. 1989;96:940–948. doi: 10.1111/j.1476-5381.1989.tb11905.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolstykh G, Belugin S, Mifflin SW. Responses to GABA(A) receptor activation are altered in NTS neurons isolated from chronic hypoxic rats. Brain Research. 2004;1006:107–113. doi: 10.1016/j.brainres.2004.01.060. [DOI] [PubMed] [Google Scholar]
- Trapani G, Altomare C, Liso G, Sanna E, Biggio G. Propofol in anesthesia. Mechanism of action, structure-activity relationships, and drug delivery. Current Medical Chemistry. 2000;7:249–271. doi: 10.2174/0929867003375335. [DOI] [PubMed] [Google Scholar]
- Travagli RA, Hermann GE, Browning KN, Rogers RC. Brainstem circuits regulating gastric function. Annual Reviews Physiology. 2006;68:279–305. doi: 10.1146/annurev.physiol.68.040504.094635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban BW, Bleckwenn M, Barann M. Interactions of anesthetics with their targets: Non-specific, specific or both? Pharmacology and Therapeutics. 2006;111:729–770. doi: 10.1016/j.pharmthera.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Urbanski RW, Sapru HN. Putative neurotransmitters involved in medullary cardiovascular regulation. Journal of the Autonomic Nervous System. 1988;25:181–193. doi: 10.1016/0165-1838(88)90023-9. [DOI] [PubMed] [Google Scholar]
- Wang X, Huang ZG, Gold A, Bouairi E, Evans C, Andresen MC, Mendelowitz D. Propofol modulates gamma-aminobutyric acid-mediated inhibitory neurotransmission to cardiac vagal neurons in the nucleus ambiguus. Anesthesiology. 2004;100:1198–1205. doi: 10.1097/00000542-200405000-00023. [DOI] [PubMed] [Google Scholar]
- Wasserman AM, Ferreira M, Jr, Sahibzada N, Hernandez YM, Gillis RA. GABA-mediated neurotransmission in the ventrolateral NTS plays a role in respiratory regulation in the rat. American Journal of Physiology, Regulatory Integrative and Comparative Physiology. 2002;283:R1423–R1441. doi: 10.1152/ajpregu.00488.2001. [DOI] [PubMed] [Google Scholar]
- Yeung JY, Canning KJ, Zhu G, Pennefather P, MacDonald JF, Orser BA. Tonically activated GABAA receptors in hippocampal neurons are high-affinity, low-conductance sensors for extracellular GABA. Molecular Pharmacology. 2003;63:2–8. doi: 10.1124/mol.63.1.2. [DOI] [PubMed] [Google Scholar]