

Abstract
Background
Brain-derived neurotrophic factor (BDNF) plays an important role in neural plasticity in the adult nervous system and has been suggested as a target gene for antidepressant treatment. The neurotrophic hypothesis of depression suggests that loss of BDNF from the hippocampus contributes to an increased vulnerability for depression, whereas up-regulation of BDNF in the hippocampus is suggested to mediate antidepressant efficacy.
Methods
We have used a viral-mediated gene transfer approach to assess the role of BDNF in subregions of the hippocampus in a broad array of behavioral paradigms including depression-like behavior and antidepressant responses. We have combined the adeno-associated virus (AAV) with the Cre/loxP site-specific recombination system to induce the knockout of BDNF selectively in either the CA1 or dentate gyrus (DG) subregions of the hippocampus.
Results
We show that the loss of BDNF in either the CA1 or the DG of the hippocampus does not alter locomotor activity, anxiety-like behavior, fear conditioning or depression-related behaviors. However, the selective loss of BDNF in the DG, but not the CA1 region, attenuates the actions of desipramine and citalopram in the forced swim test.
Conclusions
These data suggest that the loss of hippocampal BDNF per se is not sufficient to mediate depression-like behavior. However, these results support the view that BDNF in the DG may be essential in mediating the therapeutic effect of antidepressants.
Keywords: BDNF, hippocampus, viral mediated gene transfer, behavior, animal model, antidepressant
Introduction
Mood disorders are a prevalent form of mental illness. Major depression is one of the most common mental illnesses with a morbid risk of over 10% of the general population (1–3). Recent data suggest that 40–50% of the individual risk for depression is genetic, indicating a clear biological vulnerability to the disease (4, 5). However, no specific gene has been conclusively identified as a causative factor for depression.
Fortunately, several effective treatments for depression have emerged over recent years. These include serotonin and norepinephrine reuptake inhibitors, monoamine oxidase inhibitors, and electroconvulsive therapy (ECT) (6). However, the precise mechanism of action underlying the therapeutic effectiveness of these treatments remains unknown. Recent studies have identified adaptations of intracellular signaling proteins and target genes that could contribute to antidepressant treatment (7). Three principal lines of evidence have linked brain-derived neurotrophic factor (BDNF) as a target gene in antidepressant treatment (8, 9). First, chronic antidepressant treatment increases BDNF expression in the hippocampus (10, 11), a brain region implicated in the pathophysiology and treatment of depression (12). Second, upregulation of BDNF occurs in response to chronic but not acute antidepressant treatment, consistent with the time course for the therapeutic action of antidepressants (10, 11). Third, local infusions of BDNF into the midbrain (13) and more recently the hippocampus (14) have been shown to produce antidepressant effects in the learned helplessness paradigm and the forced swim test, two animal models of depression-like behavior and antidepressant action.
Conversely, BDNF expression is decreased in the hippocampus by acute and chronic exposure to stress (15, 16). Several studies have implicated a role for stress in the development of depressive symptoms in some individuals (17). This downregulation of BDNF may further contribute to the atrophy of hippocampal neurons observed following stress (8). Interestingly, the combination of chronic antidepressants and stress results in an additive effect on BDNF expression; namely, no change in BDNF levels (10). Collectively, these data led to the ‘neurotrophic hypothesis of depression’ (8), which states that the loss of BDNF may contribute to hippocampal alterations that underlie aspects of depression (18, 19), while antidepressants may mediate some of their therapeutic effects by increasing BDNF in this brain region (8, 20, 21).
Despite the numerous studies suggesting a link between BDNF in the hippocampus and depression-like behavior or the therapeutic effect of antidepressants, there has been little direct evidence examining the role of endogenous BDNF in these processes. BDNF homozygous null (−/−) knockouts do not survive past postnatal day 10–14 (22), and studies with BDNF heterozygote (−/+) mice have produced conflicting behavioral results regarding the role of BDNF in depression (23–27). To more directly examine the role of endogenous BDNF in depression-related behavior and antidepressant efficacy, we generated inducible BDNF knockout (KO) mice in which the deletion of BDNF was restricted to forebrain regions, as well as two different conditional BDNF KO’s with distinct patterns of BDNF loss in forebrain areas, and demonstrated that all three of these independent lines have normal ‘depression-like behavior’ but fail to respond to antidepressant drugs (28, 29). While these studies support the view that endogenous forebrain BDNF may be essential in mediating antidepressant efficacy, they do not directly examine the role of BDNF selectively in the hippocampus in mediating these effects.
Viral-mediated gene transfer is a method that can be used to introduce a protein of interest into a specific brain region (30). The advantage of this system is that it results in overexpression of the gene product of interest in regions confined to the site of injection. We are using the adeno-associated virus (AAV) system to express Cre recombinase to create a targeted knockout in a subregion-selective manner. A previous study using this system has shown that AAV-mediated expression of Cre recombinase occurs in a high percentage of cells in an injected region, occurs exclusively in neurons, and is associated with minimal surgical injury (31).
We selectively deleted BDNF in the dentate gyrus (DG) of the hippocampus since this subregion has increased BDNF levels following chronic antidepressant treatment (10, 14). We also deleted BDNF in the CA1 region of the hippocampus, in a separate group of mice as a comparison, since chronic antidepressant treatment does not alter BDNF levels in this subregion of the hippocampus. These experiments allow us to examine whether endogenous BDNF in specific subregions of the hippocampus is involved in mediating depression-related behavior and responses to antidepressants.
Materials and Methods
Floxed BDNF Mice
The floxed BDNF mice have previously been described (32). In prior studies, we crossed these floxed BDNF mice with an NSE-tTA x TetOp-Cre line to create an inducible deletion of BDNF in the brain as well as a targeted deletion of BDNF in the ventral tagmental area (28, 31). These studies demonstrated that BDNF is only deleted in the presence of Cre recombinase in the floxed mice. For the present study, only male floxed BDNF mice between the ages of 3–5 months were used for the AAV injection. All experiments were approved by the UT Southwestern Medical Center Animal Care and Use Committee.
Stereotaxic Surgery
Mice were anesthetized with ketamine (100 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.) then mounted on a sterotaxic apparatus. The skull was exposed and holes were drilled bilaterally above the target injection sites. The coordinates relative to Bregma for CA1 and DG were as follows: CA1, anteroposterior = −2.0mm, lateral = +1.6mm, dorsoventral = −1.5mm; and DG, anteroposterior = −1.9mm, lateral = +1.2mm, dorsoventral = −2.2mm at a 10° angle. Using a Hamilton syringe with a 33 gauge needle, a total of 1 μl of virus was bilaterally infused over 4 minutes. The syringe was left in place for an additional 5 minutes to ensure diffusion of the virus.
All mice were either injected with AAV-GFP (control; CTL) or AAV-Cre (knockout; KO), which expresses a fusion construct of Cre recombinase and GFP. The use of GFP allowed the visualization of the infected neurons. Previous work had demonstrated that the GFP did not interfere with Cre recombinase activity (31).
Fluorescent In Situ Hybridization (FISH)
After the completion of the behavioral tasks the mice were sacrificed by rapid decapitation, the brains dissected out, rapidly frozen on dry ice and stored at −80°C. The brains were sectioned at 14 mm thickness on a cryostat, collected on either superfrost slides or PEN membrane coated slides (Leica, Bannockburn, IL), and subjected to fluorescent in situ hybridization (FISH) or QPRT-PCR, respectively. BDNF and Cre recombinase probes were prepared by in vitro transcription and labeled with digoxigenin and fluorescein, respectively. The probe sequence information and detailed experimental conditions were performed as previously described (31). The BDNF and Cre recombinase probes were detected separately by anti-digoxigenin and anti-fluorescein antibodies conjugated to HRP (1:200 dilution, Dako, Carpinteria, CA). The signals were amplified by HRP with a tyramide signal amplification system (PerkinElmer, Boston, MA). The BDNF and Cre recombinase probes were visualized by Cy3 and fluorescein epifluorescence, respectively. The FISH technique allowed us to examine the injection sites and confirm whether the placements were in the correct location. If a placement was not correct bilaterally for an injection, the behavioral data of the animal was disregarded.
Quantitative Reverse Transcription PCR (QRT –PCR)
To determine the relative amount of BDNF expression in the CA1 or DG after stereotaxic injections, we used a QRT-PCR approach. Sections were collected as described above, briefly dehydrated in 70, 90, and 100% ethanol, and then subjected to laser microdissection using an AS LMD system (Leica, Bannockburn, IL). The entire CA1 or DG region containing GFP positive cells was dissected from each section. Eight to nine sections were pooled to extract total RNA using a PicoPure RNA isolation kit (Arcturus, Mountain View, CA). Each section was 140 μm apart; thus, encompassing the majority of the AAV infusion site in the dorsal hippocampus. The conditions for cDNA construction, amplification of BDNF, Cre, and β-actin, and sequences for the primers were described previously (31). For data analysis, the fold change in Cre and BDNF expression relative to actin was calculated as mean ± SEM.
Behavioral Overview
Mice were housed three to four per cage on a 12-hour light/dark cycle with ad libitum food and water. All behavioral testing was done on male BDNF floxed mice starting two weeks after stereotaxic surgery. There was no difference in the age of the AAV-GFP or AAV-Cre injected mice. The order of the behavioral tests was as follows: the elevated plus maze, locomotor activity, sucrose preference, fear-conditioning, and forced swim test with or without desipramine treatment. A second group of animals was injected in the DG with either AAV-GFP or AAV-Cre and tested in same manner as the previous group except that the sucrose preference test was excluded and the forced swim test was conducted with or without citalopram. Prior to all testing, mice were allowed to habituate in the behavioral room for one hour. Data was analyzed by Student’s t-test unless otherwise specified and presented as mean ± SEM. Significance was set at p < 0.05.
Locomotor activity
Locomotor activity was measured as described previously (28). Data were analyzed with repeated analysis of variance (ANOVA).
Elevated Plus Maze
Elevated plus maze was carried as described previously (29). The behavior of the mice was monitored for 5 minutes. The time spent in the closed and open arms, as well as the number of explorations of open-arms was determined using a video tracking system (Ethovision).
Fear Conditioning
The fear conditioning paradigm was performed as previously described [28]. Briefly for the training, mice were placed in individual chambers for two minutes followed by a loud tone (90 dB) for 30 sec then immediately followed by a 0.8 mA footshock for 2 seconds. Mice remained in the box for one minute at which time they again received the same tone-paired footshock. Context-dependent fear conditioning was assessed 24 hours after the training when mice were placed back in the same boxes for 5 minutes without a tone or shock. The amount of time the animal spent freezing was scored every 10 sec by an observer blind to the genotype of the mice. Freezing behavior was defined as no movement except for respiration. Four hours later, the cue test was performed. Mice were placed in a novel environment without tone or shock for three minutes followed by three minutes of the tone and the amount of time spent freezing was assessed.
Sucrose Preference
The sucrose preference test was conducted over a 48-hour period using a two-bottle test, one with 2% sucrose solution the other with water. The position of the bottles was changed after 24 hours to prevent potential location preference of drinking. The amount of the sucrose solution or water consumed was determined by weighing the bottles. Sucrose preference was calculated as the percentage of sucrose solution ingested relative to the total amount of liquid consumed.
Forced swim test
The forced swim test is a depression model that is sensitive to antidepressant treatment (33). Mice were placed in a 4000 ml Pyrex glass beaker containing 3000 ml of water at 24 ± 1ºC for six minutes. Water was changed between subjects. All test sessions were recorded by a video camera positioned on the side of the cylinders. The videotapes were analyzed and scored by an observer blind to the genotype. Immobility was measured during the last four minutes of the test. Saline, desipramine, or citalopram (10 mg/kg; 10 mg/kg; 20 mg/kg) was administered subchronically by i.p. injections 24, 4, and 1 hour before the test as previously described (28, 29).
Results
Localized Regional Specific Deletion of BDNF in hippocampus
We generated a localized selective deletion of the BDNF gene in the CA1 or DG using a floxed BDNF mouse, in which exon 6, the single coding region of the BDNF gene, is flanked by loxP sites (32). The BDNF floxed mice were bilaterally injected with AAV-Cre into CA1 or DG to induce the localized knockout. We have previously demonstrated that this AAV-Cre construct mediates recombination in the brain following injection into BDNF floxed mice (31). As a control, floxed BDNF mice were bilaterally injected into the CA1 or DG with AAV-GFP.
To evaluate the expression of Cre recombinase and BDNF levels, we carried out double fluorescent in situ hybridization. In animals receiving AAV-GFP, BDNF mRNA was strongly present in both the CA1 and DG (Figure 1A, B; top panels). As expected, no detectable expression of Cre recombinase was detected in AAV-GFP injected animals (Figure 1A, B). In the AAV-Cre injected mice, the localized expression of Cre mRNA was observed in either the CA1 or DG region depending on the injection site (Figure 1A, B; bottom panels). In regions expressing Cre recombinase, levels of BDNF mRNA expression were almost absent. Overlays of BDNF and Cre recombinase epifluorescence resulted in virtually no overlapping expression suggesting the loss of BDNF was due to Cre-mediated recombination (data not shown). To ensure that the decreased expression of BDNF was not due to aberrant damage of the tissue after the virus injection, we examined the injected site by nuclear staining with DAPI and did not observe any change in cell density (Figure 1A, B).
Figure 1.

Region Specific Deletion of BDNF in Subregions of the Hippocampus. (A, B) BDNF and Cre recombinase expression was analyzed by double fluorescent in situ hybridization in mice injected with AAV-GFP or AAV-Cre virus into either CA1 (A) or DG (B). Probes for BDNF and Cre recombinase were coupled to Cy3 and FITC epifluorescence, respectively. In AAV-GFP injected mice, BDNF was strongly present in the CA1 and DG subregions of the hippocampus while no detectable expression of Cre recombinase was detected. In contrast, in the AAV-Cre injected mice, the localized expression of Cre was observed in either the CA1 or DG depending on the injection site, and in these regions the levels of BDNF expression was reduced. DAPI staining was used to show that the loss of BDNF expression was not due to changes in cell number. (C, D) The entire region of the CA1 (C) and DG (D) were laser micro-dissected out from brain sections of AAV-GFP and AAV-Cre injected mice. The sections were subjected to QRT-PCR analyses for quantitation of Cre and BDNF mRNA levels. Cre expression was virtually absent in AAV-GFP injected mice but significantly increased in AAV-Cre injected animals. AAV-Cre injected animals in both CA1 and DG had significant reduction (52% and 62%, respectively) in BDNF mRNA compared to AAV-GFP mice (CA1, AAV-GFP, n = 6; AAV-CRE, n = 7: DG, AAV-GFP, n = 10; AAV-Cre, n = 13).
To quantitate the total amount of Cre and BDNF expression in the CA1 or DG following AAV injection, we used QRT-PCR. The CA1 or DG regions were laser-microdissected out from brain sections where AAV-GFP or AAV-Cre were injected as identified by GFP epifluorescence. Expression of Cre in AAV-GFP injected mice was virtually absent, while bilateral injection of AAV-Cre into the CA1 or DG resulted in strong induction of Cre mRNA (Figure 1C, D). In these animals, the levels of BDNF expression in the CA1 and DG were 52% and 62%, respectively, lower than those from AAV-GFP injected mice (Figure 1C, D). However, within an individual cell that expressed Cre recombinase, BDNF expression was virtually non-existent (data not shown).
It is important to note that after completion of the behavioral experiments, the selective injection of the virus into the CA1 or DG region was verified in each animal. If the injection was off the target region, the behavioral data from that animal was removed from analysis. We did not observe any difference in weight between AAV-GFP and AAV-Cre injected animals (data not shown).
Locomotor Activity
Locomotor activity was assessed in the AAV-GFP and AAV-Cre injected mice for 120 minutes. There was no significant difference in locomotor activity as assessed in 5-minute increments or in total overall activity (see insert) between AAV-GFP and AAV-Cre mice injected in the CA1 (Figure 2A). The level of locomotor activity was also unaltered in animals with localized deletion of BDNF in the DG region compared to AAV-GFP injected mice (Figure 2B).
Figure 2.
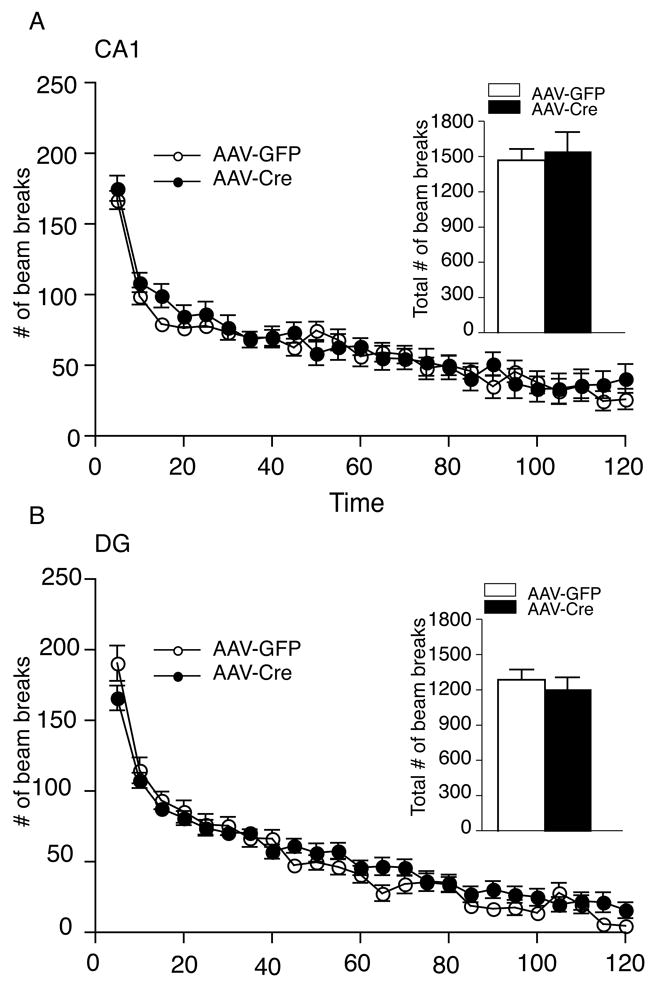
Localized Deletion of BDNF in the CA1 and DG Subregions Does Not Alter Locomotor Activity. (A) Male mice injected into the CA1 with either AAV-Cre or AAV-GFP exhibited no significant difference in locomotor activity (ambulation), as assessed by a consecutive horizontal beam break, over a two-hour period in either 5 minute increments or over the entire two-hour period (see insert; AAV-GFP, n = 12; AAV-Cre, n = 11). (B) The male mice injected into the DG region with either AAV-Cre or AAV-GFP exhibited a similar level of locomotor activity during the testing period whether analyzed as 5 minute increments or total number of beam breaks over the entire two-hour period (see insert; AAV-GFP, n = 23; AAV-Cre, n = 24).
Anxiety-Related Behavior
We examined whether the localized deletion of BDNF in the CA1 or DG alters anxiety-related behavior. Localized deletion of BDNF in the either the CA1 or DG did not alter anxiety-like behavior as assessed in the elevated plus test compared to AAV-GFP injected animals (Figure 3A–B).
Figure 3.
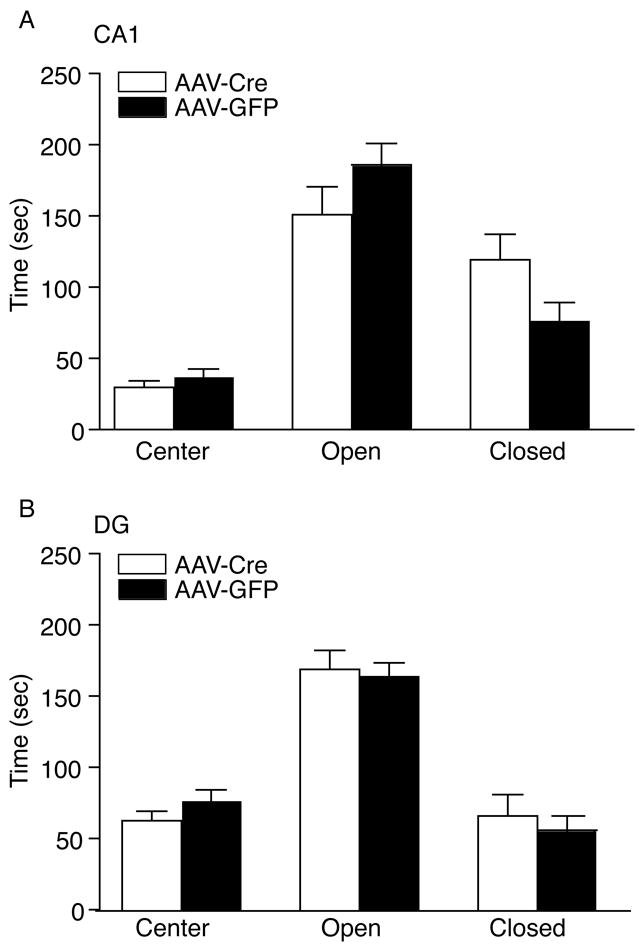
Localized Deletion of BDNF in the CA1 and DG Does Not Alter Anxiety Related Behavior. (A) The CA1 injected mice with AAV-Cre displayed a similar level of anxiety, as determined by the time spent in the center, dark side, and light side of the elevated plus maze compared to AAV-GFP injected mice (AAV-GFP, n = 12; AAV-Cre, n = 11). (B) DG injected mice with AAV-Cre exhibited normal anxiety, as assessed in the elevated plus maze, compared to AAV-GFP controls (CTL) (AAV-GFP, n = 21; AAV-Cre, n = 22).
Fear Conditioning Test
We assessed whether the selective loss of BDNF in the CA1 or DG region of the hippocampus would alter context or cue dependent fear conditioning. The selective deletion of BDNF in the CA1 did not alter context or cue-dependent fear conditioning compared to AAV-GFP injected mice (Figure 4A). AAV-Cre or AAV-GFP injected animals into the DG region also had indistinguishable levels of context and cue-dependent fear conditioning (Figure 4B). Baseline levels of freezing were similar between AAV-GFP and AAV-Cre injected animals in the CA1 or DG region.
Figure 4.
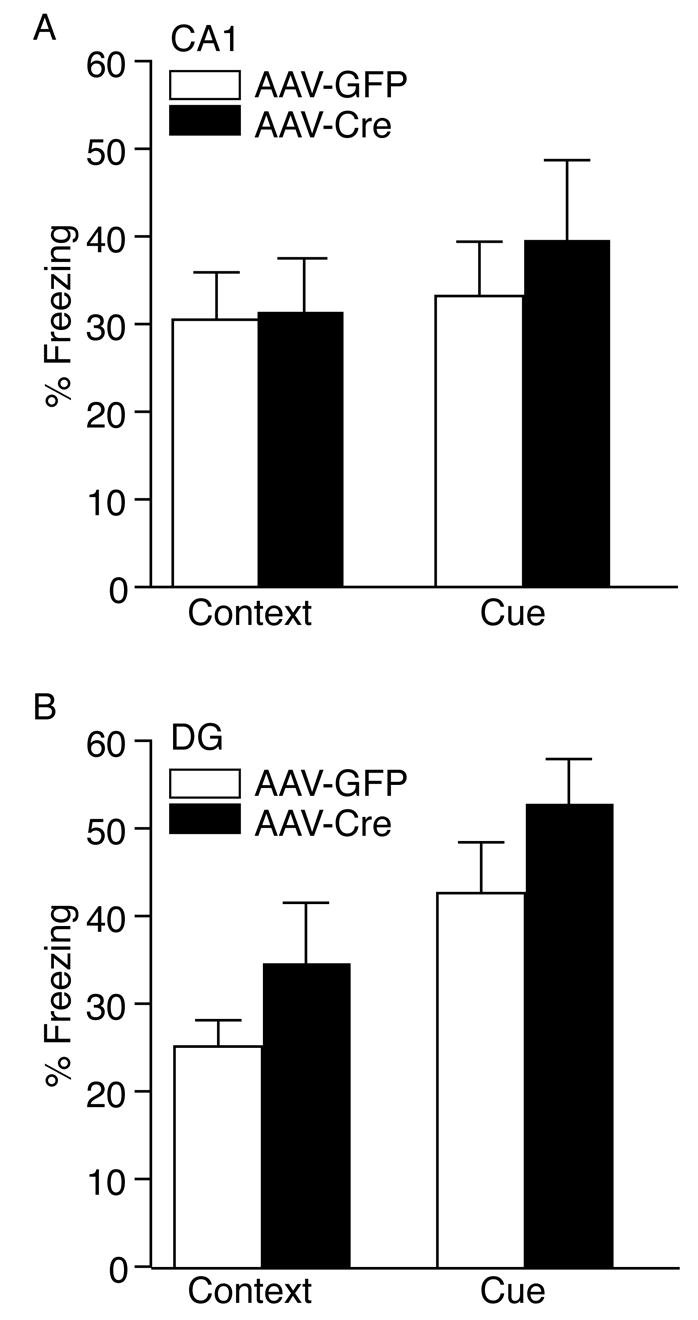
Context and Cue-Dependent Fear Conditioning is Unaltered by the loss of BDNF Selectively in the CA1 or DG region. (A) The selective loss of BDNF in the CA1 did not result in any difference in context-dependent fear conditioning 24 hours after training compared to AAV-GFP injected mice. No significant difference was observed in baseline freezing behavior. Cue-dependent fear conditioning was also indistinguishable in AAV-Cre or AAV-GFP injected mice (AAV-GFP, n = 12; AAV-Cre, n = 11). (B) The loss of BDNF in the DG did not produce any significant difference in context-dependent or cue-dependent fear conditioning compared to AAV-GFP injected animals (AAV-GFP, n = 17; AAV-Cre, n = 16). No significant difference was observed in baseline freezing behavior.
Depression-Related Behavior
To examine whether the loss of BDNF in particular subregions of the hippocampus exerts an effect on depression-related behavior, we tested the AAV-Cre and AAV-GFP mice in the sucrose preference test, a paradigm that measures an animal’s responsiveness to a natural reward (34). A loss of sensitivity to reward has been suggested to model anhedonia, one important feature of human depression. The localized deletion of BDNF in the CA1 did not result in any difference in sucrose intake compared to AAV-GFP injected controls (Figure 5A). The AAV-Cre localized deletion in the DG region also did not alter preference for the sucrose solution compared to AAV-GFP injected mice (Figure 5B).
Figure 5.
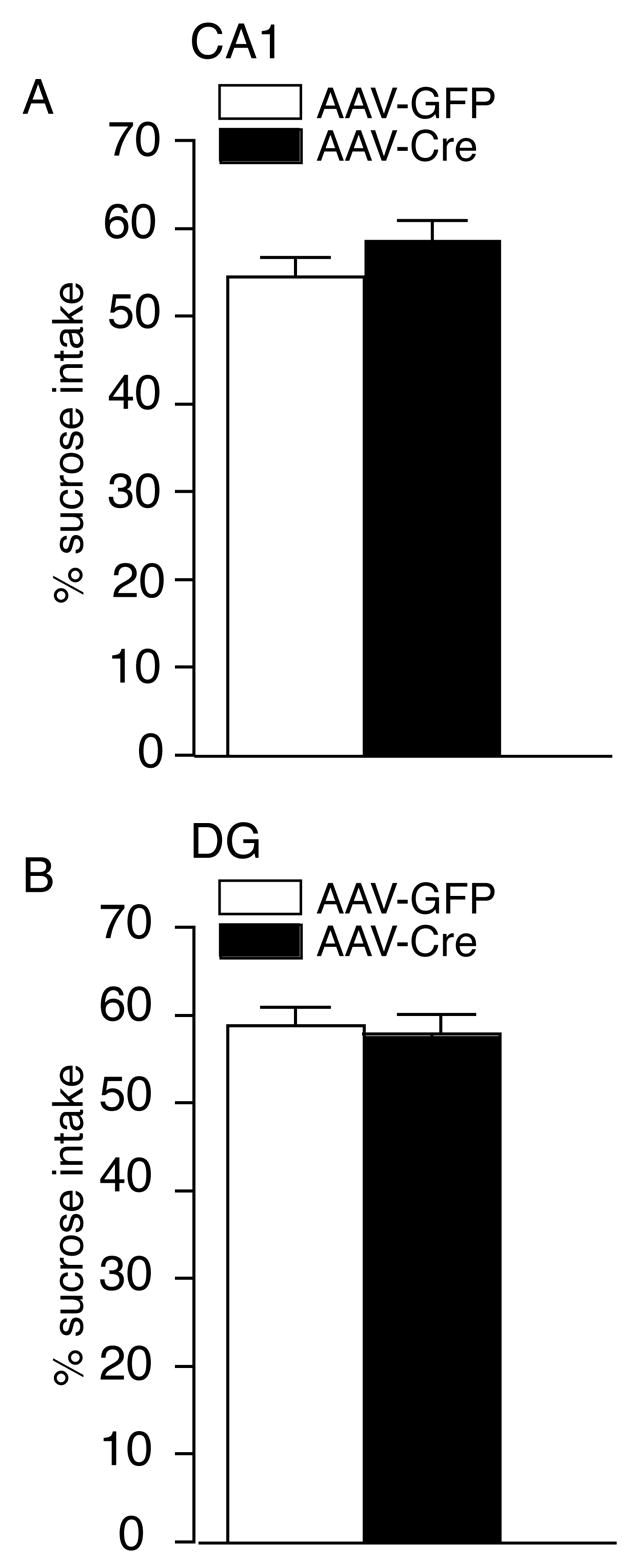
The Localized Deletion of BDNF in the CA1 or DG Region does not Alter Preference for a Natural Reward in the Sucrose Preference Paradigm. (A) Male floxed BDNF mice injected with either AAV-Cre or AAV-GFP in the CA1 display a similar preference for the sucrose solution (AAV-GFP, n = 12; AAV-Cre, n = 11). (B) Male floxed BDNF mice with a localized deletion of BDNF in the DG region have indistinguishable levels of intake of the sucrose solution compared to injected AAV-GFP mice. The total intake of liquid (water plus sucrose solution) was unchanged between the groups (p > 0.05). Results are presented as mean of sucrose preference (the percentage of sucrose solution ingested relative to the total amount of liquid consumed) ± SEM (AAV-GFP, n = 12; AAV-Cre, n = 13).
The AAV-Cre localized BDNF KO’s were next evaluated in the forced swim test (FST) to more directly test whether the loss of BDNF selectively in subregions of the hippocampus may contribute to alterations that underlie aspects of ‘depression-like’ behavior or antidepressant efficacy. In the absence of antidepressant treatment, we did not observe a significant difference in immobility in the FST between the AAV-Cre and the AAV-GFP injected CA1 or DG mice (Figure 6A–B). In the localized CA1 KO animals a significant decrease in immobility following desipramine treatment was observed that was similar to that seen in AAV-GFP mice (Figure 6A). In contrast, while the AAV-GFP injected mice in the DG showed the expected significant decrease in immobility following desipramine treatment, the AAV-Cre injected mice had no significant decrease in immobility (Figure 6B). To more closely examine this effect with another antidepressant we injected a separate group of floxed BDNF mice with either AAV-GFP or AAV-Cre in the DG. In the FST, the AAV-GFP mice showed the expected significant decrease in immobility following citalopram treatment; however, localized DG KO animals had no significant decrease in immobility, similar to what was observed with desipramine (Figure 6C).
Figure 6.
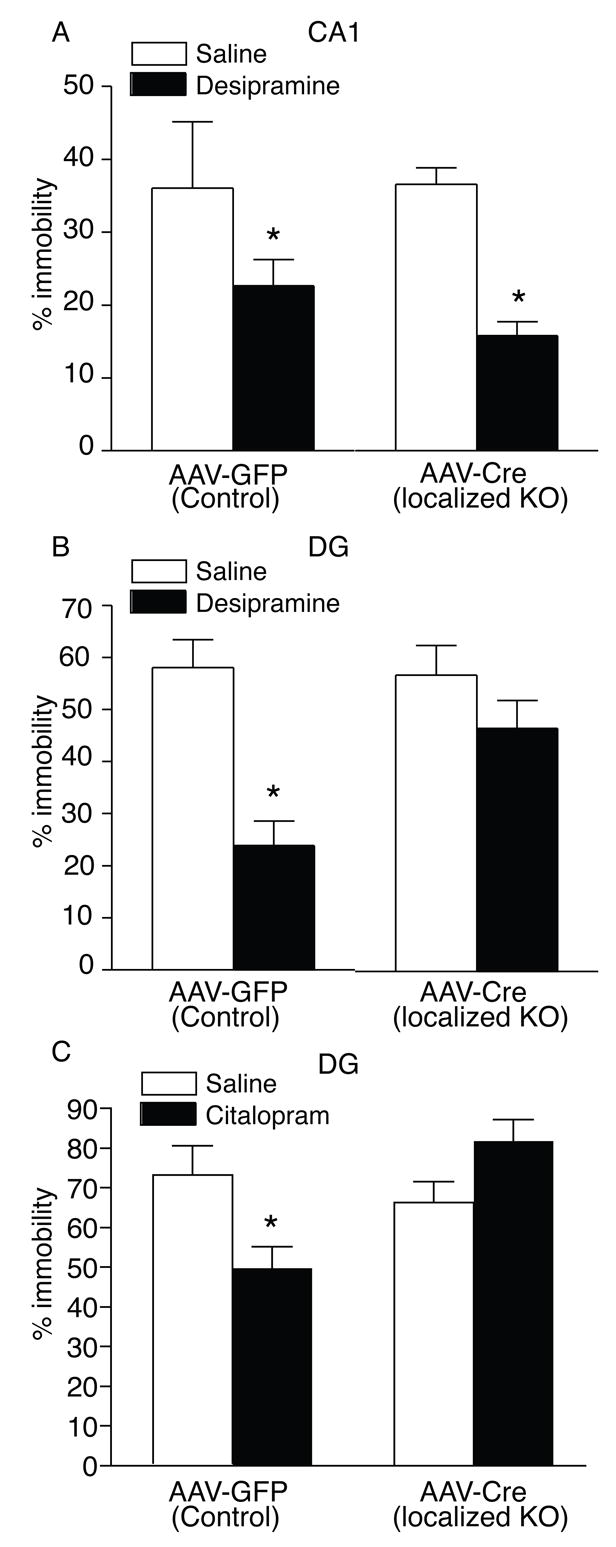
The Loss of BDNF in the DG Attenuates Antidepressant Responses in the Forced Swim Test. (A–C) Male floxed BDNF mice injected with AAV-Cre in the DG or CA1 subregion of the hippocampus display a similar % of immobility in the FST as compared to AAV-GFP mice (CTL). (A) Antidepressant treatment with desipramine significantly reduced immobility time in AAV-GFP mice (p < 0.05) as well as mice with a selective reduction of BDNF in the CA1 subregion of the hippocampus (p < 0.05). (AAV-GFP, n = 12; AAV-Cre, n = 11). (B) Desipramine treatment significantly reduced immobility time in AAV-GFP mice (p < 0.05) but not in the mice with a selective loss of BDNF in the DG (AAV-GFP, n = 12; AAV-Cre, n = 13). (C) Using the serotonin-selective reuptake inhibitor, citalopram, we found that this antidepressant also significantly reduced immobility time in AAV-GFP mice (p < 0.05) but not in the mice with a selective loss of BDNF in the DG (AAV-GFP, n = 11; AAV-Cre, n = 11).
Discussion
Results of this study demonstrate that the loss of BDNF selectively in the DG results in an attenuation of antidepressant efficacy. In contrast, the selective loss of BDNF in the CA1 subregion of the hippocampus did not alter the response to antidepressants. Mice with a selective loss of BDNF in either the CA1 or DG region of the hippocampus have normal locomotor activity, anxiety-like behavior, fear-conditioning, and depression-like behavior. This data suggests that the loss of BDNF in either the CA1 or DG subregions of the hippocampus does not alter a broad range of behavioral characteristics. However, BDNF expressed in DG appears to be necessary for mediating antidepressant responses.
In this study, we targeted the knockout of BDNF bilaterally into either the CA1 or DG region of adult male mice. In these animals, the levels of BDNF expression in the CA1 and DG subregions were 52% and 62%, respectively, lower than those from AAV-GFP injected mice. However, within an individual cell that expressed Cre recombinase, BDNF expression was virtually non-existent (data not shown). These data suggest that the loss of BDNF is in effect complete within a cell that is infected with Cre recombinase, however, the AAV is not infecting every cell within the subregion thus the deletion of BDNF was not entirely complete within the area.
The localized deletion of BDNF in either the CA1 or DG does not alter locomotor activity compared to floxed BDNF mice injected with AAV-GFP. This behavioral profile is similar to data obtained from our inducible BDNF KO’s in which we deleted BDNF in adult animals, suggesting that increased locomotor activity reported in heterozygous BDNF mice and conditional BDNF mice is not an acute outcome of BDNF loss, but rather due to a developmental effect (28). We also did not observe alterations in measures of anxiety-like behavior, which are in agreement with similar findings we obtained with inducible and conditional BDNF KO’s (28, 29).
We found normal context and cue dependent fear conditioning following the selective loss of BDNF in the CA1 or DG. This data was somewhat surprising in that we had observed a deficit in context-dependent fear conditioning in the inducible BDNF KO’s (28) and BDNF has been suggested to play a role in the cellular form of learning and memory, long-term potentiation (LTP) (35–43). However, an independent BDNF conditional KO line with a different forebrain deletion pattern did not show alterations in context-dependent fear conditioning although other learning deficits were observed (44). Closer examination of the role of BDNF in LTP provides a conflicting role of this neurotrophic factor in subregions of the hippocampus. One study demonstrated that BDNF constitutive knockouts have LTP alterations in the Schaffer collateral synapses but that this deficit could be rescued by BDNF overexpression in the CA1 region (45). In contrast, another group found that the selective loss of BDNF in the CA1 region is not involved in LTP (43). These data highlight the complex role of BDNF dependent cellular processes in subregions of the hippocampus. However, our ability to detect context-dependent deficits in fear-conditioning in the inducible KO’s and not the AAV selectively targeted deletions in this study, using the same behavioral parameters, suggest that the loss of BDNF selectively in either the DG or CA1 region is not sufficient to mediate deficits in this learning and memory process.
The loss of BDNF selectively in the CA1 or DG did not alter ‘depression-like’ behavior as assessed by the FST and sucrose preference test. This data is in agreement with our previous data showing that broad forebrain loss of BDNF is not sufficient to mediate an increase in depression-like behavior per se (28, 29). We used the FST since it has been used to examine depression-like and antidepressant-related behavior in numerous genetic models (46). We have previously attempted to examine conditional BDNF mice in the learned helplessness paradigm, another animal model to examine depression/antidepressant related responses (29). Unfortunately, we have not been able to generate a ‘helpless’ response in this mouse line presumably because of the genetic background of the floxed BDNF mice. Therefore, in this study we chose to examine sucrose preference as a model of anhedonia-like behavior. The loss of BDNF in either the CA1 or DG region did not produce alterations in depression-like behavior as assessed by either test. The one caveat is that we were only able to reduce BDNF levels by ~50% in these subregions of the hippocampus so this amount of BDNF reduction may not be sufficient to see deficits in depression-like behavior, although similar data was obtained with inducible and conditional BDNF KO’s which had broad forebrain reductions of 70 and 60% respectively (28, 29).
We demonstrated that the loss of BDNF in the DG, but not the CA1 region, attenuates the actions of the antidepressant desipramine, a tricylic that inhibits the reuptake of norepinephrine, or citalopram, a serotonin selective reuptake inhibitor, in the FST. The findings with both desipramine and citalopram suggest that the attenuated response to these antidepressants in the localized DG KO’s is sensitive to both alterations in norepinephrine and serotonin. Results from this study complement and extend previous data showing that the antidepressant effects of BDNF are observed with infusions into the DG granule cell layer but not the CA1 region of the hippocampus suggesting a regional specificity to these behavioral effects (14). This selective requirement of BDNF in the DG is intriguing and suggests that the actions of this neurotrophic factor are important in the cellular and behavioral aspects of antidepressant responses, at least those mediated by the FST. It is intriguing that the BDNF effect in mediating antidepressant efficacy is specific for the DG, the region of the hippocampus in which antidepressant treatment significantly increases neurogenesis (47). Accumulating evidence suggests that neurogenesis is important for the behavioral effects of antidepressants (48). Recent studies have shown that BDNF heterozygous mice or TrkB dominant negative mice have normal cell proliferation but decreased survival of newborn neurons (49). Future studies will be necessary to assess whether the selective deletion of BDNF in the adult DG impacts survival of newborn neurons as predicted from the previous study.
In summary, we have demonstrated that the selective loss of BDNF in the DG subregion of the hippocampus results in an attenuated response to antidepressants that is not observed following BDNF deletion in the CA1 region. The loss of BDNF in either of these subregions does not alter depression-like behavior, fear conditioning, locomotor activity, or anxiety-related behavior. These results suggest that this growth factor in the DG may be essential for mediating aspects of antidepressant treatment and highlight the regional specificity of this effect.
Acknowledgments
We thank F. Gioia, A. Tipton, S. Yarbrough, and M. Barromeo for help with the sectioning. This work was supported by grant MH070727 and from a NARSAD Young Investigator Award (L.M.M). The authors report no biomedical financial interests or potential conflicts of interest.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kessler RC, Berglund P, Demler O, Jin R, Koretz D, Merikangas KR, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R) Jama. 2003;289:3095–3105. doi: 10.1001/jama.289.23.3095. [DOI] [PubMed] [Google Scholar]
- 2.Murray CJ, Lopez AD. Global mortality, disability, and the contribution of risk factors: Global Burden of Disease Study. Lancet. 1997;349:1436–1442. doi: 10.1016/S0140-6736(96)07495-8. [DOI] [PubMed] [Google Scholar]
- 3.Wong ML, Licinio J. Research and treatment approaches to depression. Nat Rev Neurosci. 2001;2:343–351. doi: 10.1038/35072566. [DOI] [PubMed] [Google Scholar]
- 4.Fava M, Kendler KS. Major depressive disorder. Neuron. 2000;28:335–341. doi: 10.1016/s0896-6273(00)00112-4. [DOI] [PubMed] [Google Scholar]
- 5.Sanders AR, Detera-Wadleigh SD, Gershon ES. Molecular genetics of mood disorders. In: Nestler EJ, Charney DS, Bunney BS, editors. Neurobiology of Mental Illness. New York: Oxford; 1999. pp. 299–316. [Google Scholar]
- 6.Frazer A. Pharmacology of antidepressants. J Clin Psychopharmacol. 1997;17(Suppl 1):2S–18S. doi: 10.1097/00004714-199704001-00002. [DOI] [PubMed] [Google Scholar]
- 7.Manji HK, Moore GJ, Rajkowska G, Chen G. Neuroplasticity and cellular resilience in mood disorders. Mol Psychiatry. 2000;5:578–593. doi: 10.1038/sj.mp.4000811. [DOI] [PubMed] [Google Scholar]
- 8.Duman RS, Heninger GR, Nestler EJ. A molecular and cellular theory of depression. Arch Gen Psychiatry. 1997;54:597–606. doi: 10.1001/archpsyc.1997.01830190015002. [DOI] [PubMed] [Google Scholar]
- 9.Duman RS, Malberg J, Thome J. Neural plasticity to stress and antidepressant treatment. Biol Psychiatry. 1999;46:1181–1191. doi: 10.1016/s0006-3223(99)00177-8. [DOI] [PubMed] [Google Scholar]
- 10.Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. 1995;15:7539–7547. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Russo-Neustadt A, Beard RC, Cotman CW. Exercise, antidepressant medications, and enhanced brain derived neurotrophic factor expression. Neuropsychopharmacology. 1999;21:679–682. doi: 10.1016/S0893-133X(99)00059-7. [DOI] [PubMed] [Google Scholar]
- 12.Henninger GR, Charney DS. Mechanisms of action of antidepressant treatments: implications for the etiology and treatment of depressive disorders. In: Meltzer HY, editor. Psychopharmacology: the third generation of progress. New York: Raven; 1987. pp. 535–544. [Google Scholar]
- 13.Siuciak JA, Lewis DR, Wiegand SJ, Lindsay RM. Antidepressant-like effect of brain-derived neurotrophic factor (BDNF) Pharmacol Biochem Behav. 1997;56:131–137. doi: 10.1016/S0091-3057(96)00169-4. [DOI] [PubMed] [Google Scholar]
- 14.Shirayama Y, Chen AC, Nakagawa S, Russell DS, Duman RS. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci. 2002;22:3251–3261. doi: 10.1523/JNEUROSCI.22-08-03251.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nibuya M, Nestler EJ, Duman RS. Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J Neurosci. 1996;16:2365–2372. doi: 10.1523/JNEUROSCI.16-07-02365.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith MA, Makino S, Kvetnansky R, Post RM. Stress and glucocorticoids affect the expression of brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus. J Neurosci. 1995;15:1768–1777. doi: 10.1523/JNEUROSCI.15-03-01768.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tafet GE, Bernardini R. Psychoneuroendocrinological links between chronic stress and depression. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:893–903. doi: 10.1016/S0278-5846(03)00162-3. [DOI] [PubMed] [Google Scholar]
- 18.Bremner JD, Narayan M, Anderson ER, Staib LH, Miller HL, Charney DS. Hippocampal volume reduction in major depression. Am J Psychiatry. 2000;157:115–118. doi: 10.1176/ajp.157.1.115. [DOI] [PubMed] [Google Scholar]
- 19.Sheline YI, Sanghavi M, Mintun MA, Gado MH. Depression duration but not age predicts hippocampal volume loss in medically healthy women with recurrent major depression. J Neurosci. 1999;19:5034–5043. doi: 10.1523/JNEUROSCI.19-12-05034.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Altar CA. Neurotrophins and depression. Trends Pharmacol Sci. 1999;20:59–61. doi: 10.1016/s0165-6147(99)01309-7. [DOI] [PubMed] [Google Scholar]
- 21.Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. 2006;59:1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 22.Ernfors P, Lee KF, Jaenisch R. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature. 1994;368:147–150. doi: 10.1038/368147a0. [DOI] [PubMed] [Google Scholar]
- 23.Kernie SG, Liebl DJ, Parada LF. BDNF regulates eating behavior and locomotor activity in mice. Embo J. 2000;19:1290–1300. doi: 10.1093/emboj/19.6.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Linnarsson S, Bjorklund A, Ernfors P. Learning deficit in BDNF mutant mice. Eur J Neurosci. 1997;9:2581–2587. doi: 10.1111/j.1460-9568.1997.tb01687.x. [DOI] [PubMed] [Google Scholar]
- 25.Lyons WE, Mamounas LA, Ricaurte GA, Coppola V, Reid SW, Bora SH, et al. Brain-derived neurotrophic factor-deficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormalities. Proc Natl Acad Sci U S A. 1999;96:15239–15244. doi: 10.1073/pnas.96.26.15239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.MacQueen GM, Ramakrishnan K, Croll SD, Siuciak JA, Yu G, Young LT, Fahnestock M. Performance of heterozygous brain-derived neurotrophic factor knockout mice on behavioral analogues of anxiety, nociception, and depression. Behav Neurosci. 2001;115:1145–1153. doi: 10.1037//0735-7044.115.5.1145. [DOI] [PubMed] [Google Scholar]
- 27.Saarelainen T, Hendolin P, Lucas G, Koponen E, Sairanen M, MacDonald, et al. Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioral effects. J Neurosci. 2003;23:349–357. doi: 10.1523/JNEUROSCI.23-01-00349.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Monteggia LM, Barrot M, Powell CM, Berton O, Galanis V, Gemelli T, et al. Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proc Natl Acad Sci U S A. 2004;101:10827–10832. doi: 10.1073/pnas.0402141101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Monteggia LM, Luikart B, Barrot M, Theobold D, Malkovska I, Nef S, et al. Brain-derived neurotrophic factor conditional knockouts show gender differences in depression-related behaviors. Biol Psychiatry. 2007;61:187–197. doi: 10.1016/j.biopsych.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 30.Buning H, Nicklin SA, Perabo L, Hallek M, Baker AH. AAV-based gene transfer. Curr Opin Mol Ther. 2003;5:367–375. [PubMed] [Google Scholar]
- 31.Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, et al. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–868. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- 32.Rios M, Fan G, Fekete C, Kelly J, Bates B, Kuehn R, et al. Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol Endocrinol. 2001;15:1748–1757. doi: 10.1210/mend.15.10.0706. [DOI] [PubMed] [Google Scholar]
- 33.Porsolt RD, Le Pichon M, Jalfre M. Depression: a new animal model sensitive to antidepressant treatments. Nature. 1977;266:730–732. doi: 10.1038/266730a0. [DOI] [PubMed] [Google Scholar]
- 34.Barrot M, Olivier JD, Perrotti LI, DiLeone RJ, Berton O, Eisch AJ, et al. CREB activity in the nucleus accumbens shell controls gating of behavioral responses to emotional stimuli. Proc Natl Acad Sci U S A. 2002;99:11435–11440. doi: 10.1073/pnas.172091899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen G, Kolbeck R, Barde YA, Bonhoeffer T, Kossel A. Relative contribution of endogenous neurotrophins in hippocampal long-term potentiation. J Neurosci. 1999;19:7983–7990. doi: 10.1523/JNEUROSCI.19-18-07983.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- 37.Kang H, Welcher AA, Shelton D, Schuman EM. Neurotrophins and time: different roles for TrkB signaling in hippocampal long-term potentiation. Neuron. 1997;19:653–664. doi: 10.1016/s0896-6273(00)80378-5. [DOI] [PubMed] [Google Scholar]
- 38.Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci U S A. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Minichiello L, Korte M, Wolfer D, Kuhn R, Unsicker K, Cestari V, et al. Essential role for TrkB receptors in hippocampus-mediated learning. Neuron. 1999;24:401–414. doi: 10.1016/s0896-6273(00)80853-3. [DOI] [PubMed] [Google Scholar]
- 40.Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- 41.Patterson S, Pittenger C, Morozov A, Martin KC, Scanlin H, Drake C, Kandel ER. Some forms of cAMP-mediated long-lasting potentiation are associated with release of BDNF and nuclear translocation of phospho-MAP kinase. Neuron. 2001;32:123–140. doi: 10.1016/s0896-6273(01)00443-3. [DOI] [PubMed] [Google Scholar]
- 42.Xu B, Gottschalk W, Chow A, Wilson RI, Schnell E, Zang K, et al. The role of brain-derived neurotrophic factor receptors in the mature hippocampus: modulation of long-term potentiation through a presynaptic mechanism involving TrkB. J Neurosci. 2000;20:6888–6897. doi: 10.1523/JNEUROSCI.20-18-06888.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zakharenko S, Patterson SL, Dragatsis I, Zeitlin SO, Siegelbaum SA, Kandel ER, Morozov A. Presynaptic BDNF required for a presynaptic but not postsynaptic component of LTP at hippocampal CA1-CA3 synapses. Neuron. 2003;39:975–990. doi: 10.1016/s0896-6273(03)00543-9. [DOI] [PubMed] [Google Scholar]
- 44.Gorski JA, Balogh SA, Wehner JM, Jones KR. Learning deficits in forebrain-restricted brain-derived neurotrophic factor mutant mice. Neuroscience. 2003;121:341–354. doi: 10.1016/s0306-4522(03)00426-3. [DOI] [PubMed] [Google Scholar]
- 45.Korte M, Griesbeck O, Gravel C, Carroll P, Staiger V, Thoenen H, Bonhoeffer T. Virus-mediated gene transfer into hippocampal CA1 region restores long-term potentiation in brain-derived neurotrophic factor mutant mice. Proc Natl Acad Sci U S A. 1996;93:12547–12552. doi: 10.1073/pnas.93.22.12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cryan JF, Markou A, Lucki I. Assessing antidepressant activity in rodents: recent developments and future needs. Trends Pharmacol Sci. 2002;23:238–245. doi: 10.1016/s0165-6147(02)02017-5. [DOI] [PubMed] [Google Scholar]
- 47.Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci. 2000;20:9104–9110. doi: 10.1523/JNEUROSCI.20-24-09104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science. 2003;301:805–809. doi: 10.1126/science.1083328. [DOI] [PubMed] [Google Scholar]
- 49.Sairanen M, Lucas G, Ernfors P, Castren M, Castren E. Brain-derived neurotrophic factor and antidepressant drugs have different but coordinated effects on neuronal turnover, proliferation, and survival in the adult dentate gyrus. J Neurosci. 2005;25:1089–1094. doi: 10.1523/JNEUROSCI.3741-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]