

Abstract
Multiphoton microscopy has enabled biologists to collect high-resolution images hundreds of microns into biological tissues, including tissues of living animals. While the depth of imaging exceeds that possible from any other form of light microscopy, multiphoton microscopy is nonetheless generally limited to depths of less than a millimeter. Many of the advantages of multiphoton microscopy for deep tissue imaging accrue from the unique nature of multiphoton fluorescence excitation. However, the quadratic relationship between illumination level and fluorescence excitation makes multiphoton microscopy especially susceptible to factors that degrade the illumination focus. Here we examine the effect of spherical aberration on multiphoton microscopy in fixed kidney tissues and in the kidneys of living animals. We find that spherical aberration, as evaluated from axial asymmetry in the point spread function, can be corrected by adjustment of the correction collar of a water immersion objective lens. Introducing a compensatory positive spherical aberration into the imaging system decreased the depth-dependence of signal levels in images collected from living animals, increasing signal by up to 50%.
Keywords: Multiphoton microscopy, two-photon microscopy, spherical aberration, objective correction collar, imaging depth
1. Introduction – The role of intravital microscopy in biomedical research
Intravital microscopy has become an essential tool in biomedical research. It has become increasingly appreciated that biological processes must be evaluated in the context of the whole organism. In the past, such studies have involved analysis of fixed tissues. While many aspects of structure may be deduced from studies of fixed tissues, these studies can only indirectly address function and cannot characterize dynamic processes. In addition, it is frequently unclear how well the true structure of biological tissues and distribution of molecular components are preserved in fixed tissues. For this reason, various techniques of “in vivo imaging” have been developed, such as positron emission tomography (PET), single photon emission computed tomography (SPECT), and magnetic resonance imaging (MRI). However, the spatial and temporal resolution of these techniques are frequently too poor to characterize processes occurring within cells at sub-second timescales[1].
Multiphoton microscopy has given researchers the ability to image inside living organs at micron resolution and timescales of seconds or less. [2–10] Additionally, because multiphoton microscopy is a fluorescence technique, it has the ability to localize multiple specific molecules simultaneously. Multiphoton microscopy has also been shown to allow extended observation of even highly sensitive processes without detectable damage. For example, hamster embryos were repeatedly imaged every 15 minutes over a period of 24 hours without adversely affecting their development. [11]
Unfortunately, signal levels in multiphoton microscopy rapidly attenuate with depth into tissues. Consequently, multiphoton microscopy is capable of collecting images only at relatively shallow depths of less than a millimeter. [4, 5, 8, 10, 12–14] Signal attenuation results from both reduced excitation and collection of fluorescence as one images deeper into tissues. Both the light used to excite fluorescence and the fluorescence emissions themselves are scattered and absorbed by biological tissues. The role of scattering and absorption in signal attenuation in multiphoton microscopy has been amply described, characterized and modeled. [4, 8, 10, 12, 14–17] Here we describe the role of an additional factor in signal attenuation: spherical aberration resulting from refractive index mismatch.
2. Background – In vivo fluorescence microscopy
Fluorescence microscopy is a powerful, important tool in biomedical research that has enabled scientists to image in vivo with spatial and temporal resolution orders of magnitude better than other in vivo techniques. Common in vivo techniques include PET with spatial resolution ~mm, SPECT with spatial resolution >mm, and MRI with spatial resolution >mm. [1] However to achieve these high resolutions, these techniques require signal integrations on the order of minutes. These long integrations are incompatible with the rapid dynamics of many physiological processes and, ironically, also compromise the resolution of all but the most immobile tissues. In contrast, fluorescence microscopy has a spatial resolution of less than 1 micron on a timescale of seconds or less. [1] The temporal resolution and spatial resolution of fluorescence microscopy give it the capability to resolve dynamics of even subcellular events. [2, 18, 19]
Fluorescence microscopy is a technique that uses light to excite fluorescence in the sample and collects that emitted fluorescent light to form an image. Using a variety of techniques, investigators can fluorescently label specific molecules in a tissue, and then collect images of the distribution and behavior of those specific molecules. Fluorescent molecules have the property that when excited by a small range of wavelengths of light (excitation spectra), they emit light at a small range of longer wavelengths (emission spectra) spherically in all directions. The difference between the excitation and emission wavelengths (also known as the Stokes’ shift) is the basis for the extraordinarily high contrast of fluorescence microscopy. Light from a high energy light source, such as a mercury arc lamp, is passed through a barrier filter designed to pass only those wavelengths that will optimally excite the particular fluorophore, and then reflected onto the sample, where it stimulates fluorescence. This fluorescence, which is very dim relative to the amount of light applied to the sample, is then collected by the objective lens, and passed through optical filters that selectively pass only the wavelengths of light in the range of the fluorescence emissions. In this way, only the dim fluorescence, but not the very bright light used to excite the fluorescence is passed on to the eyepiece or camera. This results in an image of the distribution of the fluorophore against an essentially black background.
An additional advantage of fluorescence microscopy is the capability to detect multiple components in a sample. By specifically tagging multiple targets in a tissue with different, spectrally distinct fluorophores, researchers can collect images of the distributions of multiple molecules in the same sample, allowing them to compare their spatial relationships. Samples are sequentially illuminated with different wavelengths of light used to specifically excite each probe, and fluorescence emissions are collected using filters optimized for that probe alone. With judicious choice of spectrally distinct fluorophores, researchers can collect 2, 3, or more different fluorescent images of a sample, and then deconvolve the composite spectrum into its individual components.
Conventional fluorescence microscopy, as described above, works well for thin specimens, but for thick specimens, especially in the case of in vivo imaging, fluorescence from out-of-focus planes increases background signal, reducing contrast and degrading image quality to the point where features cannot be distinguished. For this reason, conventional fluorescence microscopy is limited to examination of the most superficial layers of biological tissue. Confocal fluorescence microscopy was developed to solve this problem. In confocal microscopy, a pinhole is placed in the emission path in a plane conjugate with the objective focal plane to reject out-of-focus fluorescence, so the detector preferentially collects light from a single point in the specimen. An image is formed by scanning a focused beam of light across the sample, and “de-scanning” the emitted fluorescence onto a photomultiplier tube, which effectively builds up the image point by point. This makes image formation substantially different from conventional wide-field fluorescence microscopy where the entire specimen is illuminated and the eye or detector, typically a CCD camera, directly images the resulting fluorescence from the entire volume. In order to acquire images in reasonable periods of time, each point in the sample is imaged in a very brief interval, on the order of microseconds. In order to capture sufficient signal during this brief interval, very intense illumination is used, typically from a laser. A corollary benefit of confocal microscopy is that each image is an optical section of a thick sample, and these optical sections can be combined to form a three-dimensional image of the tissue.
Multiphoton microscopy (MPM) is another form of fluorescence microscopy that is also capable of optical sectioning, but unlike confocal microscopy, which utilizes a confocal pinhole to eliminate out-of-focus fluorescence, the optical sectioning of MPM arises from the localized excitation of fluorescence. MPM is based upon a nonlinear process where two (or more) photons are absorbed simultaneously (via a virtual state) by a molecule causing a transition to the excited state. In the case of two-photon microscopy, this requires that the summed energy of the two photons equal the energy required to make the transition. Because energy is inversely proportional to wavelength, the wavelength of the two photons will be approximately twice that of a single photon capable of exciting the molecule, typically 700–1000nm. For these two low energy photons to be absorbed “simultaneously,” they must arrive within approximately 10−16s of each other, the lifetime of the virtual state. [10] The probability that two photons will be absorbed, such that excitation may occur, depends quadratically on the excitation power, or photon density, and is very low under normal conditions. [20] The probability is increased by focusing light to a point with a high numerical aperture (NA) objective. Due to the conical geometry of the illuminating beam, average photon density will, on first principals, decrease with the square of axial distance from the focal plane. This combined with the quadratic dependence of two-photon excitation results in fluorescence excitation falling off with the fourth power of axial distance from the focus. As a result, fluorescence is stimulated only in a sub-femtoliter volume at the focus, the only place with a photon density high enough for two-photon excitation to occur.
However, the photon density required to stimulate two-photon excitation is so high that it would rapidly destroy biological samples. This problem is avoided by using a pulsed laser system. By rapidly, but briefly pulsing the laser, a modest laser can generate a peak power sufficient to excite two-photon fluorescence, but an average power low enough to avoid harming the sample. In this way, a typical MPM system generates a peak photon flux approximately a million times that at the surface of the sun, but at an average photon flux low enough that it results in negligible heating of most samples. [21]
Since excitation is localized to a single point in the sample, image formation in MPM requires scanning, as with confocal microscopy. However, unlike confocal microscopy, the localized excitation of fluorescence means that MPM can accomplish optical sectioning without the need of a confocal pinhole.
3. Signal Attenuation in Multiphoton Microscopy
Although confocal and multiphoton microscopy make optical sectioning possible, deep tissue imaging is still a challenge due to the fact that signal levels attenuate with depth. [4, 5, 8, 10, 12–14] Fluorescence signals can attenuate with depth due to reduced excitation or reduced collection of fluorescence emissions. On first principles, both excitation and detection of fluorescence will decrease with depth due to the effects of scattering and absorption by the tissue. In addition, signal levels can decline with depth when the refractive index of the tissue is not matched to that of the immersion fluid of the objective. [22–25] This “refractive index mismatch” results in spherical aberration, a condition in which paraxial light rays and peripheral light rays focus to different places, broadening the focal point of the objective lens. While both confocal and multiphoton microscopy are susceptible to each of these factors, the magnitude of their effects is much reduced by the unique design of the multiphoton microscope, as described below.
Several factors may impede the ability to collect fluorescence stimulated at depth in biological tissue. Fluorescence photons may be absorbed by the tissue, or may be scattered away from the detector. The effects of scattering on light propagation in tissue are typically ten to one hundred times more significant than absorption. [26, 27] Calculations indicate that the magnitude of scattering is such that nearly all of the fluorescence arising from a point 100 microns into tissue is scattered prior to exiting the tissue. [4, 13] Since deep-tissue microscopy typically involves imaging into a tissue whose average refractive index does not match that of the objective immersion medium, the single point of fluorescence stimulated in a confocal or MPM system will form an image broadened by spherical aberration.
Since scattering and broadening of the image of the focal spot results in the rejection of fluorescence by the confocal pinhole, scattering and spherical aberration significantly reduce the collection of fluorescence in confocal microscopy, leading to declining signal collection with depth. However, since a confocal pinhole is not required in MPM, large-area detectors can be used to collect fluorescence emissions, even in highly scattering samples and/or in samples inducing spherical aberration. Thus the fluorescence detection system of a multiphoton microscope makes it much less susceptible to the effects of both scattering and spherical aberration on signal collection.
Scattering and absorption also may reduce fluorescence excitation. To some degree the impact of these factors is minimized in MPM, which uses near-infrared light to excite fluorescence, unlike confocal microscopy which uses either ultraviolet or visible light. Due to the fact that Rayleigh scattering decreases with the fourth power of wavelength, near-infrared light is less susceptible to scattering. In addition, because proteins absorb ultraviolet and visible light, and water absorbs higher wavelengths of light, there is an “optical window” at 600–1000nm, the range commonly used in MPM, where absorption is minimal. [1, 5]
Thus, the use of near-infrared light to excite fluorescence, and the use of largearea detectors to collect fluorescence gives MPM significant advantages over confocal microscopy for deep-tissue imaging, minimizing many of the issues of light absorption, scattering, and spherical aberration that hamper confocal microscopy.
4. The role of spherical aberration in deep-tissue multiphoton microscopy
While the use of near-infrared light gives MPM an advantage over confocal microscopy for deep-tissue fluorescence excitation, the quadratic dependence of fluorescence excitation may make MPM more sensitive to depth, in some respects. First, any attenuation in the amount of light at the focus quadratically reduces fluorescence excitation, which may offset the benefits of using near-infrared light for excitation to some extent. Second, MPM will be much more sensitive to any factor in tissue that broadens the focused spot. Consistent with this, studies in model systems have shown MPM is highly sensitive to spherical aberration [12, 22, 24, 25].
Spherical aberration may be expected to be a particular problem for MPM in biomedical research, since the refractive index of biological tissues seldom matches that of the immersion medium used in microscopy. Dong et al. [28] examined skin tissue and found signal to be increased by 10% at depth in the dermis layer when imaging with an oil immersion objective versus a comparable water immersion objective. This is likely due to better matching of refractive index since the stratum corneum of the epidermis has an average refractive index of 1.47 which is much closer to that of oil than water.
Of the various factors limiting MPM at depth, the quality of the focused illumination volume, which is critically important to efficient multiphoton fluorescence stimulation, may be an issue that can be addressed to significantly improve MPM at depth. Whereas scattering and absorption are inevitable characteristics of the tissue, the amount of spherical aberration induced by tissue can be manipulated in various ways.
Probably the simplest way of manipulating spherical aberration is to simply avoid it, by using low numerical aperture objectives that are generally less susceptible to the effects of spherical aberration. Consistent with this, Tung et al. [29] found that low numerical aperture objectives out-perform high numerical aperture objectives for MPM of test samples and biological tissues at depth.
The potential of manipulating spherical aberration in high-numerical aperture objectives in order to improve fluorescence imaging at depth was demonstrated nearly 20 years ago by the laboratory of John Sedat and David Agard. Hiraoka et al. [30] demonstrated that using an oil-immersion objective to collect images of objects mounted in glycerol (n=1.417) resulted in images distorted by positive spherical aberration. However, by using an immersion medium with a slightly higher refractive index than oil, they could collect aberration-free images at particular depths in the sample. In effect, the authors introduced a negative spherical aberration into the system that, when combined with the positive spherical aberration induced by the sample, brought the focus of the peripheral and paraxial beams to a single point in the sample.
Spherical aberration can also be manipulated optically. While this may be accomplished via separate optical elements [31], the most common example of this is in the correction collars used in high numerical aperture water immersion objectives. Since correction for spherical aberration in these objectives critically depends upon coverslip thickness, they are equipped with adjustable collars that move the objective lens elements such that the paraxial and peripheral rays of light form a tight focus after traveling through a glass coverslip of defined thickness. Conversely, the collar can be used to manipulate the spherical aberration in a particular sample. Taking this approach, Lo et al. [32] found that small, but significant improvements in imaging depth into skin and rat tail tendon could be obtained through adjustment of the correction collar of a water immersion objective.
5. Spherical aberration in deep-tissue multiphoton microscopy of kidney tissue
The kidney is a complex organ, consisting of a variety of cell types organized into a complicated network of capillaries and renal tubules. While the surface of the kidney is easily imaged by conventional optical microscopy, imaging into the kidney is best approached by MPM. [2, 3] However, we have found that MPM is capable of collecting images from only the first two hundred microns into the kidney. Here we evaluate the role of spherical aberration in signal attenuation with depth into fixed and living kidney tissues, and evaluate a simple solution to improve deep-tissue MPM.
The effects of imaging through biological tissue are shown in Figure 1, which shows xz-cross-sectional images of 1 micron red fluorescent beads collected at a depth of 50 microns into an aqueous medium (A) or 50 microns into fixed kidney tissue (B). Imaging was conducted using the Zeiss LSM-510 Meta Confocal/Multiphoton Microscope System with a water immersion objective (Zeiss, 63x C-Apochromat, NA 1.2). Fluorescence excitation was provided by a titanium-sapphire laser (Spectraphysics, Mountain View, CA) at a wavelength of 800nm. The shapes of these images are illuminating. First, as expected, the bead imaged in water forms a compact, vertically symmetrical image, somewhat elongated in z, as expected from theory. In contrast, the bead imaged in tissue forms a severely elongated, broadened image with rings on the side closest to the microscope objective. This vertical asymmetry in the image suggests negative spherical aberration, a condition in which the peripheral rays emanating from the microscope objective focus deeper into the sample than the paraxial rays, resulting in an elongated focus with pronounced rings projecting up towards the lens. This aberration is to be expected when using an optical system corrected for an aqueous medium to collect images from a sample whose refractive index is higher than water, as when imaging biological tissues.
Fig. 1.
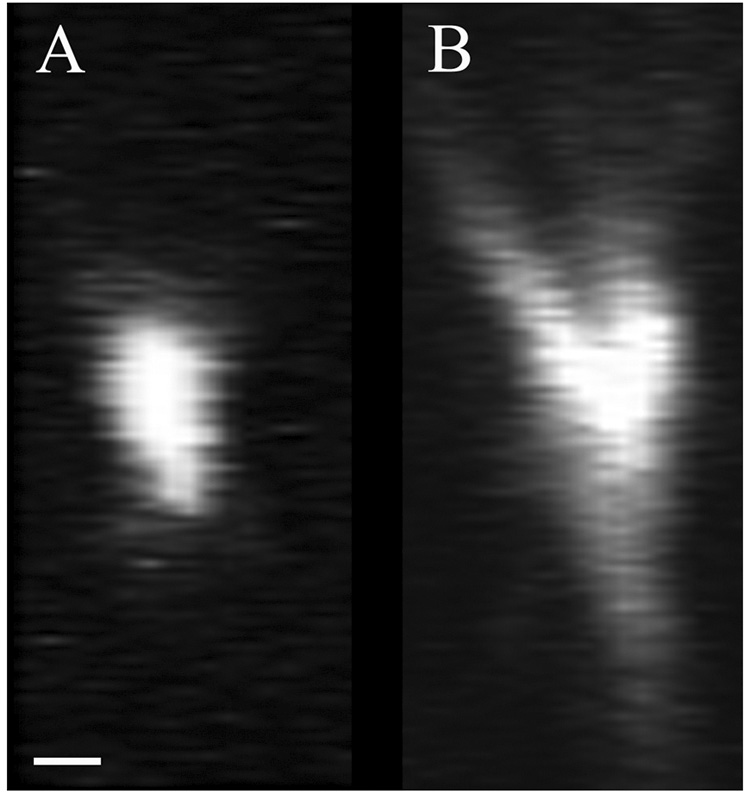
Comparison of XZ cross sectional images of 1micron fluorescent microsphere collected below 50 microns of aqueous solution (A) and below 50 microns of fixed kidney tissue (B). Scalebar = 1 micron, x=z.
This process is demonstrated in Figure 2, which shows xz cross-sectional images of sub-resolution red fluorescent microspheres collected with a water immersion objective using different correction collar settings. Imaging was conducted using the Olympus FV1000 confocal microscope system that has been adapted for two-photon microscopy by the Indiana Center for Biological Microscopy. A Mai Tai Ti:Sapphire laser (Spectraphysics, Mountain View, CA) provided the excitation light at wavelength 800nm. Image volumes were collected using a water immersion objective (Olympus, 60x Plan Apochromat, NA 1.2). In this figure, the images shown in the top row were collected with the collar adjusted for a nominal 0.13 mm thickness (A), 0.17 mm thickness (B), and 0.21 mm thickness (C). Since the coverslip was measured to be 0.18 mm thick, it is not surprising that the best results were obtained using the nominal 0.17 mm collar setting, which generated compact and vertically symmetrical point spread functions. As expected, adjusting the collar to 0.13 mm introduced negative spherical aberration into the imaging system, as reflected in the asymmetrical formation of rings projecting up towards the objective lens. Likewise, adjusting the collar to 0.21 mm resulted in positive spherical aberration, resulting in the formation of rings projecting away from the objective lens.
Fig. 2.
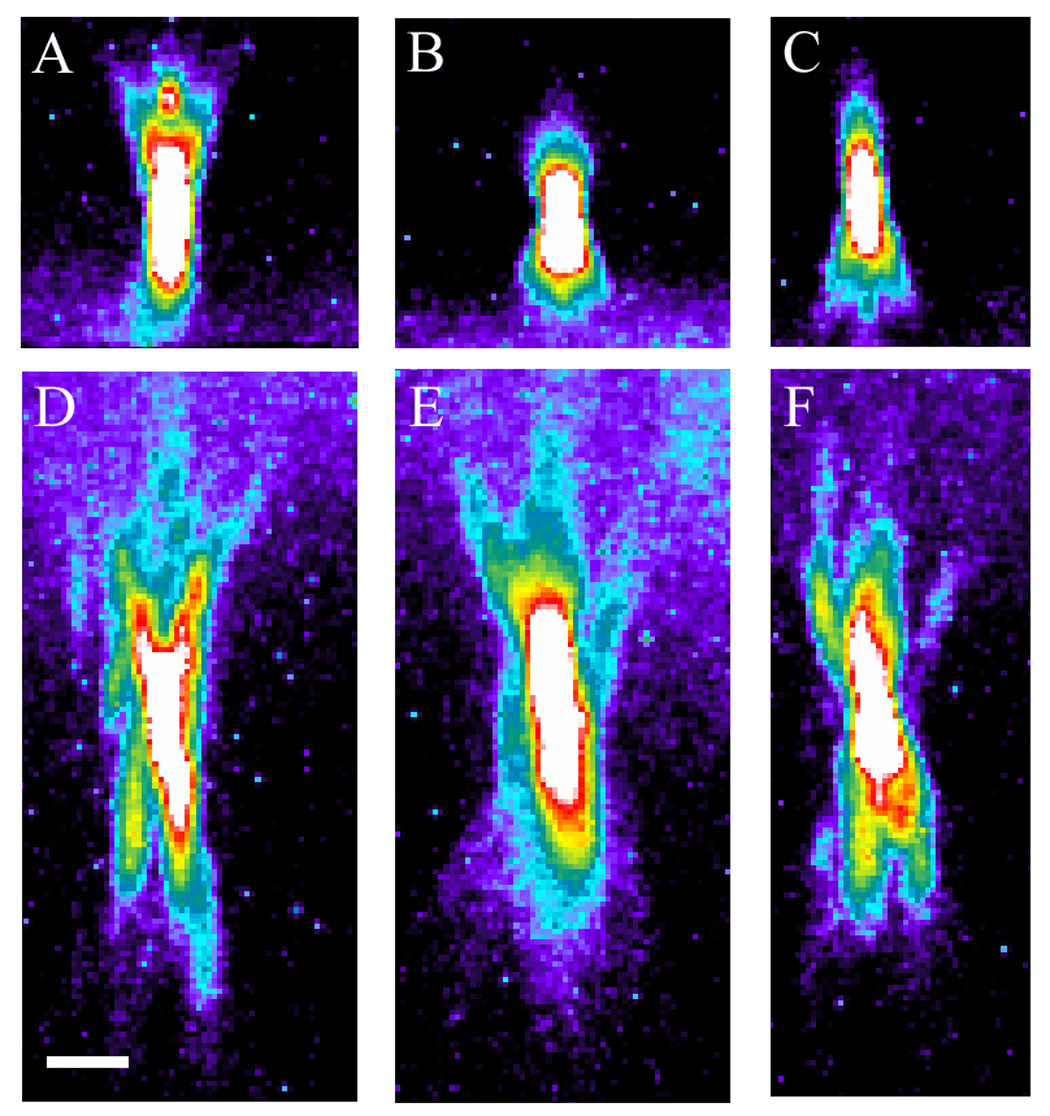
Effect of correction collar adjustments on the point spread functions of fluorescent microspheres. XZ cross section of 0.5 micron fluorescent microspheres mounted immediately below the coverslip with collar settings (A) 0.13, (B) 0.17, and (C) 0.21 mm. XZ cross section of 0.5 micron fluorescent microspheres mounted below 50 microns of fixed kidney tissue with collar settings (D) 0.13, (E) 0.17, and (F) 0.21 mm. Pseudocolor based on intensity. Scalebar = 2 microns, x=z.
In order to determine whether this approach could be used to correct for spherical aberration in multiphoton fluorescence images collected at depth in kidney tissue, we collected images of sub-resolution red fluorescent microspheres mounted under 50 microns of fixed kidney tissue. Fluorescence images were collected as in the above study, and xz cross-sectional images are shown in Figure 2 with the objective lens collar adjusted to nominal settings of 0.13 mm (D), 0.17 mm (E), and 0.21 mm (F), using a glass coverslip measured at a thickness of 0.18 mm.
Kidney tissue is predicted to have an average refractive index of approximately 1.44. [33] Thus one would expect that an optical system adjusted for an aqueous sample would generate images with negative spherical aberration. As expected, images collected with the collar adjusted to 0.17 mm, appropriate for an aqueous sample, demonstrated significant negative spherical aberration, as reflected by asymmetrical point spread functions with rings projecting up towards the objective lens. Image results were worsened when the collar was adjusted to 0.13 mm, which in effect added additional negative spherical aberration to the system. However, symmetrical point spread functions were obtained when the collar was adjusted to 0.21 mm. This adjustment, which introduced a positive spherical aberration into the system, as evidenced in Figure 2F, appeared to compensate for the negative spherical aberration induced by the tissue. Thus while the point spread functions are still significantly distorted by the tissue, the spherical aberration induced by the refractive index mismatch of the tissue can be compensated though adjustment of the correction collar of the objective lens.
In order to evaluate whether this approach could be used to ameliorate the effects of spherical aberration on signal levels collected at depth in living tissues, we conducted a quantitative analysis of multiphoton fluorescence images collected from the kidney of a living rat. Using the methods described in Dunn et al. (2002), the nuclei of renal cells were labeled via intravenous injection of Hoechst 33342, a blue-fluorescing, DNA-intercalating probe that labels all of the nuclei of the kidney independent of depth. Three-dimensional image volumes were then collected from within the kidney of the living animal, using a Bio-Rad MRC-1024MP Laser Scanning Confocal/Multiphoton scanner (Hercules, CA) attached to a Nikon Diaphot inverted microscope (Fryer Co, Huntley, IL) with a water immersion objective (Nikon, 60x Plan Apochromat, NA 1.2). Fluorescence excitation was provided by a titanium-sapphire laser (Spectraphysics, Mountain View, CA) at a wavelength of 800nm. The correction collar of the water immersion objective was adjusted to collar settings of 0.14mm, 0.17mm, and 0.19 mm. The mean Hoechst fluorescence from each plane was then measured and plotted as a function of depth, as shown in Figure 3. Similar to the previous study, the best results were obtained in the volume collected with the objective collar set to the largest value. While all three functions showed a similar shape, the images collected with a collar setting of 0.19 had a more consistent signal over a broader range of depths. In addition, signal levels were improved by approximately 50% at a depth of 35 microns. These results are again consistent with the hypothesis that the negative spherical aberration induced by using a water immersion objective to image into a sample whose refractive index is larger than that of water, can be compensated by adjusting the correction collar to introduce a positive spherical aberration into the system.
Fig. 3.
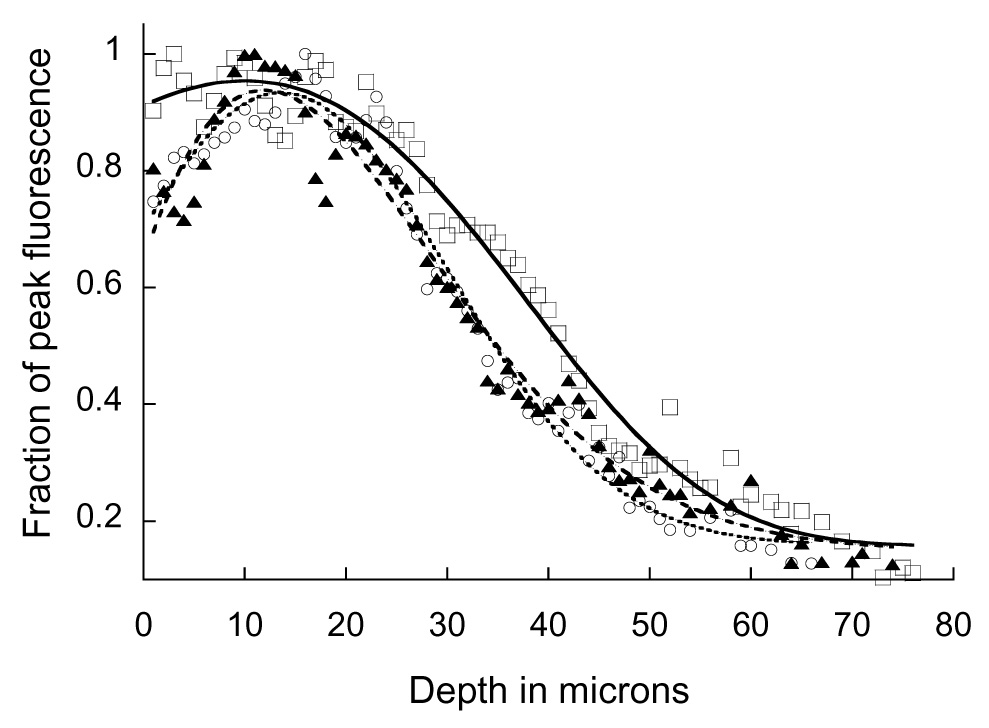
Effect of correction collar adjustment on fluorescence intensity as a function of depth in kidney tissue. Multiphoton fluorescence images of nuclei labeled with Hoechst 33342 were collected at various depths from the kidney of a living rat, using collar settings of 0.14 (▲), 0.17 (○), 0.19 mm (□). Mean fluorescence intensity of each plane is plotted, normalized to the peak of each plot. Curves included to guide the eye.
6. Conclusion
Although MPM has many advantages over confocal microscopy for deep tissue imaging, signal attenuation is still a problem limiting imaging depth to less than a millimeter. Many factors contribute to the attenuation of signal with depth, including scattering of illumination and signal, absorption of illumination and signal and optical aberrations.
Attenuation of illumination profoundly affects multiphoton fluorescence excitation, due to the quadratic dependence of fluorescence excitation on illumination power. To some degree losses of illumination can be compensated with increased laser power. However, it has been suggested that this may increase the focal volume, thereby degrading resolution. [10] The amount of power that may be added may also be limited by the laser, or more frequently by the effect of additional power on the tissue surface. One solution to this problem is to reduce the average power of the laser by reducing pulse frequency. It has been demonstrated in brain tissue that by reducing repetition frequency using a regenerative amplifier, imaging depth can be improved by two-thirds. [8] However, even in the absence of damage, the signal that may be reclaimed through increased power is limited by the quantity of fluorescence stimulated at the surface. [8, 14, 34–36] As laser power is increased to compensate for light losses to the focal point, it can reach levels sufficient to stimulate fluorescence at the surface, resulting in background fluorescence that approaches the levels of fluorescence stimulated at the deep focal point.
Previous studies have demonstrated that spherical aberration can also contribute to signal attenuation with depth in MPM. This has been indirectly demonstrated by the fact that both signal level [12, 24] and resolution [12] are significantly improved by more closely matching sample refractive index to that of the objective immersion medium.
Here we have combined analyses of point-spread functions with quantifications of signal attenuation with depth to demonstrate that spherical aberration significantly reduces signal levels collected at depth in kidney tissues. In addition, we have demonstrated that negative spherical aberration that results from using a water immersion objective to collect images from living and fixed kidney tissue can be partially corrected by introducing a compensatory positive spherical aberration into imaging system, through adjustment of the objective correction collar. The effectiveness of this approach may vary with tissue type; correction collar compensation produced a more modest benefit when applied to MPM of skin [32], perhaps due to the complex stratification of the skin. Additional methods for manipulating spherical aberration, including use of different immersion media, different thickness coverslips, and adaptive lens systems, may increase the range of tissues and depths that can be accommodated by spherical aberration compensation. The most complete (and most elaborate) solution to the problem of aberration of the focus may lie in adaptive optics systems, which have been demonstrated to improve resolution and signal level in refractive index mismatched samples [22, 25] and in living tissue [37].
Acknowledgements
This work was supported by a George M. O’Brien award from the NIH (P50-DK61594) and conducted at the Indiana Center for Biological Microscopy. The authors would like to thank Ruben Sandoval for collecting the intravital microscopy data summarized in Figure 3.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gillies RJ. In vivo molecular imaging. J Cell Biochem Suppl. 2002;39:231–238. doi: 10.1002/jcb.10450. [DOI] [PubMed] [Google Scholar]
- 2.Dunn KW, et al. Functional studies of the kidney of living animals using multicolor two-photon microscopy. Am J Physiol Cell Physiol. 2002;283(3):C905–C916. doi: 10.1152/ajpcell.00159.2002. [DOI] [PubMed] [Google Scholar]
- 3.Dunn KW, Sandoval RM, Molitoris BA. Intravital imaging of the kidney using multiparameter multiphoton microscopy. Nephron Exp Nephrol. 2003;94(1):e7–e11. doi: 10.1159/000070813. [DOI] [PubMed] [Google Scholar]
- 4.Helmchen F, Denk W. Deep tissue two-photon microscopy. Nat Methods. 2005;2(12):932–940. doi: 10.1038/nmeth818. [DOI] [PubMed] [Google Scholar]
- 5.Konig K. Multiphoton microscopy in life sciences. J Microsc. 2000;200(pt2):83–104. doi: 10.1046/j.1365-2818.2000.00738.x. [DOI] [PubMed] [Google Scholar]
- 6.Tanner GA, Sandoval RM, Dunn KW. Two-photon in vivo microscopy of sulfonefluorescein secretion in normal and cystic rat kidneys. Am J Physiol Renal Physiol. 2004;286(1):F152–F160. doi: 10.1152/ajprenal.00264.2003. [DOI] [PubMed] [Google Scholar]
- 7.Tanner GA, et al. Micropuncture gene delivery and intravital two-photon visualization of protein expression in rat kidney. Am J Physiol Renal Physiol. 2005;289(3):F638–F643. doi: 10.1152/ajprenal.00059.2005. [DOI] [PubMed] [Google Scholar]
- 8.Theer P, Hasan MT, Denk W. Two-photon imaging to a depth of 1000 microm in living brains by use of a Ti:Al2O3 regenerative amplifier. Opt Lett. 2003;28(12):1022–1024. doi: 10.1364/ol.28.001022. [DOI] [PubMed] [Google Scholar]
- 9.Yu W, Sandoval RM, Molitoris BA. Quantitative intravital microscopy using a Generalized Polarity concept for kidney studies. Am J Physiol Cell Physiol. 2005;289(5):C1197–C1208. doi: 10.1152/ajpcell.00197.2005. [DOI] [PubMed] [Google Scholar]
- 10.Zipfel WR, Williams RM, Webb WW. Nonlinear magic: multiphoton microscopy in the biosciences. Nat Biotechnol. 2003;21(11):1369–1377. doi: 10.1038/nbt899. [DOI] [PubMed] [Google Scholar]
- 11.Squirrell JM, et al. Long-term two-photon fluorescence imaging of mammalian embryos without compromising viability. Nat Biotechnol. 1999;17(8):763–767. doi: 10.1038/11698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gerritsen HC, De Grauw CJ. Imaging of optically thick specimen using two-photon excitation microscopy. Microsc Res Tech. 1999;47(3):206–209. doi: 10.1002/(SICI)1097-0029(19991101)47:3<206::AID-JEMT6>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 13.Oheim M, et al. Two-photon microscopy in brain tissue: parameters influencing the imaging depth. J Neurosci Methods. 2001;111(1):29–37. doi: 10.1016/s0165-0270(01)00438-1. [DOI] [PubMed] [Google Scholar]
- 14.Theer P, Denk W. On the fundamental imaging-depth limit in two-photon microscopy. J Opt Soc Am A Opt Image Sci Vis. 2006;23(12):3139–3149. doi: 10.1364/josaa.23.003139. [DOI] [PubMed] [Google Scholar]
- 15.Centonze VE, White JG. Multiphoton excitation provides optical sections from deeper within scattering specimens than confocal imaging. Biophys J. 1998;75(4):2015–2024. doi: 10.1016/S0006-3495(98)77643-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dunn AK, Wallace Vincent P, Coleno Mariah, Berns Michael W, Tromberg Bruce J. Influence of optical properties on two-photon fluorescence imaging in turbid samples. Applied Optics. 2000;39(7):1194–1201. doi: 10.1364/ao.39.001194. [DOI] [PubMed] [Google Scholar]
- 17.Wallace VP, Dunn Andrew K, Coleno Mariah L, Tromberg Bruce J. Two-Photon Microscopy in Highly Scattering Tissue. In: Periasamy A, editor. Methods in Cellular Imaging. Oxford University Press; 2001. pp. 180–199. [Google Scholar]
- 18.Sandoval RM, Molitoris BA. Gentamicin traffics retrograde through the secretory pathway and is released in the cytosol via the endoplasmic reticulum. Am J Physiol Renal Physiol. 2004;286(4):F617–F624. doi: 10.1152/ajprenal.00130.2003. [DOI] [PubMed] [Google Scholar]
- 19.Babbey CM, et al. Rab10 regulates membrane transport through early endosomes of polarized Madin-Darby canine kidney cells. Mol Biol Cell. 2006;17(7):3156–3175. doi: 10.1091/mbc.E05-08-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diaspro AaCJRS. Two-Photon Excitation Fluorescence Microscopy. In: Diaspro A, editor. Confocal and Two-Photon Microscopy: Foundations, Applications, and Advances. New York: Wiley-Liss, Inc; 2002. [Google Scholar]
- 21.Dunn KW, Young PA. Principles of multiphoton microscopy. Nephron Exp Nephrol. 2006;103(2):e33–e40. doi: 10.1159/000090614. [DOI] [PubMed] [Google Scholar]
- 22.Booth MJ, Wilson T. Refractive-index-mismatch induced aberrations in single-photon and two-photon microscopy and the use of aberration correction. J Biomed Opt. 2001;6(3):266–272. doi: 10.1117/1.1382808. [DOI] [PubMed] [Google Scholar]
- 23.de Grauw CJ, Frederix PLTM, Gerritsen HC. Aberrations and Penetration in In-Depth Confocal and Two-Photon-Excitation Microscopy. In: Diaspro A, editor. Confocal and Two-Photon Microscopy: Foundations, Applications, and Advances. Wiley-Liss, Inc; 2002. pp. 153–169. [Google Scholar]
- 24.Jacobsen HP, Hanninen P, Soini E, Hell SW. Refractive-index-induced aberrations in two-photon confocal fluorescence microscopy. Journal of Microscopy. 1994;176(3):226–230. [Google Scholar]
- 25.Neil MA, et al. Adaptive aberration correction in a two-photon microscope. J Microsc. 2000;200(Pt 2):105–108. doi: 10.1046/j.1365-2818.2000.00770.x. [DOI] [PubMed] [Google Scholar]
- 26.Cuccia DJ, et al. In vivo quantification of optical contrast agent dynamics in rat tumors by use of diffuse optical spectroscopy with magnetic resonance imaging coregistration. Appl Opt. 2003;42(16):2940–2950. doi: 10.1364/ao.42.002940. [DOI] [PubMed] [Google Scholar]
- 27.Doornbos RMP, Lang R, Aalders MC, Cross FW, Sterenborg HJCM. The determination of in vivo human tissue optical properties and absolute chromophore concentrations using spatially resolved steady-state diffuse reflectance spectroscopy. Physics in Medicine and Biology. 1998;44(1999):967–981. doi: 10.1088/0031-9155/44/4/012. [DOI] [PubMed] [Google Scholar]
- 28.Dong CY, et al. Performances of high numerical aperture water and oil immersion objective in deep-tissue, multi-photon microscopic imaging of excised human skin. Microsc Res Tech. 2004;63(1):81–86. doi: 10.1002/jemt.10431. [DOI] [PubMed] [Google Scholar]
- 29.Tung CK, et al. Effects of objective numerical apertures on achievable imaging depths in multiphoton microscopy. Microsc Res Tech. 2004;65(6):308–314. doi: 10.1002/jemt.20116. [DOI] [PubMed] [Google Scholar]
- 30.Hiraoka Y, Sedat JW, Agard DA. Determination of three-dimensional imaging properties of a light microscope system. Partial confocal behavior in epifluorescence microscopy. Biophys J. 1990;57(2):325–333. doi: 10.1016/S0006-3495(90)82534-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Monk C. Optical Spherical Aberration Correction. G.I.T. Imaging and Microscopy. 2004;6(3):48–49. [Google Scholar]
- 32.Lo W, et al. Spherical aberration correction in multiphoton fluorescence imaging using objective correction collar. J Biomed Opt. 2005;10(3):034006. doi: 10.1117/1.1924614. [DOI] [PubMed] [Google Scholar]
- 33.Biwas TK, Gupta AK. Retrieval of true color of the internal organ of CT images and attempt to tissue characterization by refractive index: Initial experience. Indian Journal of Radiology and Imaging. 2002;12(2):169–178. [Google Scholar]
- 34.Leray A, Mertz Jerome. Rejection of Two-photon fluorescence background in thick tissue by differential aberration imaging. Optics Express. 2006;14(22):10565–10573. doi: 10.1364/oe.14.010565. [DOI] [PubMed] [Google Scholar]
- 35.Ying J, Liu Feng, Alfano RR. Spatial distribution of two-photon-excited fluorescence in scattering media. Applied Optics. 1999;38(1):224–229. doi: 10.1364/ao.38.000224. [DOI] [PubMed] [Google Scholar]
- 36.Ying J, Liu Feng, Alfano RR. Effect of scattering on nonlinear optical scanning microscopy imaging of highly scattering media. Applied Optics. 2000;39(4):509–514. doi: 10.1364/ao.39.000509. [DOI] [PubMed] [Google Scholar]
- 37.Rueckel M, Mack-Bucher JA, Denk W. Adaptive wavefront correction in two-photon microscopy using coherence-gated wavefront sensing. Proc Natl Acad Sci U S A. 2006;103(46):17137–17142. doi: 10.1073/pnas.0604791103. [DOI] [PMC free article] [PubMed] [Google Scholar]