

Abstract
Mice infected with coxsackievirus B1 Tucson (CVB1T) develop chronic, post-viral myopathy (PVM) with clinical manifestations of hind limb muscle weakness and myositis. The objective of the current study was to establish the genetic basis of myopathogenicity in CVB1T. Using a reverse genetics approach, full attenuation of PVM could only be achieved by simultaneously mutating four sites located at C706U in the 5′ untranslated region (5′ UTR) and at Y87F, V136A, and T276A in the VP1 capsid. Engineering these four myopathic determinants into an amyopathic CVB1T variant restored the ability to cause PVM. Moreover, these same four determinants controlled PVM expression in a second strain of mice, indicating that the underlying mechanism is operational in mice of different genetic backgrounds. Modeling studies predict that C706U alters both local and long-range pairing in the 5′ UTR, and that VP1 determinants are located on the capsid surface. However, these differences did not affect viral titers, temperature stability, pH stability, or the antibody response to virus. These studies demonstrate that PVM develops from a complex interplay between viral determinants in the 5′ UTR and VP1 capsid and have uncovered intriguing similarities between genetic determinants that cause PVM and those involved in pathogenesis of other enteroviruses.
Keywords: coxsackievirus, enterovirus, post-viral myopathy, myositis, muscle, weakness, inflammation, pathogenesis, viral genetics, VP1 capsid
Introduction
The type B coxsackieviruses (CVB, serotypes 1–6) are members of the enterovirus genus in the family Picornaviridae. CVB are nonenveloped viruses comprised of an icosahedral capsid enclosing a single-stranded, positive-sense RNA genome of approximately 7.4 Kb (Muckelbauer et al., 1995). Infection begins with virus binding to the coxsackievirus and adenovirus receptor (CAR), the primary cellular receptor for CVB (Bergelson et al., 1997; Carson et al., 1997; Tomko et al., 1997). CAR facilitates CVB internalization and uncoating through interactions with the canyon surrounding the 5-fold axis of symmetry on the viral capsid (He et al., 2001). A variety of alternate or co-receptors have also been described including decay accelerating factor (DAF; CD55) (Bergelson et al., 1995) and heparan sulfate (Zautner et al., 2003). Following cell entry, viral RNA is translated through a cap-independent mechanism involving the viral internal ribosome entry site (IRES) and host proteins (reviewed by (Belsham and Sonenberg, 2000)). The resulting polyprotein is processed through a series of cleavages by viral proteases 2A and 3CD into four capsid proteins encoded by genomic region P1 and seven nonstructural proteins in regions P2 and P3. Replication ensues via production of negative-strand RNA that serves as a template for the synthesis of large amounts of positive strands to be translated or encapsidated into new virions. The 5′ UTR plays an important role in controlling the processes of viral replication and translation (reviewed by (Bedard and Semler, 2004)).
CVB infections are one of the most frequent causes of myocarditis and its sequela, dilated cardiomyopathy, and may also be a factor in the development of type I diabetes (reviewed by (Hyoty, 2002; Tam, 2006)). Consistent with their known myotropism, CVB and related enteroviruses have also been implicated in pathogenesis of chronic muscle diseases such as chronic fatigue syndrome, fibromyalgia, and myositis ((Chia and Chia, 2007; Clements et al., 1995; Douche-Aourik et al., 2003) and reviewed by (Chia, 2005)). Although they are primarily cytopathic, enteroviruses persist in some chronic fatigue syndrome patients (Chia and Chia, 2007; Galbraith et al., 1997) in a form where the ratio of positive and negative viral RNAs is approximately equal (Cunningham et al., 1990). Despite these and other supportive studies, a number of investigations have failed to link these viruses to chronic myositis or fatigue syndromes (Leon-Monzon and Dalakas, 1992; Lindh et al., 1996; McArdle et al., 1996), raising questions regarding differences in the timing, geographic location, and myopathic potential of enterovirus infections.
The possibility that CVB infection is a factor in the pathogenesis of chronic myopathies is demonstrated by an experimental model where acute cytopathic infection of newborn mice with CVB1T causes long-term sequelae including inflammation of proximal hind limb skeletal muscle and changes in gait, referred to as weakness (Ray et al., 1979; Strongwater et al., 1984). Expression of clinical weakness is asymmetric, associated with increased muscle necrosis, and does not vary once established (Jongen et al., 1996; Tam et al., 1994). By comparison, myositis peaks around 1 month post-infection (PI) and gradually dissipates by 6 months (Tam et al., 1994). Affected muscle displays a characteristic histopathology that includes focal mononuclear inflammatory cell infiltrates, permanent loss of muscle mass with fatty replacement, myofiber hypertrophy, endomysial and perimysial fibrosis, and increased numbers of mast cells (Tam et al., 1994). Infectious virus is cleared by 2 weeks PI but viral RNA persists in the hind limb muscle of some mice for as long as 12 months (Tam et al., 1994). At 1 month PI, viral RNA can be seen in areas coincident with inflammation and tissue damage as well as in histologically normal areas as a “silent” infection (Tam et al., 1991; Tam et al., 1994). Similar to patient studies, roughly equivalent amounts of positive and negative strands persist in a double-stranded conformation, indicating that viral replication is altered (Tam and Messner, 1999).
Successive passage of CVB1T through BGMK cells produces variants that cause a robust acute infection but are attenuated for PVM, indicating that PVM is controlled by distinct viral determinants (Tam and Messner, 1997). Sequence comparisons suggest that five mutations—C706U in the 5′ UTR, Y87F, V136A, and T276A in capsid protein VP1, and K53E in the viral 3C protease—are prime candidates for PVM attenuation (Tam et al., 2003b). Additional genetic differences include six translationally silent mutations that segregate with myopathogenicity. In the current study, we conclusively identified the viral determinants responsible for PVM and delineated their relationship to pathogenesis of chronic weakness and inflammation. Predicted structural changes caused by mutations in the 5′ UTR and VP1 capsid have provided further insight into how enteroviruses evolve to produce a diverse spectrum of diseases (reviewed by (Tracy et al., 2006)).
Results
Myopathogenicity of CVB1T is encoded by viral genetic determinants in the 5′ UTR and VP1
Previous studies of viral chimeras constructed from cloned viruses MP1.24 and AMP2.17 indicated that more than one region of the viral genome contributes to the development of chronic hind limb weakness and inflammation following infection (Tam et al., 2003b). In addition, comparative sequencing of MP1.24, AMP2.17, additional virus clones, and the parental strains from which they were derived identified eleven mutations that segregate with the myopathic or amyopathic phenotype of the virus including one in the 5′ UTR, four that cause coding changes, and six that are translationally silent. The initial objective of the current study was to determine if the ability of MP1.24 to cause PVM was strictly encoded within five determinants located at C706U in the 5′ UTR, Y87F, V136A, and T276A in the VP1 capsid protein, and K53E in the 3C protease. An attenuation strategy was employed in which the MP1.24 infectious myopathic clone was mutated to the amyopathic nt at one or more of the five putative determinants. Myopathogenicity was based on the severity of clinical hind limb weakness and histological grading of myositis in the hamstring muscle at 1 month after infection (Fig. 1), which is on average two weeks after infectious virus can be recovered. AMP2.17-infected mice resembled the uninfected control except for a few cases with grade 1 weakness or grade 1 inflammation, consistent with previous comparisons of myopathic and attenuated strains (Tam and Messner, 1997; Tam et al., 2003b). ANOVA followed by post-hoc comparisons of each mutagenized construct with the myopathic MP1.24 and amyopathic AMP2.17 prototype viruses was used to characterize the pathogenic phenotype of each mutant as either myopathic, amyopathic, or intermediate (Fig. 2A). This classification scheme is described in further detail in the Fig. 2 legend.
Fig 1.
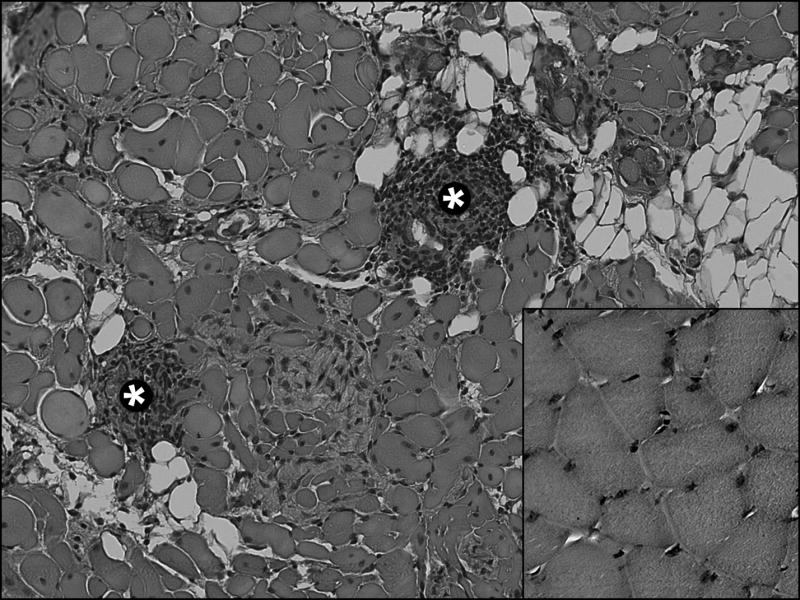
Histopathology of myositis in PVM at 1 month PI. Grade 2 myositis caused by MP1.24 infection is indicated by the presence of medium-sized mononuclear cell infiltrates (*). Other signs of pathology compared to uninfected muscle (shown in inset, lower right) include fatty replacement, interstitial fibrosis, changes in fiber diameter and shape, and the presence of central nuclei (original magnification x200).
Fig 2.

PVM phenotype in CD-1 mice infected with different mutated constructs of CVB1T at 1 month PI. (A) Statistical analysis by ANOVA with pairwise Bonferroni-Dunn post-hoc comparisons was used to compare chronic hind limb weakness and muscle inflammation grades for mutated viral constructs with the MP1.24 and AMP2.17 controls. This analysis was the basis for categorizing each construct as either myopathic (different from AMP2.17 but not MP1.24), amyopathic (different from MP1.24 but not AMP2.17), or intermediate (different than both MP1.24 and AMP2.17). The dotted lines demarcate these statistically significant differences, shown as sectors I–IX. For example, sector III contains fully myopathic viruses, sector I contains viruses that were myopathic for inflammation but amyopathic for weakness, and sector VII contains constructs that were fully attenuated. (B) Expression of weakness and inflammation in individual mice infected with MP1.24. The number of mice at each combination of grades, which extends from 0 to 3 for myositis and 0 to 6 for weakness, is shown.
Mutation of MP1.24 at all five positions in virus construct A-AAA-A (see Table 1 legend for description of viral genotype scheme) attenuated weakness and inflammation to a degree that was comparable to AMP2.17 (Table 1A and Fig. 2A), indicating that myopathogenicity was encoded within these five candidate determinants. Furthermore, mutation of the three VP1 determinants in M-AAA-M fully attenuated its ability to cause weakness. These capsid mutations also reduced but did not fully attenuate myositis, placing M-AAA-M in sector IV and indicating that at least one additional determinant was involved in pathogenesis of PVM. Both A-MMM-M and M-MMM-A were fully myopathic, and the identity of the additional determinant was resolved by engineering C706U into the M-AAA-M construct. The resulting A-AAA-M virus (sector VII) was further attenuated for inflammation compared to M-AAA-M (P = 0.003) and comparable to A-AAA-A (P > 0.05), suggesting that C706 was involved in the development of myositis even though no effect was observed for the singly-mutated A-MMM-M construct (sector III). The realization that there might be cooperative effects prompted us to examine one additional construct, M-MMA-A, because previous chimeras indicated the presence of a myopathic determinant downstream of nt 3200 (P250 in VP1) (Tam et al., 2003b) and yet, in the current study, M-MMM-A was not attenuated. Partial attenuation of M-MMA-A (sector V) was similar to what was previously observed for the viral chimera and suggested that T276A at nt 3278, either alone or in conjunction with K53E at nt 5510, was responsible.
Table 1.
Chronic hind limb weakness and proximal skeletal muscle inflammation in mice infected with CVB1T mutants
| Weakness
|
Inflammation
|
|||
|---|---|---|---|---|
| Virus | Gradea (mean ± SEM) | % (no. positive/total) | Grade (mean ± SEM) | % (no. positive/total |
| Controls: | ||||
| MP1.24 | 2.1 ± 0.2 | 72 (69/96) | 1.7 ± 0.1 | 88 (57/65) |
| AMP2.17 | 0.2 ± 0.04* | 16 (13/79)* | 0.1 ± 0.03* | 5 (4/74)* |
| PBS | 0* | 0 (0/76)* | 0* | 0 (0/38)* |
| Mutant Virus Genotype:b | ||||
| (A) | ||||
| A-AAA-A | 0.1 ± 0.1* | 10 (2/19)* | 0.1 ± 0.1* | 10 (2/19)* |
| M-AAA-M | 0* | 0 (0/25)* | 0.8 ± 0.2* | 44 (11/25)* |
| A-MMM-M | 1.7 ± 0.2 | 72 (34/47) | 1.8 ± 0.2 | 85 (33/39) |
| M-MMM-A | 1.8 ± 0.3 | 72 (29/40) | 1.7 ± 0.2 | 89 (17/19) |
| A-AAA-M | 0* | 0 (0/27)* | 0.1 ± 0.06* | 12 (3/26)* |
| M-MMA-A | 1.0 ± 0.3* | 41 (11/27)* | 0.7 ± 0.2* | 41 (11/27)* |
| (B) | ||||
| M-AMM-M | 0* | 0 (0/28)* | 0.7 ± 0.2* | 52 (13/25)* |
| A-AMM-M | 0.3 ± 0.1* | 21 (6/29)* | 0.5 ± 0.2* | 38 (8/21)* |
| M-MAM-M | 0.2 ± 0.1* | 13 (4/30)* | 1.4 ± 0.2 | 77 (20/26) |
| A-MAM-M | 0.3 ± 0.1* | 25 (7/28)* | 1.2 ± 0.2 | 83 (19/23) |
| M-MMA-M | 0.9 ± 0.2* | 50 (14/28) | 0.7 ± 0.2* | 50 (14/28)* |
| A-MMA-M | 1.0 ± 0.2* | 58 (18/31) | 0.6 ± 0.2* | 35 (11/31)* |
| (C) | ||||
| (M-MMM)-A | 1.5 ± 0.3 | 68 (17/25) | 1.3 ± 0.2 | 79 (19/24) |
Viruses causing weakness or inflammation that differed from MP1.24 at 1 month PI are indicated (*). ANOVA with Bonferroni-Dunn post-hoc testing was used to compare grades and Fisher’s exact test was used to compare incidence (adjusted for fifteen comparisons, level of significance α = 0.003).
The viral “genotype” at each of five positions is represented by an “M” for myopathic and an “A” for amyopathic. The genotype is shown in the order 5′ UTR-VP1-3C or C706U-Y87F V136A T276A-K53E, where the preceding nucleotide or residue is found in MP1.24 and the following one is in AMP2.17. Viral constructs shown in (A) and (B) were generated by site-directed mutagenesis of the MP1.24 clone to the amyopathic determinant. The revertant in (C) was produced by mutagenesis of AMP2.17 to myopathic determinants (enclosed by parentheses) in the 5′ UTR and VP1.
Individual viral determinants and their effect on chronic weakness and inflammation
Additional studies were performed to clarify the role of VP1 capsid determinants by mutating each one individually to the amyopathic form (Table 1B and Fig. 2A). Because complete attenuation of inflammation was observed only when C706U was combined with three VP1 mutations in construct A-AAA-M, each VP1 determinant was mutated singly and in combination with C706U to identify interacting determinants. Little to no weakness was observed in mice infected with MP1.24 containing Y87F or V136A in the M-AMM-M or M-MAM-M constructs, respectively. By comparison, mutation at T276A in the M-MMA-M construct only partially attenuated weakness development, and C706U had no effect on weakness when combined with any of the three capsid mutations. The pattern of involvement for individual determinants of weakness partially overlapped those responsible for myositis. As predicted by the M-AAA-M construct, full attenuation of chronic myositis could not be achieved by attenuating any one of the three capsid determinants individually. M-AMM-M and M-MMA-M showed partial attenuation of myositis while V136A in M-MAM-M had no effect. A further reduction in myositis development occurred by pairing individual capsid mutation Y87F with C706U in A-AMM-M but not in A-MAM-M or A-MMA-M. C706U reduced the mean inflammation grade from 0.7 for M-AMM-M to 0.5 for A-AMM-M. This was sufficient to shift A-AMM-M into the statistically-determined amyopathic category, even though the inflammation grades did not differ significantly when M-AMM-M and A-AMM-M were compared directly (P > 0.05) (Fig. 2A sectors IV and VII). These data reinforced a role for C706U in myositis and suggested that Y87F was the interacting determinant responsible for enhanced attenuation of the A-AAA-M construct. Noting the trend in Fig. 2A, it is also possible that the full effect of C706U is manifest in combination with all three capsid mutations as observed for A-AAA-M in sector VII. Additional comparisons of M-MMA-M with M-MMA-A reinforced the previous assumption that K53E was non-attenuating since these constructs caused similar degrees of weakness and inflammation (P > 0.05), resulting in an intermediate phenotype for both.
In the final analysis four determinants, C706, Y87, V136, and T276 were implicated in pathogenesis of PVM. To confirm their involvement, AMP2.17 was engineered to contain mutations U706C, F87Y, A136V, and A276T. Construct (M-MMM)-A reverted to a myopathic phenotype, further establishing the importance of these determinants (Table 1C and Fig. 2A sector III). The inflammation and weakness scores for (M-MMM)-A were slightly lower than other myopathic constructs, which could be due to experimental variation or may indicate that some additional determinant is lacking in the AMP2.17 genome. The incidence of affected mice mirrored what was observed by grading the severity of weakness or inflammation (Table 1). One exception was the incidence of weakness in constructs containing T276A either alone or paired with C706U or K53E. The percentage of weak mice in these three groups ranged from 41% to 58%, which was less than the 72% observed for the MP1.24 control but only significant for construct M-MMA-A. T276 was not a strong determinant of weakness which may explain its marginal effect on incidence. For inflammation, the incidence was completely concordant with the mean severity scores for each group. Notably, the incidence for A-AAA-M was 12% compared to 44% for M-AAA-M, further supporting the involvement of C706 in pathogenesis. Overall, the mapping studies indicated that Y87F was highly attenuating for weakness and moderately attenuating for inflammation unless combined with C706U. V136A was unique in that it caused strong attenuation of weakness but had no effect on inflammation, while T276A emerged as a moderate attenuator of both.
Viral determinants distinguish between pathogenesis of weakness and inflammation
Teasing out the differential involvement of individual viral determinants showed that two, V136 and C706, had distinct effects on the pathogenesis of weakness and inflammation, respectively. This relationship is apparent in Fig. 2A, where V136A (M-MAM-M) stands alone in sector I for weakness attenuation. In contrast, the absence of viral constructs in sectors VIII and IX indicates there is no combination of mutations that promotes weakness in the absence of inflammation. The distribution of disease phenotypes is recapitulated in mice infected with MP1.24 where 15 of 65 mice developed some degree of myositis in the absence of weakness, and only 4 mice showed low grade weakness in the absence of inflammation (Fig. 2B). The remaining three myopathic viruses in sector III showed a similar distribution (data not shown).
Attenuating determinants do not alter acute virulence, viral stability, or the host antibody response to virus
Acute virulence was evaluated as the percentage of mice that survived infection and the ability of virus to replicate in proximal muscle at 6 days PI (Table 2). None of the mutations caused significant differences in animal mortality or the ability of virus to replicate in muscle, similar to what has previously been observed for the prototypes MP1.24 and AMP2.17 or the original parental strains of viruses used to generate these clones, which were tested at multiple time points over the 14-day infectious period (Tam and Messner, 1997; Tam et al., 2003b). IgG antibody titers were also comparable, and thus the ability to react with MP1.24 as a target in the ELISA did not appear to be affected by the VP1 mutations. Potential changes in the physical stability of the capsid were evaluated at three different temperatures and at lowered pH (Figs. 3A and 3B). Decay of infectivity proceeded more rapidly at higher temperatures but was comparable for MP1.24 and AMP2.17. Moreover, the A-AAA-M construct behaved similarly when tested at 39°C. The characteristic stability of enteroviruses at pH 4.5 was also unaltered (Fig. 3B). To further examine possible effects of the capsid mutations on antibody recognition, serum from a randomly-selected subset of mice infected with either MP1.24 or AMP2.17 was evaluated for cross-reactivity. Serum antibodies from individual mice reacted equally well with AMP2.17 or MP1.24, whether by ELISA (Fig. 3C) or a functional virus neutralization assay (Fig. 3D), regardless of which virus was used for infection. Taken as a whole, these experiments show that the attenuating mutations do not interfere with virulence during the acute infection, do not cause capsid instability, and do not affect the specificity of antibodies involved in viral clearance.
Table 2.
Mortality, viral replication, and host antibody response to CVB1T mutants
| Survival (%)a | Virus titerb (Log TCID50/gm muscle) | Anti-viral antibody (Log U/ml) | |
|---|---|---|---|
| Controls: | |||
| MP1.24 | 84 | 7.4 ± 0.3 | 3.8 ± 0.03 |
| AMP2.17 | 89 | 7.5 ± 0.4 | 3.6 ± 0.03 |
| PBS | 91 | NDc | ND |
| Mutant Virus Genotype: | |||
| (A) | |||
| A-AAA-A | 100 | 6.7 ±0.5 | 3.6 ± 0.06 |
| M-AAA-M | 84 | 7.5 ± 0.5 | 3.5 ± 0.07 |
| A-MMM-M | 79 | 7.6 ± 0.3 | 3.7 ± 0.03 |
| M-MMM-A | 96 | 7.6 ± 0.2 | 3.6 + 0.03 |
| A-AAA-M | 83 | 7.1 ± 0.5 | 3.6 ± 0.07 |
| M-MMA-A | 97 | 8.1 ± 0.4 | 3.7 ± 0.05 |
| (B) | |||
| M-AMM-M | 100 | 7.5 ± 0.2 | 3.7 ± 0.04 |
| A-AMM-M | 100 | 6.9 ± 0.5 | 3.6 ± 0.02 |
| M-MAM-M | 100 | 7.1 ± 0.5 | 3.8 ± 0.03 |
| A-MAM-M | 100 | 6.9 ± 0.4 | 3.7 ± 0.02 |
| M-MMA-M | 97 | 6.5 ± 0.4 | 3.6 ± 0.04 |
| A-MMA-M | 92 | 6.4 ± 0.3 | 3.8 ± 0.04 |
| (C) | |||
| (M-MMM)-A | 87 | 6.8 ± 0.4 | 3.7 ± 0.04 |
Survival is the percentage of mice that survived acute infection during the first two weeks PI. Log rank comparisons with the PBS control were not significant (P > 0.05).
Values for virus titer (at 6 days PI) and anti-viral antibody (at 1 month PI) are presented as the mean ± 1 SEM. ANOVA performed on virus titers or anti-viral antibodies was not significant (P > 0.05).
ND, none detected.
Fig 3.

Viral stability and specificity of the antibody response to infection. Thermal stability of infectious virus was determined at 37°C, 39°C (A), and 47°C (B). Acid stability was determined at a pH of 4.5 (B). Serum from mice at 1 month PI that were infected with either MP1.24 or AMP2.17 was evaluated for reactivity to either virus in an ELISA (C) or in a viral neutralization assay (D). Regression analysis indicated no difference between the slopes of the fitted lines in (C) or (D) (P > 0.05). Control serum from uninfected mice was negative in both the ELISA and neutralization assay (data not shown).
Myopathic determinants control pathogenesis of PVM in BALB/c mice
Several inbred mouse strains are susceptible to CVB1T-induced PVM including A.BY/Sn, BALB/c, and DBA/2 (Tam and Messner, 1996). CD-1 mice were used in the current experiments because they were employed in earlier studies that implicated multiple determinants (Tam et al., 2003a), and switching to an inbred strain would run the risk of not being able to identify all the viral determinants. However, the question remained as to whether myopathic determinants that control PVM in CD-1 mice also dictate disease expression in other susceptible strains. BALB/c mice infected with 10, 20, or 40 TCID50 of MP1.24 showed a dose-dependent expression of PVM whereas no signs of PVM were apparent following infection with either 20 or 40 TCID50 of AMP2.17, indicating that the same determinants might be involved (Fig. 4A). The lack of PVM in mice infected with A-AAA-M and full expression of PVM in mice infected with (M-MMM)-A suggests that the same four determinants, or at the very least a subset, also control disease in BALB/c mice. As observed for CD-1 mice, virus constructs caused similar acute infections measured by virus titers in muscle and the presence of circulating antibody (data not shown). Infection with MP1.24 caused a phenotype distribution similar to that in Fig. 2B, with most mice affected by both weakness and inflammation but some displaying inflammation in the absence of weakness (Fig. 4B). Chi-square analysis indicated that this distribution did not differ between CD-1 and BALB/c mice (P = 0.2). Thus, determinants mapped in outbred CD-1 mice were equally pathogenic in BALB/c mice, indicating that the mechanism is closely tied to the genetics of the virus and is not restricted by differences in the genetic background of these two host strains.
Fig 4.

PVM phenotype of selected viral constructs in BALB/c mice at 1 month PI. (A) BALB/c mice were infected with the indicated dose (TCID50) of virus and evaluated for PVM at 1 month PI (N ≥ 9). The mean grades for weakness (hatched bars) and inflammation (solid bars) are shown (ND, none detected). At an infectious dose of 40 TCID50, weakness and inflammation scores for (M-MMM)-A were significantly different than AMP2.17 and scores for A-AAA-A differed from MP1.24 (P <0.0001). (B) Expression of weakness and inflammation in individual BALB/c mice infected with 20 or 40 TCID50 of MP1.24. The number of mice at each combination of grades is shown.
Attenuating mutations cause predicted changes in RNA structure and the capsid surface
Computer modeling of the CVB1T 5′ UTR predicted that C706U exerts local as well as long range effects on RNA secondary structure (Fig. 5). The models are presented as circular Feynman diagrams to facilitate the comparison of long range structural changes. Alignment with the current domain model of the CVB3 5′ UTR which extends from nt 1–647 (Bailey and Tapprich, 2007) but does not include the downstream spacer region indicated that domains I, II, and VI were unaffected by C706U. The most noticeable effect was observed locally with disruption of a predicted stem loop that is smaller but analogous to stem loop K (SLK) described by Yang et al. (Yang et al., 1997) and extends from nt 670–708 in MP1.24. C706U resulted in a smaller stem and hairpin loop at nt 680–693 followed by a large unpaired region spanning nt 702–732. C706U also affected long range interactions, causing new pairings between nt 641–701 and nt 229–247 in domains III and IV as well as changes that did not directly involve SLK. These included altered pairing between the 5′ basal region of domain IV (nt 250–260) with the basal stem of domain V (nt 453–462) and a new stem loop formed by internal pairing of nt 424–451, both of which involve sequences in the 5′ end of the IRES, as well as a larger stem loop upstream within domain III.
Fig 5.
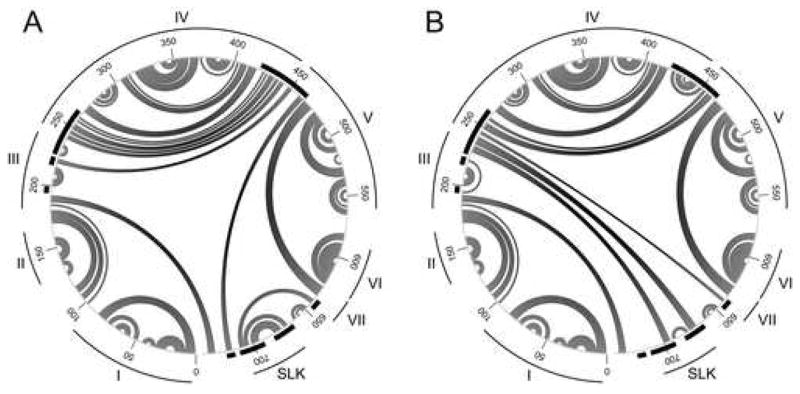
Predicted effects of the C706U determinant on RNA secondary structure of the 5′ UTR. Secondary structures of the 5′ UTR for (A) MP1.24 (dG = −244.7 kcal/mol) and (B) MP1.24 containing C706U (dG = −243.5 kcal/mol) are shown in a circular Feynman representation with paired nt connected by arcs and numbering at 50 nt intervals. Domain structures I–VII and the predicted SLK region are indicated. Regions where pairing is altered by C706U are indicated by black bars around the circle periphery.
Alignment and homology modeling of the CVB1T capsid sequences with the three dimensional coordinates of CVB3 indicated that all three VP1 determinants are likely to be exposed on the capsid surface (Fig. 6). Each of the three mutations reduced the length of the side chain, where it could potentially affect exposure of the determinant and interactions with the viral surface. Y87F is located in the βC chain two residues downstream of the BC loop and exposed on the northern edge of the CAR-binding footprint (He et al., 2001; Muckelbauer et al., 1995). V136A is located near the carboxy terminus of the DE loop, close to the five-fold axis of symmetry. This position does not appear as a surface residue in the CVB3 road map (Muckelbauer et al., 1995), but since residue 135 is visible, V136A might also be accessible (Michael Rossmann, personal communication). T276A is positioned three residues upstream of the viral 2A protease self-cleavage site and closer to the 2-fold axis of symmetry in an area rich in VP3 residues.
Fig 6.

Homology modeling of VP1 capsid determinants. A model of the capsid protomer containing VP1 (white), VP2 (light blue), VP3 (dark blue), and VP4 (yellow) was generated from the atomic structure of CVB3 using the program Modeller. The three VP1 determinants are indicated in red for (A) MP1.24 and (B) MP1.24 containing Y87F, V136A, and T276A.
Discussion
Neuromuscular disease caused by the type B coxsackieviruses has been recognized since it was first described in newborn mice infected with nonpolio enteroviruses (Godman et al., 1952; Melnick et al., 1949). While these early studies characterized acute cytopathology, our attention has focused on the sequelae of proximal muscle inflammation and hind limb weakness that persist following sublethal infection as a model of chronic PVM. In the current study, we identified the viral determinants of CVB1T that control development of chronic PVM. Mice were evaluated at 1 month PI which is on average two weeks after infectious virus can be cultured but at a time when viral RNA still persists in skeletal muscle, heart, brain, and spinal cord (Tam et al., 1994). Four viral determinants, one in the 5′ UTR at C706 and three in the VP1 capsid gene at Y87, V136, and T276, control chronic PVM development. Myopathogenicity of the (M-MMM)-A revertant of AMP2.17 further substantiates these findings and makes it unlikely that pathogenesis is tied to any of the six silent mutations previously identified by sequence comparisons of CVB1T strains (Tam et al., 2003a).
The location of PVM determinants underscores the importance of the 5′ UTR and VP1 capsid structure in enterovirus pathogenesis. In the 5′ UTR, predicted local effects of C706U include disrupting the basal stem of SLK, located downstream of the IRES core at nts 432–639 and upstream of the authentic AUG start codon at nt 743 (Liu et al., 1999). The 40s ribosomal subunit binds to the pyrimidine-rich Shine-Dalgarno-like sequence at nts 566–577 and translocates, most likely by scanning, through SLK to the AUG initiation codon (Yang et al., 2003). Structural integrity of this spacer region may be important for interactions with trans-acting regulatory factors such as La protein and polypyrimidine-tract binding protein (PTB) (Bhattacharyya and Das, 2005; Hellen et al., 1994). Taking into consideration the generally-accepted structure of domains I–VI (Bailey and Tapprich, 2007; Skinner et al., 1989; Zell and Stelzner, 1997), C706U is also predicted to cause new long-range pairings in the basal regions of domains III, IV, and V. Changes in domain III pairing include the highly conserved 5-mer 5′-232CGUUA236-3′ containing the U234C determinant of cardiovirulence attenuation (Tu et al., 1995). New sites of pairing in domain V overlap with the PTB binding site located upstream of C472U, a determinant of poliovirus attenuation (Guest et al., 2004; Gutierrez et al., 1997). Phenotypic expression of these attenuating mutations is often host cell specific, with little to no effect observed in highly permissive cell lines. Thus, the fact that amyopathic CVB1T strains are competent for acute replication in BGMK, HeLa, Neuro-2a, G8, or C2C12 cells, or in skeletal muscle (unpublished results and (Tam and Messner, 1997; Tam et al., 2003b)), does not preclude the possibility that C706U modulates the expression of immunostimulatory viral proteins involved in chronic myositis.
A shared feature of all three VP1 capsid determinants is that they appear to be exposed on the virion surface. Despite this and their proximity to known antigenic sites, there is no indication that B cell recognition and antibody production are affected by the mutations. The conserved anti-parallel eight-stranded β-barrel pocket of VP1 provides structural scaffolding whereas the loops possess greater natural variability. The BC loop of CVB3 is relatively short (residues 81–85), flanking the rim of the CAR-binding canyon (He et al., 2001). In this position, conversion of a hydrophilic to a hydrophobic residue at Y87F could have important consequences for CAR binding. In poliovirus 2-Lansing, the BC loop of VP1 is a determinant of neurovirulence in mice (Martin et al., 1988; Murray et al., 1988), where changes in flexibility affect its contact with the viral receptor (Lentz et al., 1997). Y87F is also located within two residues of a determinant linked to extended receptor usage by CVB2 (Polacek et al., 2005). Thus, Y87F is a strong candidate for altered receptor binding that is linked to pathogenesis. V136A is located in the DE loop which sits close to the fivefold axis of symmetry. Although DE loop interactions help stabilize the viral pentamer, attenuating mutations did not destabilize the viral capsid at several temperatures or low pH. Major determinants of attenuation have been mapped within the DE loop of poliovirus (Ren et al., 1991) and CVB4 (Caggana et al., 1993). Together, the BC and DE loops form a discontinuous neutralizing antigenic site in poliovirus, indicative of their close physical association (Wiegers et al., 1989). While Y87 and V136 do not appear to be part of a neutralizing antibody epitope in CVB1T, their potential for interaction may explain their interdependence and activity as a compound determinant of weakness.
T276A is located at the carboxy-terminus of VP1, three residues upstream of the VP1/2A self-cleavage site (Mulders et al., 2000). In CVB1T, this sequence is ‘ITTT/G’ with T276A occupying the P3 position. Presence of a hydrophilic amino acid at P3 is highly conserved in 2A substrates that contain a T at P2 (Muto et al., 2006). Conversion of a T276 to a hydrophobic alanine might therefore be expected to alter 2A protease activity with downstream effects on viral RNA stability, translation, and negative strand synthesis (Jurgens et al., 2006) or perhaps dystrophin cleavage (Badorff et al., 1999). A second relevant feature of T276A is its possible involvement in DAF binding, which is associated with CVB3 virulence (Stadnick et al., 2004). The DAF-binding footprint on echovirus 7 involves three regions, one of which contains VP1 residues from the carboxy terminus close to the area where T276A is exposed (He et al., 2002). Changes in DAF binding would not be unexpected, considering that the amyopathic parent AMP2 was produced by thirty sequential passages through BGMK cells (Huang et al., 2002; Tam and Messner, 1997).
These studies support a mechanism where multiple viral determinants act in concert to produce full-blown PVM. Our working model, though speculative, proposes that inflammation is driven by T cell recognition of immunodominant epitopes encoded by capsid regions containing Y87 and T276, consistent with the known involvement of T cells and MHC haplotype in PVM (Tam and Messner, 1996; Ytterberg et al., 1988). At least one of these epitopes must also be active in BALB/c (H-2d) mice. Equivalent antibody responses further imply that these are not B cell or T helper cell epitopes, focusing attention on cytotoxic T cells as possible effectors. Because of its position in the 5′ UTR, C706 may act as a modulator of chronic myositis by controlling the expression of pathologic VP1 epitopes, which conforms with the localization of inflammatory cells around muscle fibers containing persistent viral RNA (Tam et al., 1991). Promotion of myositis by C706 might be less prominent when T cell epitopes are strong yet play a larger role when they are weak, accounting for levels of inflammation that are high for A-MMM-M but intermediate for M-AAA-M. Notably, muscle inflammation is linked to but not sufficient for weakness development as demonstrated by M-MAM-M. The location of Y87, V136, and T276 in sites associated with receptor binding suggests that the inflammatory response could influence weakness indirectly through upregulation of CAR (Ito et al., 2000) or other secondary receptors on satellite cells, causing enhanced cytopathogenicity and a loss of regenerative capacity that leads to permanent myofiber loss, fatty replacement, and fibrosis (Tam et al., 1994). These studies reinforce and extend previous work, linking pathogenesis of weakness and inflammation to four viral genetic determinants that point to immunogenicity and viral tropism as prime candidates for further mechanistic studies of PVM.
Materials and Methods
Viruses and cell culture
MP1.24 (AY186745) and AMP2.17 (AY186747) are infectious cDNA clones of viruses MP1 and AMP2, respectively, that were derived from the original CVB1T stock SY8 and sequenced as described previously (Tam and Messner, 1997; Tam et al., 2003b). BGMK cells were cultured in DMEM (Mediatech, Herndon, VA) containing 10% FBS (Atlanta Biologicals, Norcross, GA)(DMEM-10) at 37°C in a humidified atmosphere containing 5% CO2. Virus was prepared by transfecting 1 μg intact plasmid in serum-free DMEM into BGMK cells using 10 μl Effectene (Qiagen, Valencia, CA) according to the manufacturer’s protocol. Virus was harvested when the culture attained 100% CPE by freeze-thawing the plate three times followed by centrifugation at 2500g for 20 minutes. Virus-containing supernatants were stored at −80°C. Titers of viruses used for injection were determined by adding two-fold dilutions of virus to 1 × 104 BGMK cells per well in a 96-well plate. Eight replicate wells were plated for each virus dilution, and the TCID50/ml was calculated using the 50% end point (Reed and Muench, 1938). Titers of virus recovered from muscle were determined similarly but using ten-fold dilutions of clarified muscle homogenate.
Mice
Specific pathogen-free ICR (CD-1) mice were purchased at late-term gestation (Harlan, Indianapolis, IN), and BALB/c mice (Jax, Bar Harbor, ME) were bred in-house. Dams with litters were housed in BSL-2 barrier conditions in the Research Animal Resources facilities at the University of Minnesota. For CD-1 mice, litters of newborn pups were pooled and redistributed randomly to their original litter size among the dams. Pups were injected intraperitoneally with 20 TCID50 of virus in 50 μl PBS within 48 hours after birth. Each mutant virus was tested in at least three litters of CD-1 mice. Control litters were injected with MP1.24, AMP2.17, or PBS in parallel with every batch of mutant viruses that was evaluated. At the experimental endpoint, mice were weighed, exsanguinated, and sacrificed by cervical dislocation while under ketamine/xylazine anesthesia, in accordance with Public Health Service policy on the humane care and use of laboratory animals and the research protocol approved by the Institutional Animal Care and Use Committee of the University of Minnesota. Moribund mice were euthanized. With the exception of sham-infected controls, only mice that had measurable titers of serum antibody to CVB1T were included to ensure that all mice used in the final analysis had undergone a productive acute infection. The overall success rate for infection was similar for all viruses and averaged 90%.
Site-directed mutagenesis
Single nucleotide site-directed mutagenesis was performed using mutagenic oligonucleotides and the GeneEditor system (Promega, Madison, WI) according to the manufacturer’s protocol. The five mutagenic primer sequences, with the targeted nt shown as myopathic/amyopathic followed by the location of the corresponding nucleotide (nt) and amino acid changes in parentheses, were as follows: 5′-CATCACGAATTTTA-C/T-AACACTAAACTAC-3′ (C706U); 5′-CAAAGAAGGGAT-A/T-CGCGGAATGGG-3′ (A2712U, Y87F); 5′-GATGCACCCG-T/C-CCAAACCCACC-3′ (U2859C, V136A); 5′-CGATTGAATATT-A/G-CAACAACTGGAG (A3278G, T276A); 5′-GAATGACCAG-A/G-AGGTAGGCGTG-3′ (A5510G, K53E). To minimize the possibility of secondary, non-target mutations, smaller subgenomic fragments of viral cDNA containing the region of interest were subcloned, mutagenized, completely sequenced using Big Dye chemistry followed by analysis on an ABI3130xl (Applied Biosystems, Foster City, CA) at the University of Minnesota Biomedical Genomics Center, and ligated back into the parent clone using compatible restriction enzyme sites. Restriction site junctions in reformed infectious clones were verified by sequencing. Sequencing results and alignments were processed with LaserGene 6.0 (DNASTAR, Madison, WI).
Clinical weakness and muscle histopathology
Motor dysfunction in the hind limbs, scored as weakness, was determined as described previously (Tam et al., 1991). In brief, mice were placed into the bottom half of a large cage and allowed to move freely while a blinded observer scored effects on leg position and gait. Each leg was graded on a scale of 0 to 3 as follows: grade 0, no disease; grade 1, leg is slightly splayed but can still be used to walk and grip a wire screen; grade 2, leg drags visibly with little or no ability to grip; grade 3, leg exhibits flexion deformity and cannot be used to walk or grip. Scores for the right and left leg were combined for a total maximum weakness grade per mouse of 6. To evaluate muscle histopathology, proximal hind limb muscle was excised as a single piece containing the hamstring, quadriceps, and femur, fixed in Zamboni’s at 4°C for a minimum of 2 weeks, decalcified, and embedded in paraffin. Cross sections were cut at 8 μm and stained with hematoxylin and eosin. The degree of inflammation was scored in the right hamstring muscle and graded on a scale ranging from none (grade 0) to severe (grade 3) as described previously (Tam et al., 2003b).
Anti-viral antibodies
The level of anti-viral IgG in serum was measured by ELISA as previously described with MP1.24 as the target (Tam and Messner, 1996). The ELISA serum standard consisted of serum pooled from infected mice averaging an A405 of 1.0 at a 1/500 dilution and assigned a value of 5,000 U/ml. A standard curve was included on each 96-well ELISA plate with all samples assayed in triplicate. Titers of anti-viral antibody in test sera were calculated using the standard curve and expressed in U/ml. Sera were considered as positive if they had values >750 U/ml which is 3 SD above the historical mean for uninfected mice. Neutralizing antibody titers were determined by performing an initial 1/32 dilution of mouse serum in DMEM followed by serial 1/2 dilutions in a 96 well plate, with a final volume of 50 μl diluted serum per well. Each sample was tested at minimum in triplicate. Fifty microliters of DMEM containing 50 TCID50 of infectious virus were added per well and the plate was incubated at 37°C. After 1 hour, 100 μl of DMEM-10 containing 1×104 BGMK cells were added per well, the plate was incubated for 4 days, and the wells were scored as positive or negative for CPE. The final neutralizing antibody titer was expressed as the log2 of the inverse of the highest dilution of serum that neutralized infectivity. Horse anti-CVB1 and horse preimmune serum (ATCC, Manassas, VA) were included as positive and negative controls, respectively. Uninfected wells served as an additional negative control, and serum from uninfected mice was uniformly negative for neutralizing activity.
Temperature and pH stability
Viruses were diluted to equivalent titers (TCID50/ml) in DMEM-10 and incubated in a water bath at 37°C, 39°C, or 47°C. Aliquots were withdrawn at the indicated times and stored frozen at −70°C until the virus titer was determined as described above. Stability at pH 4.5 was determined as described by Vlasak et al., (Vlasak et al., 2005).
RNA secondary structure and capsid homology modeling
Computer models of RNA secondary structure for the CVB1 5′ UTR were created using mfold version 3.2 (Zuker, 2003). Default folding parameters were used, and multiple sequences were submitted by the use of the mfold interface developed as part of this project (software available at http://www.bioinformatics.org/mfoldinterface). The circular Feynman representation was created with jViz.Rna (Wiese et al., 2005). Homology models of the wild type MP1.24 capsid and the mutant containing Y87F, V136A, and T276A were created in Modeller (Sali et al., 1995) available as a module of Insight II (Accelrys, San Diego, CA) using the atomic structure of the closely-related CVB3 (1COV.pdb) (Muckelbauer et al., 1995). The homology model was visualized with Maestro (Schrodinger, New York, NY).
Statistics
Statistical analyses were performed using StatView 5.0 (SAS Institute Inc., Cary, NC). All thirteen mutant viruses were compared to the prototype MP1.24 and AMP2.17 viruses by ANOVA with Bonferroni-Dunn post-hoc comparisons to categorize mutants as myopathic, amyopathic, or intermediate (adjusted for thirteen mutants compared to two prototypes or twenty-six comparisons, level of significance α = 0.002). Selected individual mutant viruses were compared using Student’s t-test. Additional details on statistical analyses are described in the results.
Acknowledgments
We wish to thank Stacey Orvik, Leah Randles, and Anna Hamlin for excellent technical assistance, Michael Rossman for advice on the capsid roadmap, and Michael Zuker for access to mfold. We also thank Yuk Yin Sham for assistance with Modeler and Maestro and acknowledge the support for LaserGene, Modeler, and Maestro provided by the University of Minnesota Supercomputing Institute. This work was supported by Public Health Service grant AI-51270 from the National Institute of Allergy and Infectious Diseases and a grant from the Arthritis Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Maribeth M. Sandager, Email: maribeth@umn.edu.
Jaime L. Nugent, Email: speck007@umn.edu.
Wade L. Schulz, Email: schu1321@umn.edu.
Ronald P. Messner, Email: messn001@umn.edu.
Patricia E. Tam, Email: tamxx001@umn.edu.
References
- Badorff C, Lee GH, Lamphear BJ, Martone ME, Campbell KP, Rhoads RE, Knowlton KU. Enteroviral protease 2A cleaves dystrophin: evidence of cytoskeletal disruption in an acquired cardiomyopathy. Nat Med. 1999;5:320–326. doi: 10.1038/6543. [DOI] [PubMed] [Google Scholar]
- Bailey JM, Tapprich WE. Structure of the 5′ nontranslated region of the coxsackievirus B3 genome: chemical modification and comparative sequence analysis. J Virol. 2007;81:650–668. doi: 10.1128/JVI.01327-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard KM, Semler BL. Regulation of picornavirus gene expression. Microbes Infect. 2004;6:702–713. doi: 10.1016/j.micinf.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Belsham GJ, Sonenberg N. Picornavirus RNA translation: roles for cellular proteins. Trends Microbiol. 2000;8:330–335. doi: 10.1016/s0966-842x(00)01788-1. [DOI] [PubMed] [Google Scholar]
- Bergelson JM, Cunningham JA, Droguett G, Kurt-Jones EA, Krithivas A, Hong JS, Horwitz MS, Crowell RL, Finberg RW. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science. 1997;275:1320–1323. doi: 10.1126/science.275.5304.1320. [DOI] [PubMed] [Google Scholar]
- Bergelson JM, Mohanty JG, Crowell RL, John NF, Lublin DM, Finberg RW. Coxsackievirus B3 adapted to growth in RD cells binds to decay- accelerating factor (CD55) J Virol. 1995;69:1903–1906. doi: 10.1128/jvi.69.3.1903-1906.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S, Das S. Mapping of secondary structure of the spacer region within the 5′-untranslated region of the coxsackievirus B3 RNA: possible role of an apical GAGA loop in binding La protein and influencing internal initiation of translation. Virus Res. 2005;108:89–100. doi: 10.1016/j.virusres.2004.08.020. [DOI] [PubMed] [Google Scholar]
- Caggana M, Chan P, Ramsingh A. Identification of a single amino acid residue in the capsid protein VP1 of coxsackievirus B4 that determines the virulent phenotype. J Virol. 1993;67:4797–4803. doi: 10.1128/jvi.67.8.4797-4803.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson SD, Chapman NN, Tracy SM. Purification of the putative coxsackievirus B receptor from HeLa cells. Biochem Biophys Res Commun. 1997;233:325–328. doi: 10.1006/bbrc.1997.6449. [DOI] [PubMed] [Google Scholar]
- Chia JK-S, Chia AY. Chronic fatigue syndrome is associated with chronic enterovirus infection of the stomach. J Clin Pathol. 2007 doi: 10.1136/jcp.2007.050054. (in press) [DOI] [PubMed] [Google Scholar]
- Chia JKS. The role of enterovirus in chronic fatigue syndrome. J Clin Pathol. 2005;58:1126–1132. doi: 10.1136/jcp.2004.020255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements GB, McGarry F, Nairn C, Galbraith DN. Detection of enterovirus-specific RNA in serum: the relationship to chronic fatigue. J Med Virol. 1995;45:156–161. doi: 10.1002/jmv.1890450208. [DOI] [PubMed] [Google Scholar]
- Cunningham L, Bowles NE, Lane RJ, Dubowitz V, Archard LC. Persistence of enteroviral RNA in chronic fatigue syndrome is associated with the abnormal production of equal amounts of positive and negative strands of enteroviral RNA. J Gen Virol. 1990;71:1399–1402. doi: 10.1099/0022-1317-71-6-1399. [DOI] [PubMed] [Google Scholar]
- Douche-Aourik F, Berlier W, Féasson L, Bourlet T, Harrath R, Omar S, Grattard F, Denis C, Pozzetto B. Detection of enterovirus in human skeletal muscle from patients with chronic inflammatory muscle disease or fibromyalgia and healthy subjects. J Med Virol. 2003;71:540–547. doi: 10.1002/jmv.10531. [DOI] [PubMed] [Google Scholar]
- Galbraith DN, Nairn C, Clements GB. Evidence for enteroviral persistence in humans. J Gen Virol. 1997;78:307–312. doi: 10.1099/0022-1317-78-2-307. [DOI] [PubMed] [Google Scholar]
- Godman GC, Bunting H, Melnick JL. The histopathology of Coxsackie virus infection in mice. I Morphologic observations with four different viral types. Am J Pathol. 1952;28:223–257. [PMC free article] [PubMed] [Google Scholar]
- Guest S, Pilipenko E, Sharma K, Chumakov K, Roos RP. Molecular mechanisms of attenuation of the Sabin strain of poliovirus type 3. J Virol. 2004;78:11097–11107. doi: 10.1128/JVI.78.20.11097-11107.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez AL, Denova-Ocampo M, Racaniello VR, del Angel RM. Attenuating mutations in the poliovirus 5′ untranslated region alter its interaction with polypyrimidine tract-binding protein. J Virol. 1997;71:3826–3833. doi: 10.1128/jvi.71.5.3826-3833.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Chipman PR, Howitt J, Bator CM, Whitt MA, Baker TS, Kuhn RJ, Anderson CW, Freimuth P, Rossmann MG. Interaction of coxsackievirus B3 with the full length coxsackievirus-adenovirus receptor. Nat Struct Biol. 2001;8:874–878. doi: 10.1038/nsb1001-874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Lin F, Chipman PR, Bator CM, Baker TS, Shoham M, Kuhn RJ, Medof ME, Rossmann MG. Structure of decay-accelerating factor bound to echovirus 7: A virus-receptor complex. Proc Natl Acad Sci USA. 2002;99:10325–10329. doi: 10.1073/pnas.152161599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellen CU, Pestova TV, Litterst M, Wimmer E. The cellular polypeptide p57 (pyrimidine tract-binding protein) binds to multiple sites in the poliovirus 5′ nontranslated region. J Virol. 1994;68:941–950. doi: 10.1128/jvi.68.2.941-950.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YT, Yam P, Yan H, Sun Y. Engineered BGMK cells for sensitive and rapid detection of enteroviruses. J Clin Microbiol. 2002;40:366–371. doi: 10.1128/JCM.40.2.366-371.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyoty H. Enterovirus infections and type 1 diabetes. Ann Med. 2002;34:138 – 147. [PubMed] [Google Scholar]
- Ito M, Kodama M, Masuko M, Yamaura M, Fuse K, Uesugi Y, Hirono S, Okura Y, Kato K, Hotta Y, Honda T, Kuwano R, Aizawa Y. Expression of coxsackievirus and adenovirus receptor in hearts of rats with experimental autoimmune myocarditis. Circ Res. 2000;86:275–280. doi: 10.1161/01.res.86.3.275. [DOI] [PubMed] [Google Scholar]
- Jongen PJH, Eling P, Van De Putte LBA. Predominant right leg dysfunction without asymmetric muscle inflammation in CD1 Swiss mice with coxsackievirus B1-induced myositis. Physiol Behav. 1996;59:763–768. doi: 10.1016/0031-9384(95)02155-8. [DOI] [PubMed] [Google Scholar]
- Jurgens CK, Barton DJ, Sharma N, Morasco BJ, Ogram SA, Flanegan JB. 2Apro is a multifunctional protein that regulates the stability, translation and replication of poliovirus RNA. Virology. 2006:346–357. 346. doi: 10.1016/j.virol.2005.09.067. [DOI] [PubMed] [Google Scholar]
- Lentz KN, Smith AD, Geisler SC, Cox S, Buontempo P, Skelton A, DeMartino J, Rozhon E, Schwartz J, Girijavallabhan V. Structure of poliovirus type 2 Lansing complexed with antiviral agent SCH48973: comparison of the structural and biological properties of the three poliovirus serotypes. Structure. 1997;5:961–978. doi: 10.1016/s0969-2126(97)00249-9. [DOI] [PubMed] [Google Scholar]
- Leon-Monzon M, Dalakas MC. Absence of persistent infection with enteroviruses in muscles of patients with inflammatory myopathies. Ann Neurol. 1992;32:219–222. doi: 10.1002/ana.410320215. [DOI] [PubMed] [Google Scholar]
- Lindh G, Samuelson A, Hedlund KO, Evengard B, Lindquist L, Ehrnst A. No findings of enteroviruses in Swedish patients with chronic fatigue syndrome. Scand J Infect Dis. 1996;28:305–307. doi: 10.3109/00365549609027178. [DOI] [PubMed] [Google Scholar]
- Liu Z, Carthy CM, Cheung P, Bohunek L, Wilson JE, McManus BM, Yang D. Structural and functional analysis of the 5′ untranslated region of coxsackievirus B3 RNA: In vivo translational and infectivity studies of full-length mutants. Virology. 1999;265:206–217. doi: 10.1006/viro.1999.0048. [DOI] [PubMed] [Google Scholar]
- Martin A, Wychowski C, Couderc T, Crainic R, Hogle J, Girard M. Engineering a poliovirus type 2 antigenic site on a type 1 capsid results in a chimaeric virus which is neurovirulent for mice. The EMBO Journal. 1988;7:2839–2847. doi: 10.1002/j.1460-2075.1988.tb03140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArdle A, McArdle F, Jackson MJ, Page SF, Fahal I, Edwards RH. Investigation by polymerase chain reaction of enteroviral infection in patients with chronic fatigue syndrome. Clin Sci. 1996;90:295–300. doi: 10.1042/cs0900295. [DOI] [PubMed] [Google Scholar]
- Melnick J, Shaw E, Curnen E. A virus from patients diagnosed as non-paralytic poliomyelitis or aseptic meningitis. Proc Soc Exp Biol Med. 1949;71:344–349. doi: 10.3181/00379727-71-17186. [DOI] [PubMed] [Google Scholar]
- Muckelbauer JK, Kremer M, Minor I, Diana G, Dutko FJ, Groarke J, Pevear DC, Rossmann MG. The structure of coxsackievirus B3 at 3.5 A resolution. Structure. 1995;3:653–667. doi: 10.1016/s0969-2126(01)00201-5. [DOI] [PubMed] [Google Scholar]
- Mulders MN, Salminen M, Kalkkinen N, Hovi T. Molecular epidemiology of coxsackievirus B4 and disclosure of the correct VP1/2Apro cleavage site: evidence for high genomic diversity and long-term endemicity of distinct genotypes. J Gen Virol. 2000;81:803–812. doi: 10.1099/0022-1317-81-3-803. [DOI] [PubMed] [Google Scholar]
- Murray MG, Bradley J, Yang XF, Wimmer E, Moss EG, Racaniello VR. Poliovirus host range is determined by a short amino acid sequence in neutralization antigenic site I. Science. 1988;241:213–215. doi: 10.1126/science.2838906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto S, Miyoshi H, Nishikawa H, Nakashima H. Novel recognition sequence of coxsackievirus 2A proteinase. Biochem Biophys Res Commun. 2006;348:1436–1442. doi: 10.1016/j.bbrc.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Polacek C, Ekstrom JO, Lundgren A, Lindberg AM. Cytolytic replication of coxsackievirus B2 in CAR-deficient rhabdomyosarcoma cells. Virus Res. 2005;113:107–115. doi: 10.1016/j.virusres.2005.04.021. [DOI] [PubMed] [Google Scholar]
- Ray CG, Minnich LL, Johnson PC. Selective polymyositis induced by coxsackievirus B1 in mice. J Infect Dis. 1979;140:239–243. doi: 10.1093/infdis/140.2.239. [DOI] [PubMed] [Google Scholar]
- Reed LJ, Muench H. A simple method of estimating fifty percent endpoints. Am J Hygiene. 1938;27:493–497. [Google Scholar]
- Ren R, Moss EG, Racaniello VR. Identification of two determinants that attenuate vaccine-related type 2 poliovirus. J Virol. 1991;65:1377–1382. doi: 10.1128/jvi.65.3.1377-1382.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A, Potterton L, Yuan F, van Vlijmen H, Karplus M. Evaluation of comparative protein modeling by MODELLER. Proteins. 1995;23:318–326. doi: 10.1002/prot.340230306. [DOI] [PubMed] [Google Scholar]
- Skinner MA, Racaniello VR, Dunn G, Cooper J, Minor PD, Almond JW. New model for the secondary structure of the 5′ non-coding RNA of poliovirus is supported by biochemical and genetic data that also show that RNA secondary structure is important in neurovirulence. J Mol Biol. 1989;207:379–392. doi: 10.1016/0022-2836(89)90261-1. [DOI] [PubMed] [Google Scholar]
- Stadnick E, Dan M, Sadeghi A, Chantler JK. Attenuating mutations in coxsackievirus B3 map to a conformational epitope that comprises the puff region of VP2 and the knob of VP3. J Virol. 2004;78:13987–14002. doi: 10.1128/JVI.78.24.13987-14002.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strongwater SL, Dorovini ZK, Ball RD, Schnitzer TJ. A murine model of polymyositis induced by coxsackievirus B1 (Tucson strain) Arthritis Rheum. 1984;27:433–442. doi: 10.1002/art.1780270411. [DOI] [PubMed] [Google Scholar]
- Tam PE. Coxsackievirus myocarditis: interplay between virus and host in the pathogenesis of heart disease. Viral Immunol. 2006;19:133–146. doi: 10.1089/vim.2006.19.133. [DOI] [PubMed] [Google Scholar]
- Tam PE, Fontana DR, Messner RP. Coxsackievirus B1-induced chronic inflammatory myopathy: differences in induction of autoantibodies to muscle and nuclear antigens by cloned myopathic and amyopathic viruses. J Lab Clin Med. 2003a;142:196–204. doi: 10.1016/S0022-2143(03)00108-2. [DOI] [PubMed] [Google Scholar]
- Tam PE, Messner RP. Genetic determinants of susceptibility to coxsackievirus B1-induced chronic inflammatory myopathy: Effect of host background and major histocompatibility complex genes. J Lab Clin Med. 1996;128:279–289. doi: 10.1016/s0022-2143(96)90029-3. [DOI] [PubMed] [Google Scholar]
- Tam PE, Messner RP. Coxsackievirus-induced chronic inflammatory myopathy: virus variants distinguish between acute cytopathic effects and pathogenesis of chronic disease. Virology. 1997;233:199–209. doi: 10.1006/viro.1997.8592. [DOI] [PubMed] [Google Scholar]
- Tam PE, Messner RP. Molecular mechanisms of coxsackievirus persistence in chronic inflammatory myopathy: viral RNA persists through formation of a double-stranded complex without associated genomic mutations or evolution. J Virol. 1999;73:10113–10121. doi: 10.1128/jvi.73.12.10113-10121.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam PE, Schmidt AM, Ytterberg SR, Messner RP. Viral persistence during the developmental phase of coxsackievirus B1-induced murine polymyositis. J Virol. 1991;65:6654–6660. doi: 10.1128/jvi.65.12.6654-6660.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam PE, Schmidt AM, Ytterberg SR, Messner RP. Duration of virus persistence and its relationship to inflammation during the chronic phase of coxsackievirus B1-induced murine polymyositis. J Lab Clin Med. 1994;123:346–356. [PubMed] [Google Scholar]
- Tam PE, Weber-Sanders M, Messner RP. Multiple viral determinants mediate myopathogenicity in coxsackievirus B1-induced chronic inflammatory myopathy. J Virol. 2003b;77:11849–11854. doi: 10.1128/JVI.77.21.11849-11854.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomko RP, Xu R, Philipson L. HCAR and MCAR: The human and mouse cellular receptors for subgroup C adenoviruses and group B coxsackieviruses. Proc Natl Acad Sci USA. 1997;94:3352–3356. doi: 10.1073/pnas.94.7.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracy S, Chapman NM, Drescher KM, Kono K, Tapprich WE. Evolution of virulence in picornaviruses. Curr Top Microbiol Immunol. 2006;299:193–209. doi: 10.1007/3-540-26397-7_7. [DOI] [PubMed] [Google Scholar]
- Tu Z, Chapman NM, Hufnagel G, Tracy S, Romero JR, Barry WH, Zhao L, Currey K, Shapiro B. The cardiovirulent phenotype of coxsackievirus B3 is determined at a single site in the genomic 5′ nontranslated region. J Virol. 1995;69:4607–4618. doi: 10.1128/jvi.69.8.4607-4618.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlasak M, Goesler I, Blaas D. Human rhinovirus type 89 variants use heparan sulfate proteoglycan for cell attachment. J Virol. 2005;79:5963–5970. doi: 10.1128/JVI.79.10.5963-5970.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegers K, Uhlig H, Dernick R. N-AgIB of poliovirus type 1: a discontinuous epitope formed by two loops of VP1 comprising residues 96–104 and 141–152. Virology. 1989;170:583–586. doi: 10.1016/0042-6822(89)90452-2. [DOI] [PubMed] [Google Scholar]
- Wiese KC, Glen E, Vasudevan A. jViz. Rna-A java tool for RNA secondary structure visualization. IEEE Transactions on NanoBioscience. 2005;4:212–218. doi: 10.1109/tnb.2005.853646. [DOI] [PubMed] [Google Scholar]
- Yang D, Cheung P, Sun Y, Yuan J, Zhang H, Carthy CM, Anderson DR, Bohunek L, Wilson JE, McManus BM. A Shine-Dalgarno-like sequence mediates in vitro ribosomal internal entry and subsequent scanning for translation initiation of coxsackievirus B3 RNA. Virology. 2003;305:31–43. doi: 10.1006/viro.2002.1770. [DOI] [PubMed] [Google Scholar]
- Yang D, Wilson JE, Anderson DR, Bohunek L, Cordeiro C, Kandolf R, McManus BM. In vitro mutational and inhibitory analysis of the cis-acting translational elements within the 5′ untranslated region of coxsackievirus B3: potential targets for antiviral action of antisense oligomers. Virology. 1997;228:63–73. doi: 10.1006/viro.1996.8366. [DOI] [PubMed] [Google Scholar]
- Ytterberg SR, Mahowald ML, Messner RP. T cells are required for coxsackievirus B1 induced murine polymyositis. J Rheumatol. 1988;15:475–478. [PubMed] [Google Scholar]
- Zautner AE, Korner U, Henke A, Badorff C, Schmidtke M. Heparan sulfates and coxsackievirus-adenovirus receptor: each one mediates coxsackievirus B3 PD infection. J Virol. 2003;77:10071–10077. doi: 10.1128/JVI.77.18.10071-10077.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zell R, Stelzner A. Application of genome sequence information to the classification of bovine enteroviruses: the importance of 5′- and 3′-nontranslated regions. Virus Res. 1997;51:213–229. doi: 10.1016/s0168-1702(97)00096-8. [DOI] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]