

Abstract
HLA-G is a nonclassical major histocompatibility complex class I molecule selectively expressed on cytotrophoblasts at the feto–maternal interface, where it may play an important role in maternal tolerance of the fetus. We provide direct evidence under physiological conditions that supports the role of HLA-G in protecting cytotrophoblasts against natural killer (NK) cytolysis in 6 semiallogenic combinations of maternal uterine NK cells and their own trophoblast counterparts, as well as in 20 allogenic combinations of maternal uterine NK cells and trophoblasts from different mothers. We show that, in all cases studied, this HLA-G-mediated protection was abolished by treatment of cytotrophoblasts with an HLA-G-specific mAb. The HLA class I-negative K562 cell line transfected with the predominant HLA-G1 isoform results in similar protection and abolition from maternal uterine NK lysis. Because maternal uterine NK cells express killer inhibitory receptors for HLA-G, we conclude that their interactions contribute to the survival of the fetal semiallograft by confering immunological tolerance to its tissues.
HLA-G is a nonclassical major histocompatibility complex class I molecule that is selectively expressed at the feto–maternal interface on the surfaces of cytotrophoblast cells that do not express HLA-A or HLA-B, but that do express low levels of HLA-C molecules (1, 2). The fact that the expression of HLA-G is limited to gestation has led to the hypothesis that it plays an important role in immunological tolerance of the fetus by the mother (3).
The HLA-G gene is alternatively spliced in cytotrophoblasts, resulting in at least five different HLA-G mRNAs potentially encoding five HLA-G isoforms, namely, the HLA-G1, HLA-G2, HLA-G3, and HLA-G4 membrane-bound isoforms, plus the soluble HLA-G5 form (4, 5). A 200-fold predominance of the HLA-G1 transcript over the HLA-G2 transcript has been described in trophoblasts (5). We recently demonstrated that when the membrane-bound HLA-G1 and HLA-G2 isoforms are transfected into the HLA-class I-negative K562 cell line they strongly inhibit peripheral blood natural killer (NK) cell-mediated lysis (6). This study provided information that may be especially important in understanding maternal–fetal interactions (7). Pazmany et al. (8) and Munz et al. (9) have recently shown that HLA-G expression protected an otherwise susceptible target cell line from lysis by NK clones and identified the killer inhibitory receptors (KIR) on NK cells that recognize HLA-G, namely p58.1 (CD158a), p58.2 (CD158b), and NKAT3, which belong to the KIR Ig superfamily. More recently, Perez-Villar et al. (10) have demonstrated that the CD94/NKG2 receptor complex, which belongs to the KIR lectin superfamily, is involved in the recognition of cells expressing HLA-G1.
In the present study, using freshly obtained first trimester fetal and maternal tissues, we demonstrate ex vivo the role of HLA-G molecules expressed on the trophoblast cell surface in protecting the fetus from the lytic activity of maternal uterine NK cells in all combinations tested: 6 semiallogenic combinations (maternal uterine NK cells and their own trophoblast counterparts) and in 20 allogenic combinations (maternal uterine NK cells and trophoblasts from different mothers). This protective effect was also observed with NK cells present among peripheral blood mononuclear cells (PBMC) obtained from 10 healthy adult donors. Furthering previous studies (11–13), the present work provides evidence in support of the role of HLA-G in materno–fetal tolerance by showing that (i) treatment of cytotrophoblasts with the pan-class I mAb (W6/32), which masks both HLA-G and HLA-C molecules—but not with the anti-HLA-B, -C mAb (B1.23.2)—abolishes protection from NK lysis, and (ii) the HLA class I-negative K562 cell line transfected with the predominant HLA-G1 isoform results in similar protection and abolition from NK lysis. The fact that the lytic activities of NK cells in both maternal uterine blood and allogenic peripheral blood were inhibited by the expression of HLA-G on cytotrophoblast target cells supports the hypothesis that HLA-G is the public ligand for KIR present in all individuals and expressed on most, if not all, NK cells.
MATERIALS AND METHODS
Cell Lines.
The K562 human erythroleukemia cell line (American Type Culture Collection) and the nonadult T cell leukemia, NK-mediating YT2C2-PR subclone were maintained in RPMI 1640 medium supplemented with 10% heat-inactivated fetal calf serum, 2 mM l-glutamine, 1 μg/ml gentamicin, and fungizone (Sigma) and cultured in a 37°C, 5% CO2, humidified incubator. K562 transfectants were obtained as described (6).
Preparation of Cytotrophoblasts.
Trophoblast tissue was obtained from first trimester terminations of normal pregnancies (6–12 wk) by suction curettage and used immediately. Local ethical committee approval was obtained for this study. First trimester trophoblast cells were isolated using a trypsin-DNase dispersion method followed by a Percoll gradient centrifugation step, as described by Kliman et al. (14). Briefly, the tissues were washed extensively in 0.9% NaCl at room temperature. Soft, villous material was cut away from connective tissue and vessels, minced, transferred to warmed calcium- and magnesium-free Hanks’ solution containing 25 mM Hepes, 0.125% trypsin (Sigma), and 0.2 mg/ml DNase I (Sigma) pH 7.4, and then incubated at 37°C for 30 min. The resultant cell suspension was filtered through muslin, washed, and layered over a preformed Percoll gradient made up in Hanks’ balanced salt solution. The gradient was made from 70 to 5% Percoll in 5% steps of 3 ml each. The gradient was centrifugated at 1200 × g at room temperature for 20 min. After centrifugation, three regions were identified: bottom, containing red blood cells and polymorphonuclear leucocytes; top, containing connective tissue elements, small vessels, and villous fragments; and middle, containing a relatively uniform population of trophoblast mononuclear cells. The middle layer was removed, washed, and then frozen.
Reverse Transcriptase–PCR (RT-PCR) Analysis.
Total mRNA from 107 cells was extracted, using the RNA NOW reagent (Ozyme, France) according to the manufacturer’s recommendations. The quality of RNA was checked by electrophoresis in 1.5% agarose denaturing gel. Complementary DNAs were prepared from 10 μg of total RNA, using oligo(dT)12–18 primer and Moloney murine leukemia virus reverse transcriptase (GIBCO/BRL). RT-PCR amplifications were carried out using either two HLA-G-specific primers, G.257 (exon 2) and G.1225 (3′-UT) (5), or two pan class I-specific primers, HLA-5P2 (5′-primer) and HLA-3pA (3′-primer) (15). Additional RT-PCR amplification of β-actin was carried out to evaluate the amount of RNA in all samples. PCR products were analyzed by electrophoresis in 1% agarose gel and stained with ethidium bromide. The specificity of PCR products was confirmed by alkaline blotting of the fragments in 0.4 M NaOH onto nylon membranes (Hybond N+, Amersham). Hybridization was accomplished with either an HLA-G-specific probe, G.1200 (5), an HLA-A-specific probe (5′-GGAGGACCAGACCCAGGACACG), an HLA-B-specific probe (5′-AGCTCCGATGACCACAACTGC) (16), or an HLA-C-specific probe (5′-TGTCCTAGCTGCCTAGGAG). The filters were exposed to Biomax films (Kodak) with amplifying screens for 4–16 hr at −80°C.
mAbs and Flow Cytometry.
In this study, we used the following mAbs: W6/32, IgG2a anti-HLA class I α chains associated with β2 m (Sigma); B1.23.2, IgG2b, anti-HLA-B, -C (American Type Culture Collection); GL183, IgG1 anti-p58 (CD158b) (Immunotech, Marseille, France); EB6, IgG1 anti-p58 (CD158a) (Immunotech); HP-3B1, IgG2a anti-CD94 (Immunotech); 3G8, IgG1 anti-CD16 conjugated with fluorescein (Immunotech); and B159, IgG1 anti-CD56 conjugated with phycoerythrin (Immunotech). For flow cytometry assays, cells were washed in PBS and stained with the corresponding mAb. After washing twice, cells were either directly analyzed in a flow cytometer (FACS Vantage; Becton Dickinson), if the mAb used was conjugated to fluorochrome or stained with an F(ab′)2 goat anti-mouse IgG antibody conjugated with phycoerythrin (Immunotech) prior to flow cytometer analysis. Control aliquots were stained with an isotype-matched Ab to evaluate nonspecific binding to target cells.
NK Effectors.
Maternal uterine blood was obtained from the above-mentioned first trimester terminations of pregnancy (6–12 wk) and used immediately for the isolation of uterine blood mononuclear cells (UBMC) by Ficoll/histopaque density gradient. PBMC from healthy adult volunteer donors (male and female, aged 30–60 years) were isolated by the same method. Phenotypic characterization of both UBMC and PBMC was then carried out.
Cytotoxicity Assays.
The cytolytic activities of UBMC, PBMC, and YT2C2-PR cells as effectors against cytotrophoblasts, and K562 transfectants as targets, were assessed in 4-hr 51Cr-release assays in which effector cells were mixed with 5 × 103 51Cr-labeled targets (100 μCi of 51Cr sodium chromate; 1 Ci = 37 GBq, Amersham) in U-bottomed microtiter plates. After 4hr at 37°C in a humidified 5% CO2 incubator, 50 μl of the supernatant was collected for liquid scintillation counting (Wallac 1410, Pharmacia). The percentage of specific lysis was calculated as follows: Percent specific lysis = [(cpm experimental release − cpm spontaneous release)/(cpm maximum release − cpm spontaneous release)] × 100. Spontaneous release was determined by incubation of labeled target cells with medium. Maximum release was determined by solubilizing target cells in 0.1 M HCl. In all experiments, spontaneous release was <30% of maximum release. Results are presented as the means of triplicate samples. In experiments in which mAbs were used to block HLA-G–NK interaction, target cells were incubated with the corresponding mAb, then washed and incubated with an F(ab′)2 goat anti-mouse IgG Ab (Jackson Immunoresearch) to prevent Ab-dependent cell cytotoxicity by interaction of NK cell Fc receptors with the first mAb used.
RESULTS
Characterization of Cytotrophoblasts Obtained from First Trimester Fetal Tissues.
Cytotrophoblast cells were isolated from tissues obtained from first trimester terminations of pregnancy, using a trypsin-DNase dispersion method followed by a Percoll gradient centrifugation step (14). To confirm the nature of these purified cells, we characterized their expression of membrane-bound HLA-class I molecules by flow cytometry and their transcription of HLA-class I by Southern blot analysis of RT-PCR products. A recent study has provided evidence of the expression of HLA-C in addition to HLA-G molecules by human first trimester trophoblasts (2). In agreement with that study, we show, using flow cytometry, that both HLA-G and HLA-C molecules were present on the surfaces of cytotrophoblast cells, as these cells were stained by both the pan-class I W6/32 mAb (75% of cells) and the anti-HLA-B, -C B1.23.2 mAb (65% of cells). Furthermore, as expected, the HLA-G transcripts were detected, as was the HLA-C transcript, whereas the HLA-A and HLA-B transcripts were detectable only at very low levels in three purified cytotrophoblast cells, T122, T138, and T146 (Fig. 1).
Figure 1.
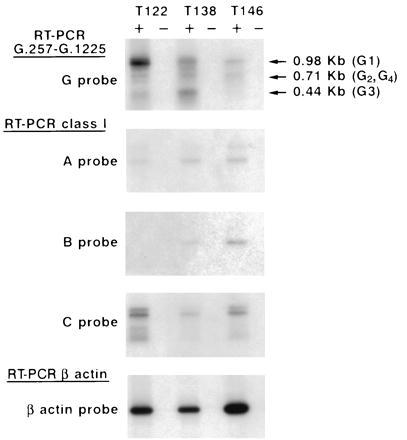
Detection of HLA class I transcripts in cytotrophoblast cells isolated in vitro from first trimester terminations of pregnancy. Reverse RNAs were amplified using either G.257 and G.1225 HLA-G-specific primers or pan class I-specific primers, and Southern blots were hybridized with either an HLA-G 32P-labeled probe (G probe), an HLA-A 32P-labeled probe (A probe), an HLA-B 32P-labeled probe (B probe), or an HLA-C 32P-labeled probe (C probe). Positive (+) and negative (−) lanes correspond to the RT+ and RT− template, as described. To control the amount of RNA in the samples, RT-PCR amplification results obtained with β-actin-specific primers and Southern blots were hybridized with a β-actin 32P-labeled probe. Note that all the cytotrophoblasts present HLA-G1, -G2, -G3, and -G4 transcripts, but at various levels.
Phenotype Characterization of Maternal Uterine NK Cells.
To characterize the maternal uterine NK cells we used, maternal UBMC were obtained from pregnant women (gestational age, 6–12 weeks). The percentage of CD56+ NK cells was determined by flow cytometry (Table 1). Our results showed that NK cells were present in large numbers in the uterine blood of these subjects during early pregnancy. These uterine NK cells were phenotypically different from classical circulating CD56+ CD16+ NK cells in that they were revealed to be CD56+ CD16− by double-color immunofluorescence analysis (data not shown). To determine KIR expression in these uterine NK cells, we used the EB6 and GL183 mAbs, which, respectively, recognize p58.1 (CD158a) and p58.2 (CD158b), known to be expressed on subsets of peripheral blood NK cells (17). We also used the HP-3B1 mAb, which recognizes the CD94 receptor, a KIR expressed on most peripheral blood NK cells (18). Our results showed that (i) CD56+ and CD94+ cells were increased among maternal uterine lymphocytes compared with peripheral blood lymphocytes obtained from healthy adult donors; (ii) CD56+ cells as well as KIR CD158a+, CD158b+, and CD94+ cells were present at various levels in the subjects’ maternal uterine lymphocytes; and (iii) there was no significant difference in the percentages of CD158a+ and CD158b+ cells in maternal uterine blood and in the peripheral blood of male and nonpregnant female subjects (Table 1).
Protection of Cytotrophoblasts from Maternal Uterine NK Cytolysis.
Cytotrophoblast cells were isolated in vitro as mentioned above and used as target cells in cytotoxicity assays of maternal uterine NK lytic activity. Experiments were carried out either in semiallogenic combinations (maternal uterine NK cells and their own trophoblast counterparts) (Fig. 2) or in allogenic combinations (uterine NK and trophoblast cells from different mothers) (Fig. 3 A and B). In both types of combination, and in all cases studied, the results showed that uterine NK lytic activity was inhibited by cytotrophoblast cells.
Figure 2.
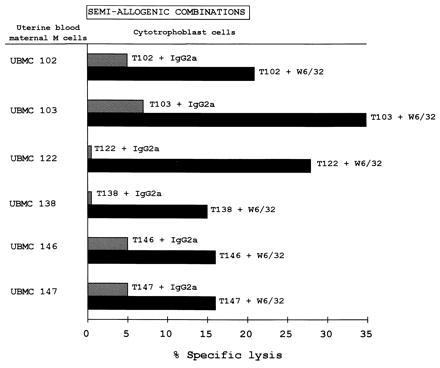
Protection of cytotrophoblasts from maternal uterine NK lysis in semi-allogenic combinations. Cytotoxicity tests were carried out using Percoll gradient-purified cytotrophoblasts as the targets (T): T102, T103, T122, T138, T146, and T147 and UBMC from mothers 102, 103, 122, 138, 146, and 147 as the effector cells (E) at a 25:1 E/T ratio. The results are expressed as the percentage of lysis recorded in a 4-hr 51Cr-release assay. The standard deviation of the mean of the triplicates was less than 5%. W6/32 and IgG2a isotype-matched control Abs were added to the assay at 10 μg/ml.
Figure 3.
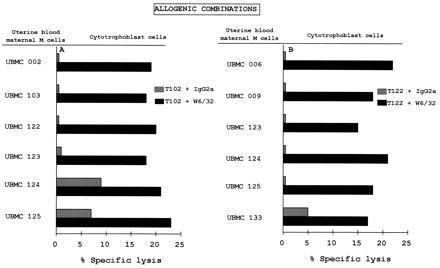
Protection of cytotrophoblasts from maternal uterine NK lysis in allogenic combinations. Cytotoxicity tests were carried out using the Percoll gradient-purified (A) T102 and (B) T122 cytotrophoblasts as targets and the indicated UBMC from different mothers as effector cells at a 25:1 E:T ratio. The tests were carried out as described in Fig. 2.
To demonstrate that the inhibition of NK lysis was due to the presence of HLA class I molecules on the cytotrophoblast cell surface, cytotoxicity assays were also carried out in the presence of target cells pre-incubated with either the pan-HLA class I W6/32 mAb or an isotype-matched control mAb. The addition of W6/32 reversed HLA class I-mediated inhibition of NK cell activity, thus restoring lysis of the cytotrophoblastic target cells in both semiallogenic (Fig. 2) and allogenic combinations (Fig. 3 A and B). Because the number of cytotrophoblasts isolated in vitro varied among the mothers, we could not test them all against all UBMC.
Protection from NK Lysis Is Mainly Mediated by HLA-G Molecules Expressed on the Cytotrophoblast Cell Surface.
Because cytotrophoblast cells of fetal origin express two HLA class I molecules, HLA-G and HLA-C, we evaluated the contribution of each to the inhibition of NK cell-mediated lysis. For this purpose, experiments were carried out using the anti-HLA-B, -C (B1.23.2) mAb and the W6/32 mAb, which masks both HLA-G and HLA-C molecules. In all the combinations tested, the addition of B1.23.2 did not reverse the protective effect against NK lysis, whereas the addition of W6/32 did, suggesting that HLA-C molecules make a minor contribution to such HLA class I-mediated NK inhibition (Fig. 4). This indicates that HLA-G molecules mainly mediate the protection of the fetus from NK lysis.
Figure 4.
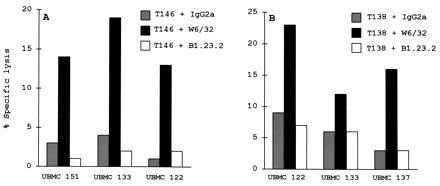
Protection of cytotrophoblasts from maternal uterine NK lysis in allogenic combinations. Cytotoxicity tests were carried out using the Percoll gradient-purified (A) T146 and (B) T138 cytotrophoblasts as targets and the indicated UBMC from different mothers as effector cells at a 25:1 E/T ratio. The tests were carried out as described in Fig. 2. W6/32, B1.23.2, and IgG2a isotype-matched control antibodies were added to the assay at 10 μg/ml.
The role of HLA-G was confirmed by transfecting the K562 cell line with HLA-G1 (the predominant HLA-G isoform expressed on cytotrophoblasts) and using it as the target for NK cytolysis. Both K562-HLA-G1 cells and cytotrophoblasts were protected against maternal uterine NK cytolysis by UBMC 122 and UBMC 138, whereas the K562 -pRc/RSV cell line transfected with the vector alone (HLA class I-negative). Both K562-HLA-G1 and cytotrophoblast cells treated with W6/32 remained sensitive to NK lytic activity. UBMC exhibited similar levels of cytotoxicity against the K562-pRc/RSV cell line and the W6/32-treated cytotrophoblasts (Table 2).
Protection of Cytotrophoblasts from Peripheral NK Cytolysis and NK-Like YT2C2-PR Subclone-Mediated Lysis.
To evaluate whether cytotrophoblast cells would also resist peripheral blood NK lysis, further experiments were carried out using polyclonal NK cells present in PBMC obtained from healthy adult donors (males and nonpregnant females). Using two PBMC (583 and 770), we show that both K562-HLA-G1 cells and cytotrophoblasts were protected against peripheral blood NK cytolysis, whereas the K562 -pRc/RSV cell line and both K562-HLA-G1 and cytotrophoblast cells treated with W6/32 remained sensitive to peripheral NK lysis (Table 2).
In addition, we found that K562-HLA-G1 and cytotrophoblast cells were not lysed by the nonadult T cell leukemia NK-like YT2C2-PR subclone, which was also used as an effector. However, in contrast to what was observed with UBMC and PBMC, the inhibition of YT2C2-PR lysis was not reversed by the use of target cells treated with the W6/32 mAb (Table 2). We have previously obtained identical results which suggested that the epitope recognized by W6/32 on HLA-G is not involved in HLA-G recognition by the putative receptor present on YT2C2 cells (6).
DISCUSSION
In this study, using freshly obtained fetal and maternal tissues, we have investigated the role of HLA-G molecules in protecting cytotrophoblast cells that express them on their surfaces from NK cell-mediated cytotoxicity. As we have previously described, membrane-bound HLA-G1 and HLA-G2 isoforms transfected into HLA class I-negative target cells inhibit NK cytolysis of polyclonal peripheral NK cells (6). In the present study, we evaluated maternal uterine NK lytic activity under physiological conditions, using polyclonal UBMC and cytotrophoblast cells, either from the same mother (semiallogenic combinations) or from another mother (allogenic combinations). As previously reported by others (19), we found that the growth of the human placenta is associated with the presence of large numbers of NK cells within the maternal decidua, where they have an unusual phenotype, CD56+ CD16−, that distinguishes them from adult peripheral blood NK cells. Our study of KIR expression on the surfaces of these uterine NK cells shows that the level of CD94+ cells is elevated in maternal uterine blood compared with the peripheral blood of nonpregnant subjects, suggesting their local recruitment in the decidua during early pregnancy. In contrast to what was previously observed by Verma et al. (20), our results showed that the percentages of p58.1+ and p58.2+ cells are not significantly different in peripheral and uterine lymphocyte populations. However, both Verma’s and our study demonstrate that those NK cells which populate the uterus during early pregnancy express KIR for polymorphic HLA-C and monomorphic HLA-G molecules present on the surfaces of cytotrophoblasts. We discuss the role of HLA-C in materno–fetal tolerance, which, unlike that of HLA-G, has not, to our knowledge, been investigated until now.
In the present work, cytotoxicity tests clearly demonstrated that cytotrophoblast cells strongly inhibited maternal uterine NK cell-mediated lysis in both semiallogenic and allogenic combinations. This inhibition was always reversed when cytotrophoblast cells were incubated with the pan-class I W6/32 mAb. This protective effect was also observed with the JEG-3 HLA-G-positive choriocarcinoma cell line as the target (data not shown). This effect was also found using PBMC from healthy adult individuals as the NK effectors. The results obtained with the anti-HLA-B, -C (B1.23.2) mAb that did not restore cytotrophoblast cell lysis show that the inhibition of NK lytic activity was predominantly exerted by HLA-G molecules present on the surfaces of cytotrophoblast target cells. It should be noted that, because the B1.23.2 mAb reacted only weakly with HLA-G (tested using the K562 cell line that had been transfected with HLA-G1; data not shown), it might not have affected the interaction between HLA-G and its KIR counterpart. However, we cannot exclude that the epitope recognized by B1.23.2 on HLA-C molecules was not involved in the HLA-C/KIR interaction, which would explain why B1.23.2-treated cytotrophoblast cells still resisted NK lysis. Because anti-HLA-C Abs could be produced by the mother during pregnancy, they could mask the presence of HLA-C molecules on the cytotrophoblast cell surface, thereby preventing their interaction with KIR on NK cells. In contrast, anti-HLA-G Abs are not supposed to be produced by the mother; this is supported by the fact that HLA-G protein exhibits very low polymorphism (to date, only two polymorphic HLA-G1 proteins have been described) compared with HLA-C class I molecules (21). The results obtained using K562-HLA-G1 transfected cells, which mimic HLA-G-positive cytotrophoblasts, further demonstrate the major role of HLA-G in protecting such cells from NK lysis. The fact that NK lytic activity in all the subjects and combinations tested was inhibited by cytotrophoblasts suggests that HLA-G could be the public ligand for NK inhibitory receptor(s) present in all individuals.
Two groups of KIR have been described as being able to interact with HLA-G. The first group belongs to the Ig superfamily and comprises p58.1, p58.2 (8), and NKAT3 (9). More recently, CD94, which is a member of the KIR C-type lectin superfamily that interacts with HLA-A, -B, and -C molecules in target cells, was involved in association with another NK lectin, NKG2, in the recognition of cells expressing HLA-G1 (10). It is worth noting that the majority of maternal uterine NK cells are CD94+. We can therefore hypothesize that HLA-G blocks the lytic activity of any NK cell, whatever KIR it may express. Interestingly, we also found that T cell leukemia YT2C2-PR clone-mediated lysis was abolished by cytotrophoblasts, although none of the known KIR capable of binding HLA-G is present on the YT2C2-PR clone, nor is CD94 (6). As we have previously shown, these results suggest that a new KIR, distinct from those already described, is involved in HLA-G recognition (6) and may be present on maternal uterine NK cells.
These findings strongly support assigning an important role for HLA-G in blastocyst development and implantation (22, 23) as well as in materno–fetal tolerance (3, 7). Thus, deletion or mutation of the HLA-G gene, as well as posttranslational modifications, could lead to loss of the fetus in early pregnancy. In this regard, recent studies of gestational pathologies have shown (i) altered expression of HLA-G on extravillous trophoblasts in patients with primary preeclampsia that may be involved in failure of invasion, leading to partial placental ischemia (24), and (ii) inhibition of intracellular HLA-G transport in placentally derived human cells infected with herpes simplex virus. This observation suggests a link between herpes simplex virus infection and spontaneous miscarriage (25).
Table 1.
Phenotypic characterization of NK effector cells
| Effector cells | % CD56+ | % CD94+ | % CD158a+ | % CD158b+ |
|---|---|---|---|---|
| UBL* | 28 ± 9† | 19 ± 7 | 4 ± 3 | 8 ± 2 |
| PBL* | 14 ± 8 | 9.7 ± 6.2 | 4.8 ± 2.6 | 7 ± 2 |
| P < 0.003 | P < 0.02 | P > 0.6 | P > 0.5 | |
| YT2C2-PR‡ | 0 | 0 | 0 | 0 |
UBL (uterine blood lymphocytes) from pregnant women and PBL (peripheral blood lymphocytes) from healthy adult subjects (M and F) were obtained as described.
Mean ± SD.
The YT2C2-PR subclone presents the CD56− and KIR− phenotypes, as we previously described (6).
Table 2.
Cytolytic activities (% specific lysis) of UBMC, PBMC, and YT2C2-PR cells against cytotrophoblasts and K562 transfectants
| Target cells* | Effector cells†
|
||||
|---|---|---|---|---|---|
| UBMC 122 | UBMC 138 | PBMC 583 | PBMC 770 | YT2C2-PR | |
| T122 + IgG2a | 2 | 5 | 10 | 0 | 0 |
| T122 + W6/32 | 15 | 17 | 34 | 17 | 0 |
| T138 + IgG2a | 7 | 8 | 0 | ND | 0 |
| T138 + W6/32 | 24 | 17 | 17 | ND | 0 |
| K562-pRc/RSV‡ | 12 | 20 | 22 | 24 | 27 |
| K562-HLA-G1 + IgG2a | 0 | 5 | 4 | 3 | 1 |
| K562-HLA-G1 + W6/32 | 9 | 14 | 16 | 24 | 2 |
Results are expressed as the percentage of specific lysis recorded in a 4-hr 51Cr-release assay. ND, not determined.
K562 transfected with either the vector alone (K562-pRc/RSV) or with HLA-G1 (K562-HLA-G1) were used as targets in addition to the Percoll gradient-purified cytotrophoblasts T122 and T138. Target cells were incubated either with the W6/32 mAb (bold characters) or the IgG2a isotype-matched control mAb at 10 μg/ml.
UBMC from mothers 122 and 138, PBMC from donors 583 and 770, and the YT2C2-PR clone were used as effector cells at a 25:1 E/T ratio.
Identical results were obtained with K562-pRc/RSV cells treated with either the IgG2a mAb or the W6/32 mAb.
Acknowledgments
We thank Dr. Luc Gourand (Hôpital des Métallurgistes, Paris) and Dr. Denis Jacob (Hôpital Lariboisière, Paris) for providing the first trimester terminations of pregnancy samples. We thank Dr. Jean-Gérard Guillet for helpful discussions and Dr. Noah Hardy for rereading and correcting the manuscript. This study was supported by grants from the Association pour la Recherche sur le Cancer.
ABBREVIATIONS
- KIR
killer inhibitory receptor
- NK
natural killer
- PBMC
peripheral blood mononuclear cells
- RT-PCR
reverse transcriptase–PCR
- UBMC
uterine blood mononuclear cells
References
- 1.Kovats S, Main E K, Librach C, Stubblebine M, Fisher S J, DeMars R. Science. 1990;248:220–223. doi: 10.1126/science.2326636. [DOI] [PubMed] [Google Scholar]
- 2.King A, Boocock C, Sharkey A M, Gardner L, Beretta A, Siccardi A G, Loke Y W. J Immunol. 1996;156:2068–2076. [PubMed] [Google Scholar]
- 3.Carosella E D, Dausset J, Kirszenbaum M. Immunol Today. 1996;17:407–409. doi: 10.1016/0167-5699(96)30055-8. [DOI] [PubMed] [Google Scholar]
- 4.Ishitani A, Geraghty D E. Proc Natl Acad Sci USA. 1992;89:3947–3951. doi: 10.1073/pnas.89.9.3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kirszenbaum M, Moreau P, Gluckman E, Dausset J, Carosella E. Proc Natl Acad Sci USA. 1994;91:4209–4213. doi: 10.1073/pnas.91.10.4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rouas-Freiss N, Marchal R, Kirszenbaum M, Dausset J, Carosella E D. Proc Natl Acad Sci USA. 1997;94:5249–5254. doi: 10.1073/pnas.94.10.5249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yokoyama Y M. Proc Natl Acad Sci USA. 1997;94:5998–6000. doi: 10.1073/pnas.94.12.5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pazmany L, Mandelboim O, Vales-Gomez M, Davis D M, Reyburn H T, Strominger J L. Science. 1996;274:792–795. doi: 10.1126/science.274.5288.792. [DOI] [PubMed] [Google Scholar]
- 9.Munz C, Holmes N, King A, Loke Y W, Colonna M, Schild H, Rammensee H-G. J Exp Med. 1997;185:385–391. doi: 10.1084/jem.185.3.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perez-Villar J J, Melero I, Navarro F, Carretero M, Bellon T, Llano M, Colonna M, Geraghty D E, Lopez-Botet M. J Immunol. 1997;158:5736–5743. [PubMed] [Google Scholar]
- 11.King A, Birkby C, Loke Y W. Cell Immunol. 1989;118:337–344. doi: 10.1016/0008-8749(89)90382-1. [DOI] [PubMed] [Google Scholar]
- 12.Chumbley G, King A, Robertson K, Holmes N, Loke Y W. Cell Immunol. 1994;155:312–322. doi: 10.1006/cimm.1994.1125. [DOI] [PubMed] [Google Scholar]
- 13.Deniz G, Christmas S E, Brew R, Johnson P M. J Immunol. 1994;152:4255–4261. [PubMed] [Google Scholar]
- 14.Kliman H J, Nestler J E, Sermasi E, Strauss J F., III Endocrinology. 1986;118:1567–1582. doi: 10.1210/endo-118-4-1567. [DOI] [PubMed] [Google Scholar]
- 15.Zemmour J, Little A-M, Schendel D J, Parham P. J Immunol. 1992;148:1941–1948. [PubMed] [Google Scholar]
- 16.McCutcheon J A, Gumperz J, Smith K D, Lutz C T, Parham P. J Exp Med. 1995;181:2085–2095. doi: 10.1084/jem.181.6.2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moretta A, Vitale M, Bottino C, Orengo A M, Morelli L, Augugliaro R, Barbaresi M, Ciccone E, Moretta L. J Exp Med. 1993;178:597–604. doi: 10.1084/jem.178.2.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Phillips J H, Chang C, Mattson J, Gumperz J E, Parham P, Lanier L L. Immunity. 1996;5:163–172. doi: 10.1016/s1074-7613(00)80492-6. [DOI] [PubMed] [Google Scholar]
- 19.King A, Loke Y W. Immunol Today. 1991;12:432–435. doi: 10.1016/0167-5699(91)90014-K. [DOI] [PubMed] [Google Scholar]
- 20.Verma S, King A, Loke Y W. Eur J Immunol. 1997;27:979–983. doi: 10.1002/eji.1830270426. [DOI] [PubMed] [Google Scholar]
- 21.Kirszembaum M, Djoulah S, Hors J, LeGall I, de Oliveira E B, Prost S, Dausset J, Carosella E D. Hum Immunol. 1997;53:140–147. doi: 10.1016/S0198-8859(97)00038-4. [DOI] [PubMed] [Google Scholar]
- 22.Jurisicova A, Casper R F, MacLusky N J, Librach C L. Fertil Steril. 1996;65:997–1002. [PubMed] [Google Scholar]
- 23.Jurisicova A, Casper R F, MacLusky N J, Mills G B, Librach C L. Proc Natl Acad Sci USA. 1996;93:161–165. doi: 10.1073/pnas.93.1.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hara N, Fujii T, Yamashita T, Kozuma S, Okai T, Taketani Y. Am J Reprod Immunol. 1996;36:349–358. doi: 10.1111/j.1600-0897.1996.tb00185.x. [DOI] [PubMed] [Google Scholar]
- 25.Schust D J, Hill A B, Ploegh H L. J Immunol. 1996;157:3375–3380. [PubMed] [Google Scholar]