

Abstract
Mutation of Bruton’s tyrosine kinase (Btk) impairs B cell maturation and function and results in a clinical phenotype of X-linked agammaglobulinemia. Activation of Btk correlates with an increase in the phosphorylation of two regulatory Btk tyrosine residues. Y551 (site 1) within the Src homology type 1 (SH1) domain is transphosphorylated by the Src family tyrosine kinases. Y223 (site 2) is an autophosphorylation site within the Btk SH3 domain. Polyclonal, phosphopeptide-specific antibodies were developed to evaluate the phosphorylation of Btk sites 1 and 2. Crosslinking of the B cell antigen receptor (BCR) or the mast cell Fcɛ receptor, or interleukin 5 receptor stimulation each induced rapid phosphorylation at Btk sites 1 and 2 in a tightly coupled manner. Btk molecules were singly and doubly tyrosine-phosphorylated. Phosphorylated Btk comprised only a small fraction (≤5%) of the total pool of Btk molecules in the BCR-activated B cells. Increased dosage of Lyn in B cells augmented BCR-induced phosphorylation at both sites. Kinetic analysis supports a sequential activation mechanism in which individual Btk molecules undergo serial transphosphorylation (site 1) then autophosphorylation (site 2), followed by successive dephosphorylation of site 1 then site 2. The phosphorylation of conserved tyrosine residues within structurally related Tec family kinases is likely to regulate their activation.
Mutation of the Bruton’s tyrosine kinase (Btk) gene produces X-linked (or Bruton’s) agammaglobulinemia in humans and X-linked immunodeficiency in mice (1–4). At the cellular level, Btk mutation is manifested by abnormal B cell responses to multiple critical factors, such as interleukin 5 (IL-5) (5–7), IL-6 (8), IL-10 (9), anti-CD38 (10, 11), and the B cell antigen receptor (BCR) (12–17). A mechanism for activation of Btk has been derived from study of endogenous receptor signaling pathways as well as through heterologous expression of Btk in fibroblasts. Src family tyrosine kinases are rapidly activated after stimulation of the BCR (18, 19), then they phosphorylate Btk at Y551 (site 1) (17, 20), a consensus Src family phosphorylation site in the Src homology type 1 (SH1) domain. This phosphorylation event dramatically increases Btk protein tyrosine kinase activity and is required for promotion of fibroblast growth in soft agar by the activated Btk allele, Btk* (17, 20–22). A second major phosphorylated tyrosine residue (Y223) is located within the Btk SH3 domain (23). Phosphorylation of Y223 (site 2) occurs by a Btk kinase-dependent mechanism, i.e., autophosphorylation (17). In contrast to site 1, site 2 phosphorylation has little discernible influence on Btk catalytic activity in vitro or in vivo. The role of the SH3 domain, however, in protein–protein interactions is well established, and site 2 corresponds to a conserved residue important for binding to proline-rich peptide sequences (24–30). Y223 phosphorylation may be a mechanism to modify such interactions.
A critical, but unresolved, feature of Btk activation is the pattern of phosphorylation of individual Btk molecules after receptor stimulation, i.e., are Btk molecules phosphorylated on both regulatory tyrosine residues or only one? Knowing the phosphorylation pattern of individual Btk molecules in the receptor-ligated B cells will clarify how Btk is activated and how it functions. For example, if the mechanism of Btk activation generates doubly phosphorylated Btk molecules, then a single Btk subpopulation would transduce the signals represented by both site phosphorylations. Alternatively, if Btk molecules are phosphorylated at only site 1 or site 2, then two distinct populations of Btk could transduce independent signals.
Polyclonal antibodies were developed to specifically detect either phosphorylated site 1 or phosphorylated site 2. Although phosphopeptide-specific antibody preparations have previously been reported for other phosphorylated proteins (31–35), most have been used to detect phosphorylation of a single site rather than alternative sites within a protein. Btk phosphopeptide-specific antibodies were used to analyze changes in the phosphorylation at Btk sites 1 and 2 after activation of three alternative receptor pathways in human B cells, murine pro-B cells, and murine mast cells. Kinetic evaluation revealed rapid, but transient, phosphorylation at sites 1 and 2 in all of these receptor systems. Tyrosine-phosphorylated Btk molecules comprised only a small fraction of the total Btk population. Importantly, the tyrosine-phosphorylated subpopulation contained both singly and doubly tyrosine-phosphorylated molecules. We conclude that Btk activation by alternative receptor pathways involves phosphorylation of individual Btk molecules at two regulatory tyrosine residues. The concerted phosphorylation of sites 1 and 2 suggests that a multistep mechanism regulates recruitment of Btk into receptor signaling pathways.
MATERIALS AND METHODS
Generation of Immune Sera.
Reagents were obtained as indicated: BSA, protease-free (Sigma), 25% glutaraldehyde (Sigma), ovalbumin (Sigma), keyhole limpet hemocyanin (Pierce), Freund’s complete and incomplete adjuvants (GIBCO), activated CH Sepharose (Pharmacia), protein A Sepharose (Pharmacia), Centricon C-30 filter (Amicon), ECL reagent (Amersham), 4G10 monoclonal antiphosphotyrosine (Upstate Biotechnology, Lake Placid, NY), and goat anti-human IgM (Southern Biotechnology Associates). Peptides and phosphopeptides were synthesized using a Model 350 Multiple Peptide Synthesizer (Advanced ChemTech). Female New Zealand White rabbits were obtained from Universal Laboratories (Bloomington, CA). MCP-5 cells, anti-dinitrophenol (DNP) IgE, DNP-human serum albumin, and anti-Lyn serum were generously provided by A. Scharenberg (Beth Israel Hospital, Harvard Medical School). IL-5 was generously provided by K. Takatsu (University of Tokyo). Phosphopeptides corresponding to Btk amino acid sequences 218–229pY223 [KVVALY(PO4)DYMPMN] and 546–557pY551 [VLDDEY(PO4)TSSVGS] were coupled to BSA at a ratio of 2.5 mg:15 mg with 0.2% glutaraldehyde using a single-step protocol (36). Female New Zealand White rabbits were maintained, immunized, and bled in accordance with institutional guidelines for laboratory animal management. Rabbits (two per phosphopeptide) received intramuscular and/or subcutaneous immunizations with the BSA-phosphopeptide conjugates (0.6–1.5 mg BSA equivalent per dose per rabbit at monthly intervals) mixed with Freund’s complete (first dose only) or incomplete adjuvants. Rabbits were bled 10–25 days after each immunization, and the sera was collected, frozen, and stored at −20°C. The Btk peptides (218–229 and Lys-546–557) and phosphopeptides (218–229pY223 and Lys-546–557pY551) were coupled to activated CH Sepharose as recommended by the manufacturer, except that the peptide was coupled at a ratio of 2.0–2.5 mg:1 ml wet gel volume. BSA was coupled at a ratio of 15–30 mg:1 ml wet gel volume. A lysine residue was added to the amino terminus of the Btk 546–557 peptide and phosphopeptide to facilitate coupling to the resin.
Purification of Antibodies by Affinity Chromatography.
Rabbit immune serum was passed through a column containing an equal volume of protein A Sepharose. The protein A Sepharose was washed with PBS, pH 7.4 (10–20 column volumes), then the bound antibodies eluted with glycine 0.1 M, pH 2.7 (2–3 column volumes), monitoring protein concentration by OD at 280 nm. The eluate was neutralized to pH 7.0–7.5 with Tris 1.5 M, pH 8.8 (5–10% of eluate volume). The total Ig fraction then was concentrated through a Centricon C-30 filter to ≈50% of initial volume. The total Ig fraction was passed through a column of Sepharose-BSA (having a volume ≈50% of the initial serum volume) to deplete antibodies reactive with BSA. The Igs present in the flow-through fraction of the Sepharose-BSA column next were passed through a column of either Sepharose-Btk 218–229 or Sepharose-Btk Lys-546–557 (having a volume of 15–25% of the initial serum volume). This column depleted the Ig fraction of antibodies recognizing Btk peptide epitopes. It is important that this step is repeated until all of the peptide-reactive antibody is removed. The Igs present in the flow-through fraction of the Sepharose-peptide columns were passed through a column of either Sepharose-Btk 218–229pY223 or Sepharose-Btk Lys-546–557pY551 (having a volume of 10–25% of the initial serum volume). This column absorbed antibodies recognizing the desired phosphopeptide epitopes. After washing the Sepharose-phosphopeptide columns with PBS (10–20 column volumes), the bound antibodies were eluted with MgCl2 4.5 M (3–5 column volumes), monitoring protein concentration at OD at 280 nm. The eluates containing phosphopeptide-specific antibodies (223PYAb and 551PYAb) were dialyzed against PBS (4 × 500 eluate volumes). The antibody preparations were concentrated by dialysis against 40% glycerol in PBS. The concentrated antibody solutions were stored at −20°C. The antibody preparations were evaluated for purity by denaturing SDS/PAGE followed by silver staining. This revealed the presence of two proteins with molecular weights consistent with Ig heavy and light chains. The recovery of 223PYAb was ≈100 μg/ml serum, whereas the recovery of 551PYAb was ≈10 μg/ml serum.
Overexpression of Btk Proteins in Fibroblasts.
Vaccinia virus expression vectors for Btk wild-type and mutant proteins (Btk Y551F and Btk Y223F) were used to infect 3T3 fibroblast cells as described (17). Cell lysates of vaccinia virus-coinfected fibroblasts were prepared with cold lysis buffer (20 mM Hepes, pH 7.4/10% glycerol/3% Triton X-100/150 mM NaCl/10 mM NaF/1 mM NaVO4/2 mM EDTA/10 μg/ml aprotinin/10 μg/ml leupeptin/1 mM phenylmethylsulfonyl fluoride). The cell lysates were centrifuged for 15 min at 400,000 × g in a Beckman table top ultracentrifuge, and the soluble cell extracts were used for immunoprecipitation.
Btk Immunoblot and Immunoprecipitation Analysis.
Btk proteins overexpressed in fibroblasts as described above were immunoprecipitated from soluble cell extracts with protein A Sepharose and affinity-purified polyclonal antibodies against the N-terminal region of Btk (3). The proteins were separated by SDS/PAGE and transferred to nitrocellulose. After blocking the nitrocellulose (5% BSA/50 mM Tris, pH 7.5/150 mM NaCl/0.1% Tween-20), immunoblot analysis was performed with the indicated antibodies (≈0.2–1 μg/ml) in a solution containing 50 mM Tris 50 at pH 7.5, 500 mM NaC, and 0.1% Tween-20. The immunoblots were developed using goat anti-rabbit IgG-horseradish peroxidase as the secondary antibody, developed with ECL reagent, and exposed to film. Btk wild-type and mutant proteins overexpressed as described were immunoprecipitated (first cycle) with protein A Sepharose and anti-Btk N-terminal antibody. The immunoprecipitates were washed with lysis buffer, then Btk proteins were denatured by addition of 50 μl of Laemmli sample buffer and heating for 10 min at 90°C. The soluble, denatured Btk proteins were diluted ≈40-fold dilution with buffer (50 mM Tris, pH 7.4/100 mM NaCl/1 mM Na3VO4/0.1 mM phenylphosphate/2% Triton X-100/0.02% SDS). A second-cycle immunoprecipitation was performed on each Btk protein with protein A Sepharose and one of the following antibodies: anti-Btk N-terminal antibody, monoclonal 4G10 anti-phosphotyrosine antibody, 223PYAb, or 551PYAb. Phenylphosphate was omitted from the 4G10 immunoprecipitation. Immunoblot analysis was performed as described.
Stimulation of Cells.
Ramos B cells grown in RPMI 1640 culture medium supplemented with 10% calf serum were washed, then incubated in serum-free RPMI medium for 60 min before stimulation. Cells (0.5 ml, 2 × 108 cells/ml) were stimulated at 37°C with goat anti-human IgM (10 μg/ml). Cold lysis buffer (2 ml) was added to the cell suspensions. After centrifugation (15 min, 400,000 × g), immunoprecipitation was performed using protein A Sepharose and anti-Btk N-terminal antibody. For second-cycle immunoprecipitation, denatured Btk protein was diluted (as above) and divided into three equal aliquots. The aliquots were subjected to second-cycle immunoprecipitation in two steps: (i) aliquots were immunoprecipitated with either preimmune serum, 551PYAb, or 223PYAb; and (ii) Btk protein in the nonbound fraction of each aliquot was quantitatively recovered by immunoprecipitation with anti-Btk N-terminal antibody. Y16 pro-B cells were grown in RPMI culture medium supplemented with IL-5 (5 units/ml), 5% fetal calf serum, and 0.05 mM 2-mercaptoethanol. The cells were washed and incubated at 37°C for 6 hr in growth medium without IL-5. Sodium vanadate (1 mM) was added to the medium during the final hour. Cells (0.4 ml, 1 × 108 cells/ml) were stimulated with IL-5 (5,000 units/ml) at 37°C, then lysed and immunoprecipitated as described. MCP-5 mast cells were grown in RPMI culture medium supplemented with 10% calf serum and IL-3 (10% WEHI-3 cell culture supernatant). The cells were resuspended in serum-free RPMI plus anti-DNP IgE (10 μg/ml). After 60 min at 37°C cells were washed to remove unbound IgE, then resuspended in RPMI (0.4 ml, 5 × 107 cells/ml) and stimulated by addition of DNP-human serum albumin (0.2 μg/ml). Lysis and immunoprecipitation were performed as described.
B Cells Overexpressing Lyn.
Ramos B cells (5 × 106 cells/ml) were infected with vaccinia virus expression vector for the Lyn tyrosine kinase for 10–12 hr (17). The cells were washed and resuspended in serum-free RPMI medium for 60 min before stimulation. Cells (0.1 ml, 1 × 108 cells/ml) were stimulated with goat anti-human IgM (10 μg/ml). Lysis and immunoprecipitation was performed as described. Lyn expression was detected by anti-Lyn immunoblot of aliquots of soluble cell extracts.
RESULTS
Phosphopeptide Antibodies Specifically Detect Phosphorylation of Btk Sites 1 and 2.
To facilitate detailed investigation of the activation of Btk, phosphopeptide-specific antibodies were generated for the two Btk regulatory tyrosine phosphorylation sites. The immunization protocol, sequential adsorption, and affinity purification of phosphopeptide site-specific polyclonal antibodies for Btk sites 1 and 2 (551PYAb and 223PYAb, respectively) are described in Materials and Methods. A tripartite panel of Btk proteins was used to evaluate specificity in an immunoblot assay. Wild-type Btk and tyrosine phosphorylation site point mutant proteins, Btk Y551F and Btk Y223F, were produced separately using vaccinia virus expression vectors. Lyn was coexpressed with each Btk form to ensure high levels of Btk tyrosine phosphorylation. Btk proteins were immunoprecipitated from cell extracts with antibody generated using a glutathione S-transferase-Btk polypeptide (amino acids 1–196). The anti-Btk N-terminal antibody recognize the amino terminal portion of wild-type and point mutant proteins equally. The proteins were loaded in groups of three for separation by SDS/PAGE then transferred to nitrocellulose for immunoblot assays.
551PYAb and 223PYAb detected phosphorylation of wild-type Btk (Fig. 1A, lanes 8 and 11) as expected. Each antibody also detected tyrosine phosphorylation of the alternate Btk point mutant protein (i.e., 551PYAb binds to Btk Y223F, Fig. 1A, lane 9). The specificity of each antibody was confirmed by the failure to recognize the corresponding Btk phosphorylation site point mutant protein (i.e., 551PYAb does not bind to Btk Y551F, Fig. 1A, lane 7). Neither 551PYAb nor 223PYAb recognized Btk K430R (data not shown), a kinase inactive mutant in which, when expressed in the absence of Lyn, both phosphorylation sites are intact but unphosphorylated (17). Control immunoblot assays demonstrate that similar amounts of all three Btk proteins are present (anti-Btk N-terminal, Fig. 1A, lanes 1–3) and that a general anti-phosphotyrosine antibody (anti-P-Tyr, Fig. 1A, lanes 4–6) cannot distinguish between the phosphorylation of sites 1 and 2. This experiment demonstrates that 551PYAb and 223PYAb accurately report the phosphorylation state of an individual regulatory tyrosine residue, either Btk Y551 or Btk Y223, whereas the standard antiphosphotyrosine antibody detects only the total tyrosine phosphorylation of Btk.
Figure 1.
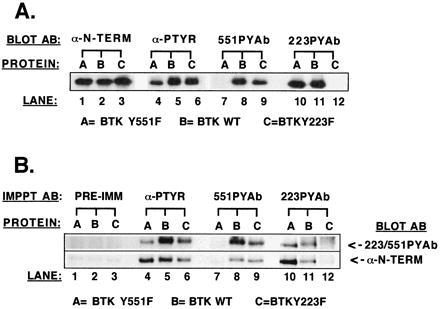
Test of phosphopeptide-specific antibodies as immunoblotting and immunoprecipitating reagents. Btk proteins (wild type, Btk Y551F, and Btk Y223F) were separately coexpressed in the presence of Lyn tyrosine kinase to maximize tyrosine phosphorylation of available sites. (A) Btk proteins were immunoprecipitated with anti-Btk N-terminal antibody, separated by SDS/PAGE, and transferred to nitrocellulose for immunoblot analysis with either anti-Btk N-terminal antibody, monoclonal 4G10 antiphosphotyrosine antibody, or phosphopeptide-specific antibody (223PYAb and 551PYAb). (B) Btk proteins first were immunoprecipitated with anti-Btk N-terminal antibody, then solubilized and immunoprecipitated in a second cycle with either Ig from a preimmune rabbit serum, monoclonal 4G10 antiphosphotyrosine antibody, or phosphopeptide-specific antibody (223PYAb and 551PYAb). After SDS/PAGE and transfer to nitrocellulose immunoblot analysis was performed with a mixture of 223PYAb and 551PYAb or the anti-Btk N-terminal antibody.
Immunoprecipitation Specificity Demonstrated with Overexpressed Wild-Type and Point Mutant Btk Proteins.
A protocol was designed to test whether or not the phosphopeptide-specific antibodies could immunoprecipitate Btk protein in a phosphotyrosine site-specific manner. Cell extracts containing either Btk wild-type, Y551F, or Y223F were prepared using vaccinia virus coexpression systems as described. The three Btk proteins were quantitatively immunoprecipitated from their cell extracts in a first-cycle immunoprecipitation by the anti-Btk N-terminal antibody. The immunoprecipitated Btk proteins were denatured, solubilized, and divided into aliquots. Second-cycle immunoprecipitation was performed on each denatured, soluble Btk protein using either a general antiphosphotyrosine antibody, 223PYAb, 551PYAb, or preimmune rabbit serum. Btk protein was visualized by immunoblot analysis with the anti-Btk N-terminal antibody or with a mixture of 551PYAb and 223PYAb (Fig. 1B). A trace amount of Btk protein could be detected in each of the preimmune immunoprecipitates (Fig. 1B, lanes 1–3). This represents the amount of Btk that nonspecifically binds to the protein A Sepharose-antibody complex. The general antiphosphotyrosine antibody recovers a significant amount of each Btk protein (Fig. 1B, lanes 4–6). The 551PYAb immunoprecipitated wild-type Btk and Btk Y223F but not Btk Y551F protein (Fig. 1B, lanes 7–9). The 223PYAb immunoprecipitated wild-type Btk and Y551F protein but not Btk Y223F (Fig. 1B, lanes 10–12). Therefore, these phosphopeptide-specific antibodies are capable of immunoprecipitating the respective populations of Btk molecules phosphorylated at a specific tyrosine residue.
Btk Y551 and Y223 Are Rapidly Phosphorylated in Distinct Receptor Signaling Pathways.
Three distinct receptor signaling pathways were evaluated to test the hypothesis that phosphorylation of the two regulatory tyrosine residues is a common element in Btk function. These receptor pathways–the BCR (14–17), the IL-5 receptor (5–7, 22), and the Fcɛ receptor (FcɛRI) (37–39)—previously have been demonstrated in genetic and biochemical tests to use Btk. To evaluate and compare the kinetics of tyrosine phosphorylation of Btk sites 1 and 2, the alternative receptor pathways were activated for increasing lengths of time (0–30 min). Btk was quantitatively immunoprecipitated with anti-Btk N-terminal antibody (i.e., using antibody sufficient to recover > 95% of the total amount of Btk). Phosphorylation of Btk at each time point was measured by immunoblot analysis with either a general antiphosphotyrosine antibody, 551PYAb, or 223PYAb.
Crosslinking of the BCR on the Ramos B cell line was previously demonstrated to enhance phosphorylation of Btk sites 1 and 2 in an experiment using radioactive [32P]orthophosphate labeling and peptide mapping techniques (17). To confirm and extend that observation, Btk from control and stimulated Ramos cells was immunopurified and evaluated as described. The total phosphotyrosine content of Btk (Fig. 2A, Top) rapidly increases after cell stimulation, reaching a maximum at 2–5 min. In comparison, immunoblot analysis with the phosphopeptide-specific antibodies (Fig. 2A, Middle) reveals a subtle distinction between the kinetics of phosphorylation of the two sites. Site 1 phosphorylation was maximal at 30 sec. The increase in site 2 phosphorylation was detected at 30 sec and maximal at 2–5 min. The magnitude of site 1 phosphorylation diminished rapidly, whereas site 2 phosphorylation appeared to decrease more slowly. Phosphorylation of each site was similar to the basal level 30 min after stimulation. These data demonstrate a rapid and transient increase in the phosphorylation of both Btk regulatory tyrosine residues after ligation of the BCR.
Figure 2.
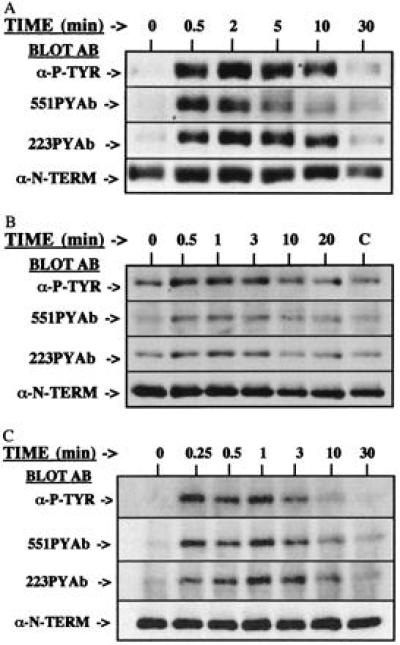
Kinetics of phosphorylation of Btk sites 1 and 2 after stimulation of alternative receptor pathways. (A) Ramos B cells were activated by crosslinking of the BCR with goat anti-human IgM for 0–30 min. (B) IL-5-dependent Y16 pro-B cells were deprived of IL-5, then stimulated 0–20 min by addition of IL-5 (lane C, 20 min, no IL-5). (C) MCP-5 mast cells were primed with anti-DNP IgE, then FcɛRI was crosslinked by addition of DNP-human serum albumin for 0–30 min. After immunoprecipitation, the phosphorylation of Btk was demonstrated by immunoblot analysis with monoclonal 4G10 antiphosphotyrosine antibody, 551PYAb, or 223PYAb. The amount of Btk protein is demonstrated by immunoblot analysis with anti-Btk N-terminal antibody.
The IL-5-dependent Y-16 murine pro-B cell line is a model cell system for analysis of the IL-5 receptor signaling pathway (5). Y-16 cells were deprived of IL-5 for 6 hr, then a saturating dose of the growth factor was added for 0–20 min to activate the IL-5 receptor signaling pathway. The general antiphosphotyrosine antibody (Fig. 2B, Top) detects an increase in total Btk tyrosine phosphorylation within 30 sec after IL-5 stimulation. The phosphopeptide-specific antibodies (Fig. 2B, Middle) demonstrate rapid and transient increase in the phosphorylation of each site. The pattern of phosphorylation of Btk regulatory tyrosine residues induced by IL-5R activation is similar to that induced by ligation of the BCR. Importantly, these alternative receptor pathways function at different stages of B cell development. Thus, two distinct receptor signaling pathways, each of which control aspects of B cell development and function, activate Btk through concerted phosphorylation of the two regulatory tyrosine residues.
Although Btk is best known for its role in B cell development and function, it also is expressed in and required for certain mast cell responses (38, 39). The high-affinity receptor for IgE, FcɛRI, is an important regulator of mast cell function (40). The MCP-5 mast cell line (41) was used to evaluate the kinetics of phosphorylation of Btk sites 1 and 2 after crosslinking of the FcɛRI for increasing lengths of time. The total antiphosphotyrosine immunoblot (Fig. 2C, Top) demonstrates a very rapid response to the crosslinking of the FcɛRI. The phosphopeptide-specific antibodies demonstrate that the response includes increased phosphorylation of both Y551 and Y223 within 15 sec of crosslinking (Fig. 2C, Middle). Phosphorylation of each site falls to nearly baseline level by 10 min after stimulation. Together, these data strongly support the conclusion that phosphorylation of these two regulatory tyrosine residues is a critical component of the regulation of Btk function in multiple receptor signaling pathways.
Influence of Lyn Dosage on BCR-Dependent Stimulation of Btk Phosphorylation.
Heterologous expression of Btk in several systems has clearly demonstrated the influence of Src family tyrosine kinases on Btk function (17, 20, 21). However, none of these studies directly evaluated whether Src family kinases mediate the receptor-dependent increased tyrosine phosphorylation of Btk. To test this hypothesis the tyrosine phosphorylation of Btk was assessed in B cells expressing basal or enhanced amounts levels of Lyn. Control Ramos B cells and cells infected with a vaccinia virus expression vector for Lyn were treated with anti-IgM to activate the BCR for 0–30 min. The total phosphotyrosine content of Btk was analyzed by immunoprecipitation followed by general antiphosphotyrosine immunoblot (Fig. 3, Top). A marked increase in BCR-mediated tyrosine phosphorylation of Btk is found in the presence of an increased dosage of Lyn. Importantly, the dosage of Btk in the cells (Fig. 3, Middle) was not influenced by the increased Lyn dosage (Fig. 3, Bottom). The data are consistent with previous results in heterologous expression systems, but also demonstrate the dose-response influence of Lyn upon Btk phosphorylation in the BCR signaling pathway. The influence of Lyn dosage upon phosphorylation of the Btk regulatory tyrosine residues also was examined by immunoblots using the phosphopeptide-specific antibodies. Phosphorylation of both site 1 and site 2 phosphorylation after BCR crosslinking is increased in the cells overexpressing Lyn (Fig. 4). This experiment confirms the direct relationship between Lyn dosage and the BCR-mediated increase in phosphorylation of Y551 and Y223.
Figure 3.
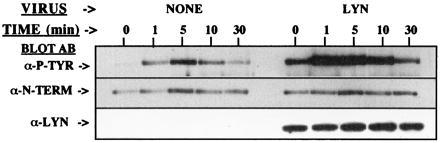
Enhanced BCR-mediated tyrosine phosphorylation of Btk after overexpression of Lyn. Ramos B cells uninfected or overexpressing Lyn were treated with or without anti-IgM for 0–30 min. After immunoprecipitation, the phosphorylation of Btk was demonstrated by immunoblot analysis with monoclonal 4G10 antiphosphotyrosine antibody. The level of Btk or Lyn expression was assessed by immunoblot analysis with anti-Btk N-terminal or anti-Lyn antibodies.
Figure 4.
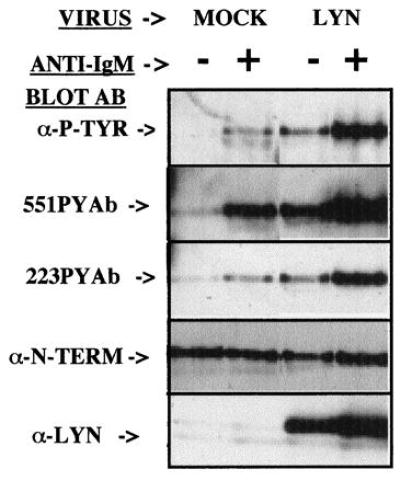
Increased Lyn dosage enhances BCR-mediated phosphorylation of Btk sites 1 and 2. Ramos B cells infected with control or Lyn-expressing virus were treated without or with anti-IgM for 5 min. After immunoprecipitation, phosphorylation of Btk was demonstrated by immunoblot analysis with monoclonal 4G10 antiphosphotyrosine antibody, 551PYAb, or 223PYAb, as indicated. The level of Btk or Lyn expression was assessed by immunoblot analysis with anti-Btk N-terminal or anti-Lyn antibodies.
A Small Fraction of the Total Btk Population Is Tyrosine-Phosphorylated After Crosslinking of the BCR on B Cells.
To determine the fraction of Btk molecules recruited into a receptor signaling pathway after maximal receptor activation, phosphorylated Btk molecules were recovered from soluble extracts of control and BCR-crosslinked Ramos B cells using a stepwise immunoprecipitation protocol that accounts for all Btk molecules present (see Materials and Methods). Btk molecules phosphorylated at each site were immunoprecipitated with 551PYAb or 223PYAb and compared with anti-Btk N terminal, which recovers the total population of Btk molecules (Fig. 5 A and B). 551PYAb immunoprecipitated nearly all of the site 1-phosphorylated molecules, as seen by comparison of Fig. 5A lanes 2 and 4. 223PYAb immunoprecipitated only a portion (≈10–15%) of the Btk molecules phosphorylated at site 2, as seen by comparison of Fig. 5B, lanes 2 and 4.
Figure 5.
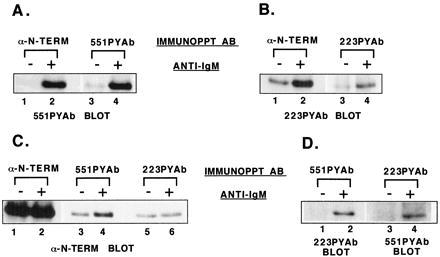
Analysis of the fraction of Btk molecules phosphorylated and the pattern of tyrosine phosphorylation. Ramos B cells were untreated or stimulated with goat anti-human IgM for 1 min. Btk protein was immunoprecipitated from cell extracts with anti-Btk N-terminal antibody, then resolubilized and diluted. Btk from untreated or stimulated samples was immunoprecipitated with 551PYAb, 223PYAb, or preimmune serum. The remaining soluble fraction of Btk then was recovered by immunoprecipitation with anti-Btk N-terminal antibody. Immunoblot analysis was performed as indicated. (A) Comparison of the amount of site 1-phosphorylated Btk recovered from untreated or stimulated cell samples by immunoprecipitation with anti-Btk N-terminal or 551PYAb. (B) Comparison of the amount of site 2-phosphorylated Btk recovered from untreated or stimulated cell samples by immunoprecipitation with anti-Btk N-terminal or 223PYAb. (C) Comparison of the amount of Btk recovered from untreated or stimulated cell samples by immunoprecipitation with 551PYAb, 223PYAb, or anti-Btk N-terminal antibody. (D) Double phosphorylation of Btk molecules recovered from untreated or stimulated cell samples by immunoprecipitation with 551PYAb or 223PYAb, detected by immunoblot analysis with the alternative phosphopeptide-specific antibody.
The fraction of Btk proteins phosphorylated at each site was estimated by anti-Btk N-terminal immunoblot analysis of 551PYAb, 223PYAb, and anti-Btk N-terminal immunoprecipitates (Fig. 5C). The total population of Btk from control and stimulated cells is represented by lanes 1 and 2 of Fig. 5C. The background level of Btk bound to protein A Sepharose in this assay is represented by the signals in Fig. 5C, lanes 3, 5, and 6 and the preimmune serum control (data not shown). The fraction of Btk phosphorylated on site 1 thus can be estimated from the strength of the signal in Fig. 5C, lane 4. Timed serial exposures of the immunoblots to film indicated that approximately 2% of the total Btk population is phosphorylated at site 1 (i.e., the specific signal in Fig. 5C, lane 4). Calculation of the fraction of Btk molecules phosphorylated at site 2 is more difficult because only 10–15% of that subpopulation was immunoprecipitated by 223PYAb, but it appears that as much as 5% of the total Btk population is phosphorylated on site 2. Although the values derived with this method are rough estimates, it is clear that the phosphorylated population of Btk molecules is a small fraction of the total cellular pool.
Btk Molecules Are Singly and Doubly Tyrosine-Phosphorylated After BCR Crosslinking.
The pattern of phosphorylation of individual Btk molecules gives important clues to how Btk is activated and how it functions in signal transduction. Importantly, receptor-mediated induction of Btk phosphorylation at two regulatory tyrosine residues can occur through alternative mechanisms: e.g., a single population of individual Btk molecules may be phosphorylated at both sites 1 and 2, or two populations of Btk molecules may be phosphorylated at one site but not both. To distinguish between the possible patterns of Btk phosphorylation after BCR crosslinking, the 551PYAb and 223PYAb immunoprecipitates were immunoblotted with the alternate phosphopeptide-specific antibody (Fig. 5D). Btk protein immunoprecipitated with 551PYAb from BCR-activated samples contains a detectable site 2 phosphorylation (Fig. 5D, lane 2). A complementary result demonstrates that the 223PYAb immunoprecipitate from BCR-activated samples contains a detectable site 1 phosphorylation (Fig. 5D, lane 4). These results establish that Btk molecules can be doubly tyrosine-phosphorylated. Based upon comparison of timed serial exposures, approximately 50% of Btk phosphorylated at site 1 also is phosphorylated at site 2 and approximately 20% of site 2 phosphorylated molecules are phosphorylated at site 1. This result indicates that approximately 1% of the total Btk population is doubly phosphorylated. Although this method of quantitation again provides only a rough estimate, if doubly phosphorylated Btk molecules were produced only through a random process, the predicted fraction of doubly phosphorylated molecules should be ≈0.1%, or 10-fold smaller than observed.
DISCUSSION
Btk is a member of the Tec family of cytoplasmic tyrosine kinases (42). This family is structurally distinct from other tyrosine kinases because its members have pleckstrin homology (PH) and Tec homology domains at the N terminus and lack a C-terminal autoinhibitory domain. These structural differences suggest that the function of Tec family tyrosine kinases is regulated by mechanisms distinct from other kinases. To study the mechanism of Btk regulation by phosphorylation at sites 1 and 2, phosphopeptide-specific antibodies were developed to specifically detect each site. Phosphorylation of these critical tyrosine residues was an early and consistent feature of Btk activation in three alternative receptor signaling pathways and was augmented by an increased dosage of Src family kinase. Btk molecules were found to be both singly and doubly phosphorylated after BCR crosslinking. Taken together with the kinetic data, we conclude that a single subpopulation of Btk transduces the distinct regulatory signals resulting from phosphorylation of both sites 1 and 2. In this model, the doubly tyrosine-phosphorylated Btk molecule represents a transient intermediate in a dynamic cycle of activation and deactivation. Thus, Btk undergoes transphosphorylation by Src family kinase to form Btk 551PY, autophosphorylation to form Btk 551PY/223PY, dephosphorylation of site 1 to form Btk 223PY, then dephosphorylation at site 2 to yield unphosphorylated Btk (Fig. 6). Btk’s function as a transducer for alternative receptor signaling pathways is likely to be influenced by other proteins. These comodulators could interact directly or indirectly with specific elements of Btk’s complex domain structure to modify the activation/deactivation cycle.
Figure 6.
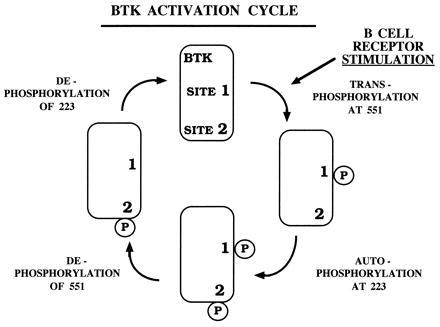
A schematic representation of a Btk activation/deactivation cycle after crosslinking of BCR, including the sequential phosphorylation and dephosphorylation of the regulatory tyrosine residues Y551 (site 1) and Y223 (site 2).
The Janus kinase family member JAK2 is a critical early component of the IL-5R signaling pathway (43, 44). In contrast, a role for Src family kinases, including Lyn, in IL-5 signaling has not yet been described. Our results suggest either that Janus family kinases can directly transphosphorylate and activate Btk, or that Src family kinases can be activated after IL-5R stimulation. A precedent for the involvement of Lyn in the erythropoietin receptor signaling pathway recently has been described (45). Lyn was required for erythropoietin-induced cell differentiation via a mechanism involving direct binding of Lyn to the erythropoietin receptor. This receptor is similar to the IL-5R in its capacity to activate JAK2 (46). Further study is required to clarify the potential mechanisms linking the IL-5R and other cytokine receptors to Btk activation.
How does the mechanism of Btk activation relate to that of other cytoplasmic tyrosine kinases? Autoinhibition of inactive Src results from multiple intramolecular interactions that distort the catalytic site (47, 48). Activation of Src family kinases is intimately related to dephosphorylation of the C-terminal inhibitory region, which is not present in Btk, and phosphorylation within the SH1 domain “activation loop” (49). It is possible that Btk is also intramolecularly wrapped in an autoinhibitory conformation, but that the requirements to release it to an active conformation are distinct from those for Src. As in Src, phosphorylation of the Btk activation loop residue (Y551) appears to activate the kinase by “unblocking” the catalytic site. An early target for this activated kinase is the autophosphorylation site Y223 within the SH3 domain. Several results suggest site 2 is critical to Btk intramolecular or intermolecular binding events. NMR structural data for a fragment of the structurally related T cell cytoplasmic tyrosine kinase, Itk, demonstrated the intramolecular binding of Itk SH3 domain to a proline-rich region within the Tec homology domain (50). Thus, this Tec homology domain/SH3 domain may modulate access of other proteins to these domains. Site 2 autophosphorylation may alter the stability of this interaction, leading to a global change in Btk conformation. This may influence binding with signaling proteins known to interact with the Btk SH3 domain, such as c-Cbl (51) and Lyn (52). Autophosphorylation of site 2 also may influence binding of proteins to other Btk domains. A 135-kDa Btk-associated protein (BAP-135) binds to the Btk PH domain (53). In a heterologous expression system, Btk-dependent phosphorylation of the Btk-associated protein BAP-135 is abrogated by the Y223F mutation, suggesting that the Btk-BAP135 interaction relies upon phosphorylation of site 2. Another possible role for Y223 phosphorylation is the creation of a binding site (-Y(P)-D-Y-M-) for a phosphotyrosine-binding protein. The Btk sequence is similar to the binding site sequence (-Y(P)-X-X-M-) for the SH2 domain of the p85 subunit of phosphatidylinositol 3′-kinase (54). However, because the Btk SH3 domain may maintain its tertiary structure after phosphorylation of Y223, the specificity for binding to this residue may not be solely determined by primary sequence at that site.
Finally, it is likely that additional elements regulating Btk activation may be present in the N-terminal region of the protein. The capacity of the PH domain to bind to the intracellular second messenger molecule phosphatidylinositol 3,4,5-trisphosphate (55) may regulate binding of Btk to the plasma membrane, locating it in close proximity to the activated receptor and associated kinases. Alteration of this PH domain-mediated lipid binding interaction may be the molecular basis for both the activating mutation, Btk*, and the loss of function mutation, X-linked immunodeficiency (55, 56). Direct binding of G protein βγ heterodimers to the PH domain can modulate Btk activity (57). Protein kinase C can phosphorylate serine residues within the PH domain (58), and a mouse strain harboring a targeted deletion of the protein kinase C-β gene has a phenotype strikingly similar to X-linked immunodeficiency (59). Finally, an intriguing feature of Btk’s structure is the presence of a Zn2+-binding site adjacent to the PH domain. Whether the role of this Zn2+ binding domain is structural or catalytic remains unknown. However, mutation of a cysteine residue involved in binding the Zn2+ results in X-linked agammaglobulinemia, demonstrating its importance in Btk function (60). Evaluation of the contributions of these and other interactions to the regulation of Btk function requires further structural and functional analysis.
Acknowledgments
We thank Dr. A. Scharenberg and Dr. J.-P. Kinet for recombinant vaccinia virus stocks and reagents for activation of MCP-5 cells, Dr. C. Turck of the Howard Hughes Medical Institute/University of California at San Francisco Protein Structure Laboratory for preparation of synthetic peptides, T. Li for assistance with rabbit sera, J.C. White for assistance with the figures, and Drs. S. Smale, D. Black, A. Berk, and L. Zipursky for their helpful comments. This work was supported by a Human Genetics Training Grant GM08243 Fellowship and the Cancer Research Fund of the Damon Runyon-Walter Winchell Foundation Fellowship, DRG-086 (M.I.W.), National Institutes of Health Physician Scientist Award AR01912 (D.J.R.), Human Frontier Science Program Fellowship (A.C.F.), Cancer Research Institute Fellowship (H.P.), and National Cancer Institute CA 12800 (Principal Investigator Dr. Randy Wall, project leader, O.N.W.). O.N.W. is an Investigator of the Howard Hughes Medical Institute.
ABBREVIATIONS
- BCR
B cell antigen receptor
- Btk
Bruton’s tyrosine kinase
- IL
interleukin
- PH
pleckstrin homology
- SH
Src homology
- DNP
dinitrophenol
- FcɛRI
Fcɛ receptor
References
- 1.Rawlings D J, Saffran D C, Tsukada S, Largaespada D A, Grimaldi J C, Cohen L, Mohr R N, Bazan J F, Howard M, Copeland N G, Jenkins N A, Witte O N. Science. 1993;261:358–361. doi: 10.1126/science.8332901. [DOI] [PubMed] [Google Scholar]
- 2.Thomas J D, Sideras P, Smith C I E, Vorechovsky I, Chapman V, Paul W E. Science. 1993;261:355–358. doi: 10.1126/science.8332900. [DOI] [PubMed] [Google Scholar]
- 3.Tsukada S, Saffran D C, Rawlings D J, Parolini O, Allen R C, Klisak I, Sparkes R S, Kubagawa H, Mohandas T, Quan S, Belmont J W, Cooper M D, Conley M E, Witte O N. Cell. 1993;72:279–290. doi: 10.1016/0092-8674(93)90667-f. [DOI] [PubMed] [Google Scholar]
- 4.Vetrie D, Vorechovsky I, Sideras P, Holland J, Davies A, Flinter F, Hammarström L, Kinnon C, Levinsky R, Bobrow M, Smith C I E, Bentley D R. Nature (London) 1993;361:226–233. doi: 10.1038/361226a0. [DOI] [PubMed] [Google Scholar]
- 5.Sato S, Katagiri T, Takaki S, Kikuchi Y, Hitoshi Y, Yonehara S, Tsukada S, Kitamura D, Watanabe T, Witte O, Takatsu K. J Exp Med. 1994;180:2101–2111. doi: 10.1084/jem.180.6.2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hitoshi Y, Sonoda E, Kikuchi Y, Yonehara S, Nakauchi H, Takatsu K. Int Immunol. 1993;5:1183–1190. doi: 10.1093/intimm/5.9.1183. [DOI] [PubMed] [Google Scholar]
- 7.Koike M, Kikuchi Y, Tominaga A, Takaki S, Akagi K, Miyazaki J, Yamamura K, Takatsu K. Int Immunol. 1995;7:21–30. doi: 10.1093/intimm/7.1.21. [DOI] [PubMed] [Google Scholar]
- 8.Matsuda T, Takahashi-Tezuka M, Fukada T, Okuyama Y, Fujitani Y, Tsukada S, Mano H, Hirai H, Witte O N, Hirano T. Blood. 1995;85:627–633. [PubMed] [Google Scholar]
- 9.Go N F, Castle B E, Barret R, Kastelein R, Dang W, Mosmann T R, Moore K W, Howard M. J Exp Med. 1990;172:1625–1631. doi: 10.1084/jem.172.6.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Santos-Argumedo L, Lund F E, Heath A W, Solvason N, Wu W W, Grimaldi J C, Parkhouse R M E, Howard M. Int Immunol. 1995;7:163–170. doi: 10.1093/intimm/7.2.163. [DOI] [PubMed] [Google Scholar]
- 11.Yamashita Y, Miyake K, Kikuchi Y, Takatsu K, Noda T, Kosugi A, Kimoto M. Immunology. 1995;2:248–255. [PMC free article] [PubMed] [Google Scholar]
- 12.Aoki Y, Isselbacher K J, Pillai S. Proc Natl Acad Sci USA. 1994;91:10606–10609. doi: 10.1073/pnas.91.22.10606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cambier J C, Pleiman C M, Clark M R. Annu Rev Immunol. 1994;12:457–486. doi: 10.1146/annurev.iy.12.040194.002325. [DOI] [PubMed] [Google Scholar]
- 14.de Weers M, Brouns G S, Hinshelwood S, Kinnon C, Schuurman R K B, Hendriks R W, Borst J. J Biol Chem. 1994;269:23857–23860. [PubMed] [Google Scholar]
- 15.Hinshelwood S, Lovering R C, Genevier H C, Levinsky R J, Kinnon C. Eur J Immunol. 1995;25:1113–1116. doi: 10.1002/eji.1830250439. [DOI] [PubMed] [Google Scholar]
- 16.Pleiman C M, D’Ambrosio D, Cambier J C. Immunol Today. 1994;15:393–398. doi: 10.1016/0167-5699(94)90267-4. [DOI] [PubMed] [Google Scholar]
- 17.Rawlings D J, Scharenberg A M, Park H, Wahl M I, Lin S, Kato R M, Fluckiger A C, Witte O N, Kinet J P. Science. 1996;271:822–825. doi: 10.1126/science.271.5250.822. [DOI] [PubMed] [Google Scholar]
- 18.Saouaf S J, Mahajan S, Rowley R B, Kut S, Fargnoli J, Burkhardt A L, Tsukada S, Witte O N, Bolen J B. Proc Natl Acad Sci USA. 1994;91:9524–9528. doi: 10.1073/pnas.91.20.9524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamanashi Y, Kakiuchi T, Mizuguchi J, Yamamoto T, Toyoshima K. Science. 1991;251:192–194. doi: 10.1126/science.1702903. [DOI] [PubMed] [Google Scholar]
- 20.Mahajan S, Fargnoli J, Burkhardt A L, Kut S A, Saouaf S J, Bolen J B. Mol Cell Biol. 1995;15:5304–5311. doi: 10.1128/mcb.15.10.5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Afar D E H, Park H, Howell B W, Rawlings D J, Cooper J, Witte O N. Mol Cell Biol. 1996;16:3465–3471. doi: 10.1128/mcb.16.7.3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li T, Tsukada S, Satterthwaite A, Havlik M H, Park H, Takatsu K, Witte O N. Immunity. 1995;2:451–460. doi: 10.1016/1074-7613(95)90026-8. [DOI] [PubMed] [Google Scholar]
- 23.Park H, Wahl M I, Afar D E, Turck C W, Rawlings D J, Tam C, Scharenberg A M, Kinet J-P, Witte O N. Immunity. 1996;4:515–525. doi: 10.1016/s1074-7613(00)80417-3. [DOI] [PubMed] [Google Scholar]
- 24.Feng S, Chen J K, Yu H, Simon J A, Schreiber S L. Science. 1994;266:1241–1247. doi: 10.1126/science.7526465. [DOI] [PubMed] [Google Scholar]
- 25.Goudreau N, Cornille F, Duchesne M, Parker F, Tocque B, Garbay C, Roques B P. Nat Struct Biol. 1994;1:898–907. doi: 10.1038/nsb1294-898. [DOI] [PubMed] [Google Scholar]
- 26.Koyama S, Yu H, Dalgarno D C, Shin T B, Zydowsky L D, Schreiber S L. Cell. 1993;72:945–952. doi: 10.1016/0092-8674(93)90582-b. [DOI] [PubMed] [Google Scholar]
- 27.Lim W A, Richards F M, Fox R O. Nature (London) 1994;372:375–379. doi: 10.1038/372375a0. [DOI] [PubMed] [Google Scholar]
- 28.Noble M E M, Musacchio A, Saraste M, Courtneidge S A, Wierenga R K. EMBO J. 1993;12:2617–2624. doi: 10.2210/pdb1shf/pdb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Terasawa H, Kohda D, Hatanaka H, Tsuchiya S, Ogura K, Nagata K, Ishii S, Mandiyan V, Ullrich A, Schlessinger J, Inagaki F. Nat Struct Biol. 1994;1:891–897. doi: 10.1038/nsb1294-891. [DOI] [PubMed] [Google Scholar]
- 30.Yu H, Chen J K, Feng S, Dalgarno D C, Brauer A W, Schreiber S L. Cell. 1994;76:933–945. doi: 10.1016/0092-8674(94)90367-0. [DOI] [PubMed] [Google Scholar]
- 31.Bangalore L, Tanner A J, Laudano A P, Stern D F. Proc Natl Acad Sci USA. 1992;89:11637–11641. doi: 10.1073/pnas.89.23.11637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Epstein R J, Druker B J, Roberts T M, Stiles C D. Proc Natl Acad Sci USA. 1992;89:10435–10439. doi: 10.1073/pnas.89.21.10435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ginty D D, Kornhauser J M, Thompson M A, Bading H, Mayo K E, Takahashi J S, Greenberg M E. Science. 1993;260:238–241. doi: 10.1126/science.8097062. [DOI] [PubMed] [Google Scholar]
- 34.Kawakatsu H, Sakai T, Takagaki Y, Shinoda Y, Saito M, Owada M K, Yano J. J Biol Chem. 1996;271:5680–5685. doi: 10.1074/jbc.271.10.5680. [DOI] [PubMed] [Google Scholar]
- 35.Tzartos S J, Kouvatsou R, Tzartos E. Eur J Biochem. 1995;228:463–472. [PubMed] [Google Scholar]
- 36.Harlowe E, Lane D. Antibodies: A Laboratory Manual. Plainview, New York: Cold Spring Harbor Lab. Press; 1988. pp. 79–85. [Google Scholar]
- 37.Dvorak A M, Miura T, Letourneau L, Ishizaka T, Kawakami T. Int Arch Allergy Immunol. 1996;111:118–125. doi: 10.1159/000237355. [DOI] [PubMed] [Google Scholar]
- 38.Kawakami Y, Yao L, Miura T, Tsukada S, Witte O N, Kawakami T. Mol Cell Biol. 1994;14:5108–5113. doi: 10.1128/mcb.14.8.5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kawakami Y, Miura T, Bissonnette R, Hata D, Khan W N, Kitamura T, Maeda-Yamamoto M, Hartman S E, Yao L, Alt F W, Kawakami T. Proc Natl Acad Sci USA. 1997;94:3938–3942. doi: 10.1073/pnas.94.8.3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Razin E, Pecht I, Rivera J. Immunol Today. 1995;16:370–373. doi: 10.1016/0167-5699(95)80003-4. [DOI] [PubMed] [Google Scholar]
- 41.Arora N, Min K-U, Costa J J, Rhim J S, Metcalfe D D. Int Arch Allergy Immunol. 1993;100:319–327. doi: 10.1159/000236432. [DOI] [PubMed] [Google Scholar]
- 42.Neet K, Hunter T. Genes Cells. 1996;1:147–169. doi: 10.1046/j.1365-2443.1996.d01-234.x. [DOI] [PubMed] [Google Scholar]
- 43.Pazdrak K, Stafford S, Alam R. J Immunol. 1995;155:397–402. [PubMed] [Google Scholar]
- 44.van der Bruggen T, Caldenhoven E, Kanters D, Coffer P, Raaijmakers J A, Lammers J W, Koenderman L. Blood. 1995;85:1442–1448. [PubMed] [Google Scholar]
- 45.Tilbrook P A, Ingley E, Williams J H, Hibbs M L, Klinken S P. EMBO J. 1997;16:1610–1619. doi: 10.1093/emboj/16.7.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Witthuhn B A, Quelle F W, Silvennoinen O, Yi T, Tang B, Miura O, Ihle J N. Cell. 1993;74:227–236. doi: 10.1016/0092-8674(93)90414-l. [DOI] [PubMed] [Google Scholar]
- 47.Sicheri F, Moarefi I, Kuriyan J. Nature (London) 1997;385:602–609. doi: 10.1038/385602a0. [DOI] [PubMed] [Google Scholar]
- 48.Xu W, Harrison S, Eck M. Nature (London) 1997;385:595–602. doi: 10.1038/385595a0. [DOI] [PubMed] [Google Scholar]
- 49.Superti-Furga G, Courtneidge S A. BioEssays. 1995;17:321–330. doi: 10.1002/bies.950170408. [DOI] [PubMed] [Google Scholar]
- 50.Andreotti A H, Bunnell S C, Feng G, Berg L J, Schreiber S L. Nature (London) 1997;385:93–97. doi: 10.1038/385093a0. [DOI] [PubMed] [Google Scholar]
- 51.Cory G O C, Lovering R C, Hinshelwood S, MacCarthy-Morrogh L, Levinsky R J, Kinnon C. J Exp Med. 1995;182:611–615. doi: 10.1084/jem.182.2.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cheng G, Ye, Zheng-S, Baltimore D. Proc Natl Acad Sci USA. 1994;91:8152–8155. doi: 10.1073/pnas.91.17.8152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang W, Desiderio S. Proc Natl Acad Sci USA. 1997;94:604–609. doi: 10.1073/pnas.94.2.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Songyang Z, Shoelson S E, Chaudhuri M, Gish G, Pawson T, Haser W G, King F, Roberts T, Ratnofsky S, Lechleider R J, Neel B G, Birge R B, Fajardo J E, Chou M M, Hanafusa H, Schaffhausen B, Cantley L C. Cell. 1993;72:767–778. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- 55.Salim K, Bottomley M J, Querfurth E, Zvelebil M J, Gout I, Scaife R, Margolis R L, Gigg R, Smith C I, Driscoll P C, Waterfiel M D, Panayotou G. EMBO J. 1996;15:6241–6250. [PMC free article] [PubMed] [Google Scholar]
- 56.Ferguson K M, Lemmon M A, Schlessinger J, Sigler P B. Cell. 1995;83:1037–1046. doi: 10.1016/0092-8674(95)90219-8. [DOI] [PubMed] [Google Scholar]
- 57.Langhans-Rajasekaran S A, Wan Y, Huang X Y. Proc Natl Acad Sci USA. 1995;92:8601–8605. doi: 10.1073/pnas.92.19.8601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yao L, Kawakami Y, Kawakami T. Proc Natl Acad Sci USA. 1994;91:9175–9179. doi: 10.1073/pnas.91.19.9175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Leitges M, Schmedt C, Guinamard R, Davoust J, Schaal S, Stabel S, Tarakhovsky A. Science. 1996;273:788–791. doi: 10.1126/science.273.5276.788. [DOI] [PubMed] [Google Scholar]
- 60.Hyvonen M, Saraste M. EMBO J. 1997;16:3396–3404. doi: 10.1093/emboj/16.12.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]