

Abstract
We have generated a mouse where the clotting factor IX (FIX) gene has been disrupted by homologous recombination. The FIX nullizygous (−/−) mouse was devoid of factor IX antigen in plasma. Consistent with the bleeding disorder, the factor IX coagulant activities for wild-type (+/+), heterozygous (+/−), and homozygous (−/−) mice were 92%, 53%, and <5%, respectively, in activated partial thromboplastin time assays. Plasma factor IX activity in the deficient mice (−/−) was restored by introducing wild-type murine FIX gene via adenoviral vectors. Thus, these factor IX-deficient mice provide a useful animal model for gene therapy studies of hemophilia B.
Hemophilia B (Christmas disease or factor IX deficiency) is a bleeding disorder that is inherited as an X chromosome-linked recessive trait. The disease affects approximately 1 in 30,000 males and is distributed equally among ethnic groups (1, 2). The clinical presentation of the disease can be mild, moderate, or severe and is clinically indistinguishable from classic hemophilia A due to factor VIII deficiency (3, 4). Hemophilia B results from the absence of normal factor IX, a plasma serine protease that converts factor X to its active form, thereby propagating the well-orchestrated coagulation cascade (5). Affected individuals are treated with repeated injections of either human plasma factor IX concentrate or recombinant factor IX protein. Because this form of treatment is neither satisfactory nor long lasting, hemophilia B has become a very attractive target for gene therapy (6).
Several investigators have used a variety of viral vectors to introduce factor IX genes in a variety of tissues in mice and hemophilic dogs. Retroviral vectors containing canine or human factor IX cDNA can efficiently transduce fibroblasts and produce biologically active factor IX protein; however, the expression is transient upon transplantation of transduced cells in the animals (7, 8). The problem of long-term expression was overcome by the use of tissue-specific enhancers when in vitro-transduced mouse myoblasts were transplanted in vivo (9). However, similar experiments failed when transduced dog myoblasts were transplanted in the hemophilic dogs (Y. Dai and I.M.V., unpublished data). Very low levels of sustained expression of factor IX could be detected in partially hepatectomized hemophilic dogs injected with canine factor IX recombinant retrovirus (10). Recombinant adenoviral vectors containing factor IX gene have been delivered directly in the muscle or liver of both nude and normal mice. The accumulated data show that although large amounts of biologically active factor IX protein is synthesized and secreted, the expression was only transient due to immunological problems (11, 12). Recently, it has been shown that by using adeno-associated viral vectors, transduced factor IX gene is expressed at therapeutic levels in mouse liver and muscle for more than 6 months (13, 14). To investigate both long-term expression and the possible immunological problems caused by viral proteins, it was highly desirable to generate a factor IX “knock-out” mouse as an experimental model system. Herein we describe the generation and the characterization of a factor IX-deficient mouse.
MATERIALS AND METHODS
Construction of the Factor IX Targeting Vector and Generation of FIX-Deficient Mice.
A 1.5-kb cDNA fragment containing human FIX gene was used to screen a 129Sv genomic library (Stratagene). One clone obtained was about 20 kb and contained an exon corresponding to exon 8 of the human FIX gene. The targeting vector was constructed by inserting the 7.2-kb XhoI–BstBI fragment into the NotI site of the pPNT (15), upstream of the PGK-neo cassette, and the 5.5-kb BamHI fragment that is downstream of the coding region into the BamHI site of the pPNT (Fig. 1A). The targeting vector was linearized with NotI and introduced into the 129Sv embryonic stem (ES) cell line by electroporation, and stable transfectants were selected as described (16). Individual ES clones were screened for homologous recombination by Southern blot analysis with an 800-bp fragment as a probe (Fig. 1A). Positive ES clones were injected into C57BL/6 blastocysts as described (16), and the resulting chimeric males were bred to C57BL/6 and 129Sv to establish an inbred line of mutant mice.
Figure 1.
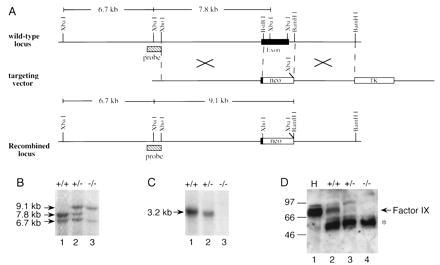
Targeting of the FIX gene by homologous recombination. (A) Schematic of the 3′ portion of the murine factor IX gene showing the last exon, the targeting vector, and the expected recombined locus. The targeting construct contains a PGK-neo cassette, flanked by DNA fragments upstream and downstream the putative exon 8. The predicted product of successful homologous recombination is shown at the bottom. (B) Southern blot analysis of XbaI-digested genomic DNA prepared from tail biopsies of FIX +/+, +/−, and −/− mice and hybridized with the probe shown in A. The probe used was the same 800 bp used to identify the homologous recombined ES clones, as shown in A. New genomic fragment of the expected size 9.1 kb is seen in FIX +/− and −/− mice. The 7.8-kb fragment is absent from FIX −/− mice. The 6.7-kb XbaI fragment is upstream of the recombination region and is present in both the normal and mutated allele. (C) Northern blot analysis of FIX RNA. Total RNA was isolated from mouse liver. The FIX probe used was a 2.7-kb BamHI fragment of a murine FIX cDNA that covers the entire coding region of FIX gene. FIX-specific RNA was undetectable in liver RNA from FIX −/− mice. (D) Western blot analysis of plasma from FIX +/+, +/−, and −/− mice. H, human plasma used as a control. The factor IX protein is indicated by an arrow. The star indicates the protein band in mouse plasma that cross-reacts with the rabbit anti-human factor IX antibody A0300 (Dako).
Genotypes of mice were established by Southern blot hybridization using tail biopsy DNA as described above.
Northern Blot Analysis.
Total RNA was prepared from mouse liver as described (17). Twenty-microgram samples were fractionated by electrophoresis on denaturing agarose gel, transferred to nitrocellulose filters (18), and hybridized to a 32P-labeled FIX probe. The FIX probe was prepared from a 2.7-kb BamHI fragment of a mouse FIX cDNA that covers the entire coding region of FIX gene.
Western Blot Analysis.
Mouse plasma was subjected to barium citrate adsorption twice (19). Four microliters of 1 M BaCl2 was added to 50 μl of mouse plasma, incubated at room temperature for 5 min, and centrifuged at 3,800 × g for 10 min. The precipitated proteins were dissolved in 25 μl of citrate–saline buffer and precipitated again by BaCl2. The pellets were dissolved in 75 μl of citrate–saline buffer, and 10-μl samples were electrophoresed through a SDS/12% polyacrylamide gel. The gel was blotted on a poly(vinylidene difluoride) membrane (Immobilon-P, Millipore), followed by sequential incubations with solutions containing rabbit anti-human factor IX antibody (A0300, Dako), and horseradish peroxidase-conjugated donkey anti-rabbit IgG antibody (Amersham). Renaissance chemiluminescence reagent (DuPont) was used as a substrate to detect antibody-bound protein bands.
Factor IX Activity Assay.
Blood samples were collected from the retroorbital plexus into 0.1 vol of 3.2% sodium citrate. After two sequential centrifugation steps (2,500 × g and 20,000 × g), plasma was stored at −70°C. Factor IX activity was determined by activated partial thromboplastin time (APTT) assays as follows. Fifty microliters of APTT reagent (Dade, Miami, FL), 50 μl of factor IX-deficient human plasma (George King Biomedical, Overland, KS), and 50 μl of a 1:10 dilution of mouse test plasma in Hepes buffer (50 mM Hepes/100 mM NaCl/0.02% NaN3, pH 7.4), were incubated at 37°C in an ST4 coagulometer (American Bioproducts, Parsippany, NJ). After 3 min, clotting was initiated by the addition of 50 μl of 33 mM CaCl2 in Hepes buffer. Factor IX activity of duplicate samples was determined from a log–log standard curve that was constructed from the APTT results for dilution (1:5 to 1:640) of pooled plasma from 15 normal (129Sv) mice (8 females and 7 males, 4–12 weeks old).
Mouse-Tail-Clip Bleeding Assays.
One centimeter was cut from the tip of the mouse tail, and then the mouse was returned to the cage. The bleeding time was measured.
Construction of Recombinant Adenoviral Vectors.
An expression cassette containing 2.7 kb of murine FIX cDNA, under the transcriptional control of cytomegalovirus enhancer/promoter and followed by human growth hormone poly(A) signal, was cloned into an adenoviral vector pΔE1sp1B (Microbix Biosystems, Ontario). The resulting construct was cotransfected with pBHG11 (Microbix Biosystems) by the calcium phosphate method into 293 cells. Recombinant adenoviral plaques were isolated and further purified by two rounds of plaque assays as described (20). The adenoviral vectors were propagated in 293 cells, purified by double CsCl banding, dialyzed, and titered as described (21).
RESULTS
Disruption of the Mouse Factor IX Gene.
Gene targeting strategy was used for FIX gene disruption, as illustrated in Fig. 1A. Briefly, genomic clones containing mouse FIX gene were isolated by screening a mouse 129/Sv genomic library using human FIX cDNA as a probe. A targeting construct was generated where the last exon (corresponding to human factor IX exon 8), coding for a large portion of the catalytic domain of factor IX, was replaced with a PGK-neo cassette (Fig. 1) (15). Thus, the catalytic site of factor IX was rendered nonfunctional. Homologous recombination between the targeting construct and the wild-type allele in ES cells leads to the loss of a 2-kb fragment of FIX gene that includes the region encoding the C-terminal 164 amino acids [amino acids 262–425 (22)] of the factor IX protein and the 3′ untranslated region. Successful targeting can be detected by Southern blot analysis of DNA isolated from ES cell clones after XbaI digestion (Fig. 1A). Five out of 700 neomycin-resistant ES clones analyzed were found to have the replacement. Chimeras were generated in which ES cells contributed to their germ line. The F1 female mice heterozygous for the targeted allele (+/−) were bred with chimeric males to generate hemizygous male and homozygous female mice. Southern blot analysis confirmed that the 6.7- and 7.8-kb XbaI diagnostic fragments could be detected in wild-type mouse DNA with the probe shown in Fig. 1A. Heterozygous female mice (+/−) were identified based on the appearance of a 9.1-kb XbaI fragment (diagnostic for recombined locus) in addition to the 6.7- and 7.8-kb fragments. In the homozygous female (−/−) and hemizygous male (−/Y) mouse DNA, only the 9.1- and 6.7-kb fragments could be detected. Because the FIX gene resides on the X chromosome, the knock-out in male is only on one X chromosome compared with both X chromosomes in female homozygous (−/−) mice. For convenience both male and female factor IX-deficient mice are referred to as (−/−) and heterozygous females (X+/X−) are referred to as (+/−).
Knock-Out Mice Do Not Express Factor IX mRNA or Plasma Factor IX.
FIX RNA was not detected in total liver RNA isolated from a homozygous (−/−) mouse (Fig. 1C, lane 3), by Northern blot hybridization using a FIX-specific probe, whereas appropriate amounts in the heterozygous (+/−; Fig. 1C, lane 2) and wild-type mice (Fig. 1C, lane 1) were easily detected when analyzed in parallel. No factor IX-specific protein could be identified by Western blot analysis of the plasma obtained from knock-out (−/−) mice with a polyclonal antibody against human factor IX (Fig. 1D, lane 4).
Factor IX-Deficient Mice Are Hemophilic.
The factor IX-deficient mice show extensive bleeding after clipping a portion of the tail and bleed to death unless the wound is cauterized. Wild-type and heterozygous mice stopped bleeding within 10–20 min, but the factor IX-deficient mice kept bleeding after 4 h. At that point we cauterized the wounds to save the mice. Additionally, in contrast to the normal mice, they also show swollen extremities and extensive hemorrhagic lesion after trauma (Fig. 2). Interestingly, the female homozygous mice (−/−) give birth without complications. To quantify the amount and activity of factor IX protein, we determined coagulant activity of mouse plasma by using APTT clotting assays. Fig. 3 shows that the factor IX activity in knock-out mice (−/−) plasma was at least 20-fold lower (<5.4%) than that observed for the wild-type mice. The heterozygous mice (+/−) plasma had half as much activity (53%). The low level of apparent factor IX activity observed in deficient mice (−/−) was probably due to procoagulant activity generated in the process of blood sampling by ocular puncture. Thus, based on biological assays, the factor IX-deficient mice have little if any factor IX activity. Also, there are no significant differences between the factor IX-deficient female homozygotes and the male hemizygotes in the above tail-clip bleeding assays and APTT assays.
Figure 2.

Bleeding disorder in a factor IX-deficient mouse. Photographs of a wild-type (Left) and a factor IX-deficient (−/−) mouse (Right) are shown after trauma generated by pulling through a mouse restrainer. Notice the swollen extremities and hematoma in the knock-out mouse (see arrow), compared with the normal mouse. (Inset) Enlargement of the swollen hemorrhagic foot of the mutant (−/−) mouse.
Figure 3.
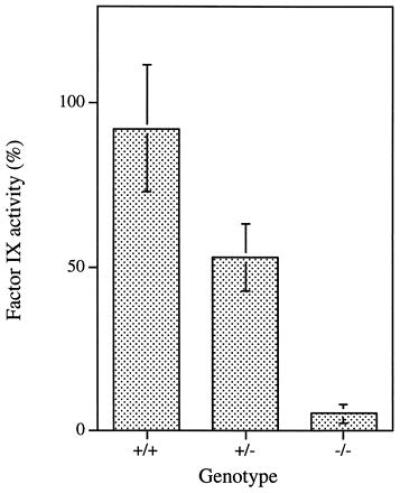
Factor IX coagulant activity in FIX +/+, +/−, and −/− mice. Factor IX activity was quantitated by using APTT assays. The factor IX activity for wild-type (+/+) mice was 92.1 ± 19.1% (n = 21), for heterozygous (+/−) mice was 52.9 ± 10.4% (n = 12), and for homozygous (−/−) mice was 5.4 ± 2.9% (n = 26).
Correction of Factor IX Deficiency.
To test whether the factor IX-deficient mice can be used as a model for gene therapy treatment, we generated recombinant adenovirus containing mouse FIX cDNA (rAd.mFIX) driven by the cytomegalovirus promoter/enhancer. Table 1 shows that intravenous injection of rAd.mFIX restored the factor IX activity of the deficient mice by day 4 to 30–155% of the levels observed in wild-type mouse. However, the E1-deleted replication-defective adenoviral vectors are known to induce only transient expression of the transduced gene, it was therefore expected, as observed, that the biological activity of factor IX decreased to much lower levels by day 7 (11, 23). Nevertheless, restoration of factor IX activity in nullizygous (−/−) mice suggest that these factor IX-deficient mice can be used as model systems for gene therapy approaches.
DISCUSSION
Successful gene therapy approaches will require extensive validation of the technology in an experimental model system prior to undertaking clinical trials. For a variety of diseases, such as cancer and AIDS, it may not always be possible to have a good animal model system. But for genetic diseases, it will be very desirable to have a mouse model system to study the efficacy of the vector and immunological problems associated with the vector or the transgene and to monitor the vector safety. Toward this goal, we report the generation of mice where the gene encoding blood coagulation factor IX has been disrupted by homologous recombination. The evidence that the mice are factor IX-deficient is provided by (i) the absence of factor IX-specific RNA in the liver, (ii) the lack of factor IX protein by Western blot analysis in mouse plasma, (iii) little or no factor IX coagulant activity in the mouse plasma, and (iv) hematomas and bleeding episodes resulting from the slightest injury to the joints (Fig. 2). Furthermore, when tail bleeds were performed, the wild-type mice stopped bleeding in 10–20 min, whereas the factor IX-deficient mice (−/−) continued to bleed past 240 min. After being injected with an adenoviral vector encoding mouse factor IX, FIX-deficient mice showed factor IX coagulant activity in plasma similar to that observed in normal mouse plasma. The ability of the recombinant adenoviruses encoding mouse factor IX to produce biologically active factor IX in plasma at levels of activity comparable to wild-type mouse suggests that this mouse can serve as an excellent model for testing gene therapy approaches using both viral and nonviral vectors. Although the engineered deletion in the FIX gene used for homologous recombination involved truncation of only 164 amino acids at the C terminus of the factor IX protein and the 3′ untranslated region, there is no evidence of truncated factor IX mRNA or protein. Furthermore, it is yet not clear whether the factor IX-deficient mice will mount an immune response to factor IX proteins. Although factor IX-deficient dogs have served as good model systems (12), the factor IX-deficient mice will likely be a more tractable model for a variety of gene therapy approaches, novel drugs, or other modalities used to treat hemophilia B, because of the ease, comparative cost, and convenience associated with mouse systems.
Table 1.
Analysis of factor IX activity in FIX(−/−) mice infected with recombinant adenovirus
| Mouse | Age, weeks | Dose of virus, pfu (×109) per animal* | Factor IX activity, %†
|
||
|---|---|---|---|---|---|
| Before | Day 4 | Day 7 | |||
| 4137 | 10 | 0.75 | 7.0 | 155.0 | 28.5 |
| 4133 | 8.5 | 1 | 6.6 | 77.0 | 29.0 |
| 4148 | 9.5 | 1 | 5.2 | 47.0 | 2.4 |
| 5001 | 7 | 1 | 2.4 | 30.0 | 7.0 |
pfu, Plaque-forming units.
Factor IX-deficient animals were injected once intravenously via the tail vein with the indicated dose of virus.
Factor IX activity in each mouse was quantitated by APTT assay.
Acknowledgments
We thank Dr. D. Stafford and colleagues of the University of North Carolina for providing us with mouse factor IX cDNA. We also thank K. Suter and Y. Marchuk from the Salk Animal Resource Department for their excellent technical support, and B. Coyne for manuscript preparation. This work is supported by grants from the National Institutes of Health, the American Cancer Society, the National Heart, Lung, and Blood Institute, and the H. N. and Frances Berger Foundation to I.M.V., as well as National Institutes of Health Grant RO1HL21544 and the Stein Endowment Fund (to J.H.G.). K.-F.L. is supported by Grants HD34534 and the Basil O’Connor Award and a Pew Scholar Award. I.M.V. is an American Cancer Society Professor of Molecular Biology.
ABBREVIATIONS
- APTT
activated partial thromboplastin time
- ES cell
embryonic stem cell
References
- 1.Hedner V, Davie E W. In: Introduction to Hemostasis and the Vitamin K-Dependent Coagulation Factors. Scriver C R, Beaudet A L, Sly W S, Valle D, editors; Scriver C R, Beaudet A L, Sly W S, Valle D, editors. New York: McGraw–Hill; 1989. pp. 2107–2134. [Google Scholar]
- 2.McGraw R A, Davis L M, Lundblad R L, Stafford D W, Roberts H R. Clin Haematol. 1985;14:359–383. [PubMed] [Google Scholar]
- 3.Pavlovsky A. Blood. 1947;2:185–188. [PubMed] [Google Scholar]
- 4.Biggs R, Douglas A A, Macfarlane R G, Davie J V, Pitney W R, Mersky C, O’Brien J. Br Med J. 1952;2:1378–1382. doi: 10.1136/bmj.2.4799.1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thompson A R. Blood. 1986;67:565–572. [PubMed] [Google Scholar]
- 6.Thompson A R. Thromb Haemostasis. 1995;74:45–51. [PubMed] [Google Scholar]
- 7.St. Louis D, Verma I. Proc Natl Acad Sci USA. 1988;85:3150–3154. doi: 10.1073/pnas.85.9.3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palmer T D, Rosman G J, Osborne W R A, Miller A D. Proc Natl Acad Sci USA. 1991;88:1330–1334. doi: 10.1073/pnas.88.4.1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dai Y, Roman M, Naviaux R K, Verma I M. Proc Natl Acad Sci USA. 1992;89:10892–10895. doi: 10.1073/pnas.89.22.10892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kay M A, Rothenberg S, Landen C N, Bellinger D A, Leland F, Toman C, Finegold M, Thompson A R, Read M S, Brinkhous K M, Woo S L C. Science. 1993;262:117–119. doi: 10.1126/science.8211118. [DOI] [PubMed] [Google Scholar]
- 11.Dai Y, Schwarz E M, Gu D, Zhang W W, Sarvetnick N, Verma I M. Proc Natl Acad Sci USA. 1995;92:1401–1405. doi: 10.1073/pnas.92.5.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kay M A, Landen C H, Rothenberg S, Taylor L A, Leland F, Wiehle S, Fang B, Bellinger D, Finegold M, Thompson A R, Read M, Brinkhous K M, Woo S L C. Proc Natl Acad Sci USA. 1994;91:2353–2357. doi: 10.1073/pnas.91.6.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Snyder R O, Miao C H, Patijn G A, Spratt S K, Danos O, Nagy D, Gown A M, Winther B, Meuse L, Cohen L K, Thompson A R, Kay M A. Nat Genet. 1997;16:270–276. doi: 10.1038/ng0797-270. [DOI] [PubMed] [Google Scholar]
- 14.Herzog R W, Hagstrom J N, Kung S H, Tai S J, Wilson J M, Fisher K J, High K A. Proc Natl Acad Sci USA. 1997;94:5804–5809. doi: 10.1073/pnas.94.11.5804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tybulewicz V L, Crawford C E, Jackson P K, Bronson R T, Mulligan R C. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- 16.Lee K F, Li E, Huber L J, Landis S C, Sharpe A H, Chao M V, Jaenisch R. Cell. 1992;69:737–749. doi: 10.1016/0092-8674(92)90286-l. [DOI] [PubMed] [Google Scholar]
- 17.Chomczynski P, Sacchi N. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 18.Ruffner H, Reis L F, Naf D, Weissmann C. Proc Natl Acad Sci USA. 1993;90:11503–11507. doi: 10.1073/pnas.90.24.11503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bajaj S P, Birktoft J J. Methods Enzymol. 1993;222:96–128. doi: 10.1016/0076-6879(93)22009-5. [DOI] [PubMed] [Google Scholar]
- 20.Graham F L, Smiley J, Russell W C, Nairn R. J Gen Virol. 1977;36:59–74. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- 21.Kanegae Y, Makimura M, Saito I. Jpn J Med Sci Biol. 1994;47:157–166. doi: 10.7883/yoken1952.47.157. [DOI] [PubMed] [Google Scholar]
- 22.Wu S-M, Stafford D W, Ware J. Gene. 1990;86:275–278. doi: 10.1016/0378-1119(90)90290-8. [DOI] [PubMed] [Google Scholar]
- 23.Yang Y, Ertl J, Wilson J M. Immunity. 1994;1:433–442. doi: 10.1016/1074-7613(94)90074-4. [DOI] [PubMed] [Google Scholar]