

Abstract
The link between recognition and replication is fundamental to the operation of the immune system. In recent years, modeling this process in a format of phage-display combinatorial libraries has afforded a powerful tool for obtaining valuable antibodies. However, the ability to readily select and isolate rare catalysts would expand the scope of library technology. A technique in which phage infection controlled the link between recognition and replication was applied to show that chemistry is a selectable process. An antibody that operated by covalent catalysis to form an acyl intermediate restored phage infectivity and allowed selection from a library in which the catalyst constituted 1 in 105 members. Three different selection approaches were examined for their convenience and generality. Incorporating these protocols together with well known affinity labels and mechanism-based inactivators should allow the procurement of a wide range of novel catalytic antibodies.
Much of the work of the biochemical world is accomplished as a result of protein–ligand binding. In the attempt to mimic proteins found in Nature, selection methods from large libraries of molecules have been extremely valuable. In this regard, the phage-display format is particularly attractive in that it duplicates in vitro the essence of the in vivo immune response by linking the fundamental processes of recognition and replication (1–5). Yet, in both of these in vitro and in vivo selection systems, the recognition event that drives replication is based on noncovalent interactions, wherein the outcome is selection based on binding rather than chemistry. When searching for antibody–antigen interactions, the precise utilization of binding energy is unimportant so long as it is sufficient to confer the function of immunological recognition. In contrast, the selection for function is especially relevant for procuring new enzymes, where a substrate must undergo a chemical transformation upon binding.
We recently developed several paradigms for the selection of catalytic antibodies on the basis of chemical reactivity. The direct selection from combinatorial libraries (6, 7) and reactive immunization (8) both afforded a subpopulation of antibodies in which chemistry was installed in the combining site. While investigation of these methods will continue to realize their full potential, the underlying concept could be refined in a design that establishes a more intimate link between chemistry and the replication process. The utilization of phage that are selectively infective (9–11) provides a means to achieve enrichment of antibody catalysts, since selection could be strictly governed by chemistry. In this way, antibodies that carry out a chemical reaction can be identified, isolated, and replicated because the chemical event distinguishes the phage-bearing antibodies from the rest of the population. Herein, we describe the implementation of this approach to select for an antibody that operates by means of covalent catalysis.
MATERIALS AND METHODS
Synthesis of Compounds.
The heterobifunctional reagents 3, 6, and 8 were constructed by coupling two subcomponents followed by additional transformations as outlined (Fig. 1). To this end, compound 1 was prepared by means of the 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDC) coupling of the 2-trimethylsilylethyl (TMSE) ester of N-Z-6-aminohexanoic acid (Z = carbobenzoxy; Sigma) with N-maleoylglycine followed by deprotection with trifluoroacetic acid (TFA). The TMSE ester of N-Z-6-aminohexanoic acid was obtained by using EDC coupling with 2-trimethylsilylethanol (Aldrich). The N-maleoylglycine was prepared by modifications of procedures found in the literature (12, 13). Compound 2 was prepared from 4-nitrophenylacetic acid (Aldrich) in two steps that entailed EDC coupling with phenol followed by hydrogenation. The final compound 3 was purified by flash chromatography (Rf = 0.22, 95/5 CH2Cl2/MeOH). Compound 4 was synthesized from the lysine derivative N-(5-Z-1-carboxypentyl)iminodiacetic acid (14) and a 10-fold molar excess of 2-trimethylsilylethanol using EDC coupling followed by hydrogenation. Compound 5 was prepared from 4-aminophenylacetic acid (Aldrich) by the four-step sequence (i) Boc protection, (ii) EDC coupling with benzyl alcohol, (iii) TFA deprotection; and (iv) glutaric anhydride, triethylamine, CH2Cl2. The tris(TMSE) ester precursor to 6 was purified by flash chromatography (Rf = 0.50, 70/30 CH2Cl2/EtOAc). Compound 7 was synthesized from 4-nitrophenylacetic acid by first forming the TMSE ester using EDC, then hydrogenation, followed by successive cycles of EDC coupling with N-Z-4-aminobutanoic acid and hydrogenation. The final compound 8 was purified by flash chromatography (Rf = 0.50, 75/25 CH3CN/H2O). In all cases, TFA deprotection of TMSE esters required only the subsequent thorough removal of volatiles with no further purification.. Similarly, compounds obtained from hydrogenations required no further purification. All other intermediates were purified by flash chromatography or crystallization.
Figure 1.

Synthesis of compounds under discussion. Reagents and conditions: (a) 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide (EDC), 4-dimethylaminopyridine (DMAP), and dimethylformamide (DMF). (b) H2 [40 psi (280 kPa)], Pd/C, MeOH, 1 hr. (c) Phenol, EDC, DMAP, and DMF. (d) Trifluoroacetic acid (TFA), 0°C, 1 hr. (e) (+)-Biotin (Aldrich), EDC, DMAP, and DMF. TMSE is 2-trimethylsilylethyl.
Construction of Δg3N Helper Phage.
The mutant helper phage Δg3N was constructed by a complete PCR amplification of the VCSM13 (Stratagene) bacteriophage genome, excluding the N-terminal domain of gene III. The primers used were labeled Δg3Nrev and Δg3Nfor. Δg3Nrev, 5′-CCACAGGGATCCCTCATAGTTAGCGTAACGAT; Δg3Nfor, 5′-GGCTCTGGATCCGGTGATTTTGATTATGAA. The PCR reaction was performed in a Perkin–Elmer Gene Amp PCR system 9600 with each cycle run for 1 min at 95°C, 1 min at 62°C, and 8.5 min at 74°C for a total of 35 cycles. The resulting fragment was digested with BamHI overnight at 37°C, self-ligated, and used to transform XL1-Blue Escherichia coli cells (Stratagene).
Construction of Phage-Display Vector pCGMT.
The vector pCGMT was derived from pComb3H (15). The polylinker, including a ribosome-binding site, a pelB leader sequence, a FLAG peptide, and an amber stop codon TAG was custom synthesized, assembled, and inserted between EcoRI and BstX I of pComb3H. Also, a strong tHP transcription terminator (16) was incorporated into the PvuII site located upstream of the lac promoter.
 |
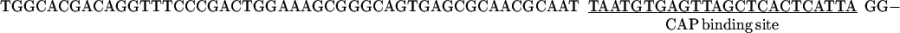 |
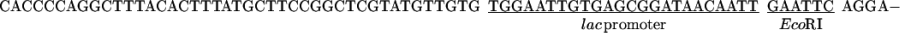 |
 |
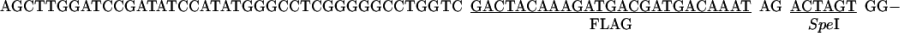 |
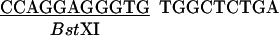 |
The combination of tHP termination, glucose repression of the lac promoter (17), and repression of lacIQ in XL1-Blue cells (18) minimized the toxicity due to leaky expression (19). The inducer isopropyl β-d-thiogalactopyranoside (IPTG) was added to activate the expression during the phage packaging.
Construction of Single-Chain Fv Antibodies scFv 21H3 and scFv 2H6.
The scFvs were engineered by inserting a linker sequence, (Gly4Ser)3, between VH and VL. The DNA assembly was done through overlap PCR by using the DNA of Fab 21H3 and Fab 2H6 as a template (20). First, the VH-linker fragment was amplified by using primers H1 and H4 for Fab 21H3 and primers H1 and H7 for Fab 2H6. In the amplification of the VL-linker, primers L2 and L3 and primers L5 and L6 were used for Fab 21H3 and Fab 2H6, respectively. The final construction was done by mixing equimolar amounts of VH-linker and VL-linker in a PCR. Both the scFv 21H3 and scFv 2H6 genes were digested with NcoI and SpeI and ligated into the phage display vector pCGMT that had been digested using the same restriction enzymes. The sequences of the primers were as follows: H1, GCCTACGGCAGCCGCTGGATTGTTATTACT; L2, GGAGCCGCCGCCGCCAGAACCACCACCACCAGAGACAGTGACCAG; L3, GGCGGCGGCGGCTCCGGTGGTGGTGGTTCTGATATCCAAATGACACAA; H4, TGACTTACTAGTTTTGTCATCGTCATCTTTGTAATCAGCCCGTTTGATTTC; L5, GGAGCCGCCGCCGCCAGAACCACCACCACCGGAGACGGTGACTGAGGTTCC; L6, GGCGGCGGCGGCTCCGGTGGTGGTGGTTCTGTGCTCACCCAGTCTCCAGCA; and H7, TGACTTACTAGTTTTGTCATCGTCATCTTTGTAATCAGTCCGTTTCAACTCCAG.
Construction and Production of Fusion Proteins.
The N1-N2-(SGCPH6) was constructed according to a previous report (9) using a VCSM13 template and Pfu DNA polymerase. Using the same approach, a His6 tail was added directly after the N1-N2 domain to afford the N1-N2-H6 construct. The DNA was subcloned into pET15b (Novagene) in which protein expression is under the control of the T7 promoter. The proteins were expressed in E. coli B834(DE3) by adding IPTG. After induction, the cells were removed by centrifugation (9,000 × g, 20 min, 4°C). The 10 liters of growth medium containing the soluble protein was concentrated to approximately 200 ml using a Hollow Fiber Concentrator (Amicon) equipped with three 10,000 NMWL Hollow Fiber Cartridges. To remove precipitated material, the solution was centrifuged (30,000 × g, 30 min, 4°C). Purification of N1-N2-(SGCPH6) and N1-N2-H6 was performed using a Ni2+-nitrilotriacetate (NTA)-agarose column (Qiagen) according to the instructions. The dimer of N1-N2-(SGCPH6) was reduced by overnight dialysis against 100 mM Bicine, 5 mM DTT, pH 8.5 at 4°C. Excess DTT was removed by gel filtration on a PD-10 column (Pharmacia) eluting with PBS (20 mM sodium phosphate/150 mM NaCl, pH 7.4). To construct N1-N2-streptavidin, the synthetic streptavidin-encoding gene was used in overlap PCR and subcloned into pET15b. The N1-N2-streptavidin was expressed as inclusion bodies in B834(DE3). The inclusion bodies were suspended in a denaturing buffer (0.58 M Tris⋅HCl/6 M guanidine⋅HCl/5.4 mM EDTA) and refolded by gentle removal of guanidine⋅HCl according to a literature method (21). The refolded sample was centrifuged (35,000 × g, 15 min, 4°C), the supernatant was adjusted to pH 11.0 with 1 M NaOH, and then the sample was purified on a 2-iminobiotin-agarose column (Pierce) according to the supplier’s instructions. SDS/PAGE indicated that the N1-N2-streptavidin was isolated as a tetrameric protein.
Production of Noninfective Monodisplayed Phage.
The helper phage Δg3N was electroporated into XL1-Blue cells containing either scFv 21H3 or scFv 2H6 phagemid. A single colony was picked out and grown until OD600 = 0.3–0.5 in superbroth medium, which contained 1% glucose, 50 μg/ml carbenicillin, 70 μg/ml kanamycin, and 10 μg/ml tetracycline. The culture was diluted 3-fold with the same medium, and IPTG was added to a final concentration of 0.25 mM. The culture was then shaken overnight at 30°C for phage production and the phage particles were precipitated twice with PEG/NaCl, resuspended into TBS (50 mM Tris⋅HCl/150 mM NaCl, pH 7.5)/8% (vol/vol) glycerol and stored at −80°C. The number of phage particles was estimated by spectrophotometry (22).
Infectivity Restored by Catalytic Activity.
Generally, a total of 1011 phage particles were used in these studies. Stock solutions of acylating reagents 3, 6, and 8 were prepared in DMF and the reactions were carried out using a final concentration of 10 μM of the reagents in 100 mM phosphate buffer, pH 7.0, with 10% DMF cosolvent at room temperature for 1 hr. The excess reagent then was removed by passing the solution through a molecular weight 100,000 cut-off Microcon (Amicon) in 100 mM NH4OAc, pH 5.5, and finally the particles were exchanged back into PBS. According to the characteristics of the modified N1-N2 domains, different processes were applied as follows. In the maleimide approach, a final concentration of 2 μM N1-N2-(SGCPH6) was added to the acylated-phage solution and excess protein was removed by using a molecular weight 100,000 cut-off Microcon. In the NTA approach, NiSO4 was added at a final concentration of 50 μM before the removal of excess reagent and the buffer exchange. The Ni2+-NTA-His6 complex was formed using a final concentration of 2 μM N1-N2-H6 in PBS. In the biotin approach, due to the difficulty in the complete removal of the tetrameric N1-N2-streptavidin, serial dilutions of fusion protein (10 μM to 1 nM) were added to the acylated-phage solution to determine the minimum level that allowed restoration of infectivity. For each N1-N2-streptavidin concentration, the phage were titered at dilutions of 0, 10, and 100 into 100 μl of XL1-Blue cells (OD600 = 0.8), kept at room temperature for 15 min, and then shaken at 37°C for 1 hr. Aliquots were subsequently plated onto Luria–Bertani agar plates containing 100 μg/ml carbenicillin. To couple the reagent 3 to N1-N2-(SGCPH6), a 20-fold molar excess of reagent was used in a 7 mg/ml solution of protein in PBS. The reaction was incubated at room temperature for 2.5 hr, then at 4°C overnight. The reaction progress was monitored by titration with Ellman’s reagent. After removal of excess 3, the acylation reaction was performed by adding N1-N2-3 at a final concentration of 10 μM into phage solution at room temperature for 1 hr. Unreacted protein was separated by a molecular weight 100,000 cut-off Microcon and the resulting phage solution was titered.
Phage ELISA.
An ELISA plate was coated with 25 μl of 10 μg/ml sec-phenethyl-4-(4-carboxylbutanamido)benzyl phosphonate (PCP)-BSA at room temperature overnight and blocked with 50 μl of Blotto (2% skim milk powder in PBS). Typically, 25 μl of phage solution was added and the plate was incubated at 37°C for 1 hr. After washing, 25 μl of a 1/1000 dilution of horseradish peroxidase/anti-M13 conjugate (Pharmacia) in Blotto was added and incubated at 37°C for 30 min. The plate was developed with TMB (3,3′,5,5′-tetramethylbenzidine) substrate (Pierce) and quenched with an equal volume of 2 M H2SO4, and the absorbance was read on an ELISA plate reader at 450 nm.
Enrichment of scFv 21H3 Phage by Using a Library Format.
The scFv 21H3 phage were diluted into scFv 2H6 phage at ratios of 1:102, 1:104, and 1:105. The mixture was allowed to react with N1-N2-3 in PBS at room temperature for 1 hr. The resulting phage solution was titered as described above. To verify the identity of the clones, the phagemid were isolated and digested with BglII, which cuts a unique site in the DNA sequence of scFv 21H3.
RESULTS AND DISCUSSION
It has been shown that the gene III protein (gIIIp) is critical for phage infectivity (23). Structural studies revealed that the gIIIp consists of three domains designated N1 (amino acids 1–81), N2 (amino acids 87–218), and C-terminal (CT) (amino acids 211–406) joined by glycine-rich linkers (9) and that removal of N1 and N2 abolished infectivity (24). In the phagemid used for conventional antibody phage-display technology, phage packaging proteins are expressed from the DNA of helper phage. Consequently, the wild-type gIIIp expressed from helper phage competes with the gene III fusion protein that is expressed from phagemid. Due to the lacZ repressor in E. coli, the amount of fusion protein is less than the wild-type gIIIp, so that the majority of phage are devoid of fusion protein and a small fraction are monodisplayed (25). To produce monodisplayed, noninfective phage, we deleted the sequence encoding the N1-N2 domain from the helper phage genome, but retained the gene for the CT protein to allow competition in the packaging process.
We used our previously described (26) catalytic mAb PCP21H3 as a paradigm for linking chemistry and infectivity. This antibody undergoes acylation with substrate phenyl esters within seconds at pH 7.0, and the acyl intermediate can be isolated at pH 5.5. For the present work, the gene sequence of PCP21H3 was converted into a scFv antibody format (scFv 21H3) and cloned into a phagemid. Cotransformation of the mutant helper phage (Δg3N) with the scFv 21H3 phagemid in E. coli afforded a noninfective phage in which scFv 21H3 was monodisplayed (Fig. 2). Because only a single clone was under investigation, this method was a convenient alternative to superinfection procedures advantageous for phage-display libraries (10). To ensure the activity of scFv 21H3, the antibody was overexpressed with a FLAG tag in yeast (27, 28) and purified by anion-exchange, affinity, and gel-filtration chromatography. The purity of scFv 21H3 was >98% according to SDS/PAGE, and the kinetic behavior for both transesterification and ester hydrolysis was similar to that of PCP21H3. Following the same strategy described above, we also constructed scFv 2H6 in noninfective-phage display. The mAb PCP2H6 was a highly active lipase antibody derived from the same hapten as PCP21H3, but it did not operate by means of covalent catalysis (29). Hence, scFv 2H6 served as an excellent control for our selectable chemistry experiments.
Figure 2.

The pCGMT vector and proposed pathway for scFv assembly on noninfective phage.
The general strategy for selecting antibody catalysts mediated by infectivity relies on bifunctional compounds in which one end selects for chemistry at the antibody combining site and the other couples to an infection-promoting N1-N2 construct (Fig. 3). Because our approach will be used in future studies to screen for a variety of catalyst activities from combinatorial antibody libraries, a robust and general design was sought. We investigated the utility of three approaches that incorporated scFv 21H3, the bifunctional panning reagents 3, 6, and 8, and three differently engineered N1-N2 domains (Fig. 4). The maleimide technique offers a functional group that is very reactive toward a cysteine sulfhydryl, and the resulting thioether bond is very stable under physiological conditions (30). We added a cysteine-containing peptide (SGCPH6) at the C terminus of the N1-N2 domain. The maleimide-cysteine protocol has been successfully used to restore infectivity through a noncovalent antibody-hapten interaction (9). An NTA group in the presence of Ni2+ and a histidine tail (His6) fused at the C terminus of a recombinant protein forms a stable complex that has been applied to protein purification (31), and was in fact utilized for purification of the modified N1-N2 in Fig. 4A. Given the strong binding interaction of the complex (1013 M, pH 8.0), the protein of interest can be purified even under strongly denaturing conditions such as 8 M urea or 6 M guanidine hydrochloride. The biotin and (strept)avidin interaction has found widespread application in many areas of research (32). Its tight binding dissociation constant (1015 M) is unaffected over a wide pH range and is also stable in 8 M guanidine hydrochloride. An N1-N2-streptavidin fusion protein was engineered and cloned into a T7 expression vector (33), as were the other N1-N2 constructs from parts A and B (Fig. 4). Based on the C-terminal attachment, all fusion proteins were purified by affinity chromatography. The modified N1-N2 proteins described above were demonstrated to be functional by blocking the infectivity of normal helper phage.
Figure 3.

Generalized schematic for linking chemistry to infectivity. A represents a functional group that can be coupled to the modified N-terminal domain of phage gIIIp. B represents an affinity label or mechanism-based inactivator for selection of a catalytic antibody displayed on noninfective (gIIIp-deficient) phage. The components A and B are joined by a chemical linker.
Figure 4.

Linking catalysis to infectivity for the selection of a covalent acyl-antibody.
To evaluate the full merit of the selectable chemistry process, namely, selection as governed by infectivity, it was critical to determine whether the N1-N2 fusion protein itself attached nonspecifically to phage particles. A very low background infectivity [1–10 cfu (colony-forming unit) per 1010 phage] was observed by previous workers employing the maleimide strategy at 4°C (9). However, we found a significantly higher level (105 cfu per 1011 phage) when scFv 21H3 phage were incubated with N1-N2-(SGCPH6) at 4°C for 1 hr. At room temperature, the background was increased by an additional factor of 3. Furthermore, we found our fusion protein readily dimerized during purification at room temperature. Pretreatment of N1-N2-(SGCPH6) with maleimidoacetic acid reduced the background infectivity below the detection limit. In our hands, N1-N2-(SGCPH6) apparently bound to phage through formation of a disulfide bond, probably with a free cysteine on a coat protein, or perhaps through disulfide interchange with the scFv. We did not observe any significant background infectivity (<5 cfu per 1011) in either the NTA or biotin protocols.
As anticipated, the infectivity of scFv 21H3-phage was restored with each of the three methods when the phage were first treated with reagent 3, 6, or 8. Both phage-ELISA and the DNA sequence of phagemid showed that these were indeed scFv 21H3-phage. The infectivity could be inhibited by the original phosphonate PCP hapten (29) as well as by maleimidoacetic acid in the maleimide approach, EDTA in the NTA approach, or free biotin in the biotin approach. Hence, the restored infectivity was directly linked to the catalytic activity of scFv 21H3. The data indicated that >105 of the phage were recovered in the maleimide system, but the specificity (cfu with reagent/cfu in background) was low due to the high background (see above). For both the NTA and biotin methods, specificities were 1,000 and 100, respectively, although the actual levels of infection (102 to 103 cfu) were less. To improve the specificity using maleimide, we first coupled reagent 3 with N1-N2-(SGCPH6) then blocked any unreacted cysteine with excess maleimidoacetic acid. The background infectivity dropped (<5 cfu per 1011) and 105 phage were recovered, affording a specificity of ≈105. Infectivity with the NTA approach using 6 was compromised because of pH restrictions. The Ni2+-NTA-His6 complex has maximal stability at pH 8, where histidine is deprotonated. However, in the present case, the acylated intermediate was hydrolyzed too rapidly at pH 8 for efficient selection. Hence, the NTA complex was formed at pH 7, which resulted in reduced stability and lower levels of infection. A priori, the strong binding interaction between biotin and streptavidin seemed to be a good approach for attachment of the N1-N2 domain. Yet the restoration of infectivity by using 8 was the poorest of the three methods. We speculated that the tetrameric structure of streptavidin caused steric hindrance between the phage particle and the F pilus during the process of DNA injection into E. coli. This hypothesis was supported by the fact that N1-N2-streptavidin inhibited infectivity as effectively as the other modified N1-N2 proteins. In addition, it was shown by others that infectivity was reduced 100-fold when β-lactamase (29 kDa) was inserted between N1-N2 and CT (9). Because scFv 21H3 (27 kDa) is comparable in size to β-lactamase, one would expect a similar result with the maleimide and NTA methods, and even a further reduction with biotin because of the large steptavidin tetramer (98 kDa). The fusion of a scFv at the N terminus of N2-CT produced infective phage by means of binding of an N1-hapten complex (9). We are currently investigating ways to improve the biotin system by using a new N1-streptavidin fusion protein.
On the basis of the restoration of infection, the maleimide approach appeared to offer the best results. However, to achieve this end, a maleimide-containing bifunctional reagent must first be coupled to N1-N2 to avoid a high background, rather than initially used as a substrate for a library of antibodies. This will be a disadvantage when activity selection is based on a mechanism-based inactivator with a high partition ratio. Given 1011 phage per ml (≈0.17 pM) in the selection, a total of 0.17 μM fusion protein coupled to the reagent would be needed for catalysts with a partition ratio of 106. Importantly, some antibody–inactivator combinations with poor partition ratios might actually turn out to be excellent catalysts. When large protein excesses are used, a recombinant protein is wasted and protein–protein interactions might be detrimental. Perhaps more significant and nontrivial, it would be necessary to thoroughly remove the excess N1-N2 protein to avoid inhibition of infectivity. With both the NTA and biotin strategies, a large excess of free bifunctional reagent can be used in the selection and then easily removed by dialysis. In such a way, a catalyst with an unfavorable partition ratio can be trapped and recovered. Additionally, only nanomolar concentrations of the modified N1-N2 domain are required to restore infectivity.
We demonstrated that our design could be used to enrich a catalyst from a library-like ensemble. By mixing scFv 21H3-phage with a large pool of the control scFv 2H6-phage, the enrichment of scFv 21H3-phage through infectivity was examined. To attain the maximal efficiency for the reaction under investigation, we used the maleimide method with precoupling (see above). As noted above, this would generally not be the most desirable strategy, but our main objective was to determine the efficiency of the selection process. When the reagent-modified N1-N2 domain was added to the mixture of scFv-phage, only the scFv 21H3-phage formed the acyl intermediate so that the infectivity was restored. As expected, infectivity with scFv 2H6-phage alone was not observed. The restored infectivity was increased with increasing amounts of scFv 21H3-phage. By examining the specific restriction enzyme digestion patterns, it was concluded that almost all the recovered clones were scFv 21H3 (12 of 12 in a 102 dilution, 11 of 12 in a 104 dilution, and 6 of 10 in a 105 dilution) from a single round of selection. Hence, given that the number of phage could be readily increased to 1012-1013 by minor changes in experimental protocols, it should be possible to extract an existing catalyst from a library having a diversity of 106-107 members.
The major challenge in the mechanism-based panning of antibody libraries is how to distinguish a catalyst(s) from high-affinity binders and nonspecific binders, particularly where the catalyst is a rare species or the sum of the others constitute the vast majority of the population. During conventional antibody–phage selection, the replication process provides no advantage for selecting clones and may sometimes even be a disadvantage by propagating more viable antibody-deletion mutants (21). Because simple ELISAs will not differentiate covalently modified antibodies from noncovalent and/or nonspecific binders, labor-intensive screening procedures and the overexpression and purification of many false-positive clones are required in the search for catalysts. The key aspect in panning noninfective antibody–phage is that catalysts will not be missed, but rather selected, and these species will dominate the selected population. The problem of nonspecific adsorption is eliminated and convenient protocols greatly reduce noncovalent selection. Our selection system has several features: (i) The affinity label or mechanism-based inactivator is used as a convenient reagent. (ii) Reactions can be performed under homogeneous conditions such that pseudo-first-order kinetics apply. (iii) In principle, any mechanism-based inactivator can be used for selection because the reagent concentration can usually be made sufficiently high to account for high partition ratios. Further rounds of selection can exclude poor catalysts by shortening reaction times. (iv) The strong chemical interaction between the N1-N2 domain and the reagent allows the use of homogeneous denaturing conditions for excellent removal of noncovalently bound phage without loss of covalently bound phage, as might occur from a solid support.
For a number of years, we have studied covalent catalysis as being exemplary of progress toward more mechanistically complex and efficient catalytic antibodies. The current work has, in essence, brought us full circle from our initial discovery of PCP21H3. In that case, the antibody was fortuitously elaborated by and isolated from the immune response, whereas here, we have purposely selected this clone from a library approaching the size of the immune repertoire. Using an iterative procedure in the panning of a noninfective-phage library in which selection is first made using the phosphonate transition-state analogue, followed by selection with one of the described reagents could, in principle, provide a clone analogous to PCP21H3. It now seems plausible that antibody catalysts can be selected based on those that add a chemical event to the transition-state instructions. Importantly, this procedure would appear suited to select for the chemistry required for even more complex reactions.
Acknowledgments
We thank Dr. Chong-Hwan Chang for providing the plasmid DNA encoding synthetic streptavidin that was constructed by Dr. P. C. Weber. The research described was supported in part by the National Institutes of Health (Grant GM 43858, K.D.J.), Program Project Grant P01CA27489–16 (R.A.L.), The Skaggs Institute for Chemical Biology (K.D.J.), and The Scripps Research Institute.
ABBREVIATIONS
- cfu
colony-forming unit
- DMF
dimethylformamide
- EDC
1-(3-dimethylaminopropyl)-3-ethylcarbodiimide
- gIIIp
gene III protein
- IPTG
isopropyl β-d-thiogalactopyranoside
- NTA
nitrilotriacetate
- PCP
sec-phenethyl-4-(4-carboxylbutanamido)benzyl phosphonate
- scFv
single-chain Fv antibody
References
- 1.Smith G P. Science. 1985;228:1315–1317. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- 2.Burton D R, Barbas C F., III Adv Immunol. 1994;57:191–280. doi: 10.1016/s0065-2776(08)60674-4. [DOI] [PubMed] [Google Scholar]
- 3.Burton D R. Acc Chem Res. 1993;26:405–411. [Google Scholar]
- 4.Barbas C F, III, Bain J D, Hoekstra D M, Lerner R A. Proc Natl Acad Sci USA. 1992;89:4457–4461. doi: 10.1073/pnas.89.10.4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lerner R A, Kang A S, Bain J D, Burton D R, Barbas C F., III Science. 1992;258:1313–1314. doi: 10.1126/science.1455226. [DOI] [PubMed] [Google Scholar]
- 6.Janda K D, Lo C-H L, Li T, Barbas C F, III, Wirsching P, Lerner R A. Proc Natl Acad Sci USA. 1994;91:2532–2536. doi: 10.1073/pnas.91.7.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Janda K D, Lo L-C, Lo C-H L, Sim M-M, Wang R, Wong C-H, Lerner R A. Science. 1997;275:945–948. doi: 10.1126/science.275.5302.945. [DOI] [PubMed] [Google Scholar]
- 8.Wirsching P, Ashley J A, Lo C-H L, Janda K D, Lerner R A. Science. 1995;270:1775–1782. doi: 10.1126/science.270.5243.1775. [DOI] [PubMed] [Google Scholar]
- 9.Krebber C, Spada S, Desplancq D, Krebber A, Ge L, Plückthun A. J Mol Biol. 1997;268:607–618. doi: 10.1006/jmbi.1997.0981. [DOI] [PubMed] [Google Scholar]
- 10.Dueñas M, Borrebaeck C A K. Bio/Technology. 1994;12:999–1002. doi: 10.1038/nbt1094-999. [DOI] [PubMed] [Google Scholar]
- 11.Gramatikoff K, Georgiev O, Schaffner W. Nucleic Acids Res. 1994;22:5761–5762. doi: 10.1093/nar/22.25.5761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keller O, Rudinger J. Helv Chim Acta. 1975;58:531–541. doi: 10.1002/hlca.19750580224. [DOI] [PubMed] [Google Scholar]
- 13.Helferich B, Wesemann W. Chem Ber. 1967;100:421–424. [Google Scholar]
- 14.Hochuli E, Döbeli H, Schacher A. J Chromatog. 1987;411:177–184. doi: 10.1016/s0021-9673(00)93969-4. [DOI] [PubMed] [Google Scholar]
- 15.Barbas C F, III, Wagner J. Methods. 1995;8:94–103. [Google Scholar]
- 16.Nohno T, Saito T, Hong J S. Mol Gen Genet. 1986;205:260–269. doi: 10.1007/BF00430437. [DOI] [PubMed] [Google Scholar]
- 17.Tagami H, Aiba H. Nucleic Acids Res. 1995;23:599–605. doi: 10.1093/nar/23.4.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ørum H, Andersen P S, Øster A, Johansen L K, Riise E, Bjornvad M, Svendsen I, Engberg J. Nucleic Acids Res. 1993;21:4491–4498. doi: 10.1093/nar/21.19.4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krebber A, Burmester J, Plückthun A. Gene. 1996;178:71–74. doi: 10.1016/0378-1119(96)00337-x. [DOI] [PubMed] [Google Scholar]
- 20.Lo C H, Gao C, Mao S, Matsui K, Lerner R A, Janda K D. Isr J Chem. 1996;36:195–198. [Google Scholar]
- 21.Maeda Y, Koga H, Yamada H, Ueda T, Imoto T. Protein Eng. 1995;8:201–205. doi: 10.1093/protein/8.2.201. [DOI] [PubMed] [Google Scholar]
- 22.Day L A. J Mol Biol. 1969;39:265–277. doi: 10.1016/0022-2836(69)90316-7. [DOI] [PubMed] [Google Scholar]
- 23.Model P, Marjorie R. In: Filamentous Bacteriophages. Calendar R, editor. Vol. 2. New York: Plenum; 1988. pp. 375–456. [Google Scholar]
- 24.Stengele I, Bross P, Garcés X, Giray J, Rasched I. J Mol Biol. 1990;212:143–149. doi: 10.1016/0022-2836(90)90311-9. [DOI] [PubMed] [Google Scholar]
- 25.Barbas C F, III, Kang A S, Lerner R A, Benkovic S J. Proc Natl Acad Sci USA. 1991;88:7978–7982. doi: 10.1073/pnas.88.18.7978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wirsching P, Ashley J A, Benkovic S J, Janda K D, Lerner R A. Science. 1991;252:680–686. doi: 10.1126/science.2024120. [DOI] [PubMed] [Google Scholar]
- 27.Knappik A, Plückthun A. BioTechniques. 1994;17:754–761. [PubMed] [Google Scholar]
- 28.Ridder R, Schmitz R, Legay F, Gram H. Bio/Technology. 1995;13:255–260. doi: 10.1038/nbt0395-255. [DOI] [PubMed] [Google Scholar]
- 29.Janda K D, Benkovic S J, Lerner R A. Science. 1989;244:437–440. doi: 10.1126/science.2717936. [DOI] [PubMed] [Google Scholar]
- 30.Smyth D G, Blumenfeld O O, Konigsberg W. Biochem J. 1964;91:589–595. doi: 10.1042/bj0910589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Janknecht R, de Martynoff G, Lou J, Hipskined R A, Nordheim A, Stunnenberg H G. Proc Natl Acad Sci USA. 1991;88:8972–8976. doi: 10.1073/pnas.88.20.8972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Savage D, Mattson G, Desai S, Nielander G, Morgensen S, Conklin E. Avidin-Biotin Chemistry: A Handbook. Rockford, IL: Pierce Chemical Co.; 1992. [Google Scholar]
- 33.Moffatt B A, Studier F W. J Mol Biol. 1986;189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]