

Abstract
Neurotoxicity induced by overstimulation of N-methyl-d-aspartate (NMDA) receptors is due, in part, to a sustained rise in intracellular Ca2+; however, little is known about the ensuing intracellular events that ultimately result in cell death. Here we show that overstimulation of NMDA receptors by relatively low concentrations of glutamate induces apoptosis of cultured cerebellar granule neurons (CGNs) and that CGNs do not require new RNA or protein synthesis. Glutamate-induced apoptosis of CGNs is, however, associated with a concentration- and time-dependent activation of the interleukin 1β-converting enzyme (ICE)/CED-3-related protease, CPP32/Yama/apopain (now designated caspase 3). Further, the time course of caspase 3 activation after glutamate exposure of CGNs parallels the development of apoptosis. Moreover, glutamate-induced apoptosis of CGNs is almost completely blocked by the selective cell permeable tetrapeptide inhibitor of caspase 3, Ac-DEVD-CHO but not by the ICE (caspase 1) inhibitor, Ac-YVAD-CHO. Western blots of cytosolic extracts from glutamate-exposed CGNs reveal both cleavage of the caspase 3 substrate, poly(ADP-ribose) polymerase, as well as proteolytic processing of pro-caspase 3 to active subunits. Our data demonstrate that glutamate-induced apoptosis of CGNs is mediated by a posttranslational activation of the ICE/CED-3-related cysteine protease caspase 3.
Glutamate, the principal excitatory amino acid neurotransmitter in the central nervous system, is also a potent excitotoxin when one (or more) of the known ionotropic glutamate receptor subtypes is overstimulated (1). Excitotoxicity, induced by excessive activation of glutamate receptors, has been postulated to underlie the neuronal death that occurs after ischemic (stroke) or traumatic brain injury (1) as well as that associated with a number of neurodegenerative disorders, including Alzheimer disease and Huntington disease (2–4). Although excitotoxicity that occurs after overstimulation of glutamate receptors is associated with an exaggerated and prolonged rise in intracellular Ca2+ (5, 6), little is known about the subsequent events that ultimately lead to cell death.
The exact mode of neuronal cell death induced by excitotoxins has remained controversial (see refs. 7 and 8 for discussion), although more recent data suggest that both necrotic and apoptotic pathways are activated after overstimulation of various glutamate receptor subtypes (9–11). Many of the genes responsible for apoptotic cell death, including those underlying neuronal apoptosis, initially were identified in the nematode Caenorhabditis elegans (C. elegans cell death or CED genes) (12). Mammalian homologs of these genes now have been identified, and to date, at least 11 members of the interleukin 1β-converting enzyme (ICE)/CED-3 protease family have been reported (13). Each of these aspartate-specific cysteine proteases is synthesized as a proenzyme that is proteolytically processed to subunits that form catalytically active heterodimers (14). Although various ICE/CED-3 proteases have been shown to mediate apoptosis in many cell types, the role of these proteases in mediating apoptosis of mammalian neurons, either that which occurs during normal brain development (15, 16) or after traumatic or toxic insult (17), is poorly understood. Very recently, however, mice deficient in the ICE/CED-3-related cysteine protease CPP32 have been generated and brain development found to be profoundly affected (18). Moreover, mice deficient in CPP32 appear to have markedly reduced apoptosis in brain regions where during normal brain development major morphogenetic change occurs (18).
We recently have cloned a rat homolog of CPP32 and found expression of its mRNA to be profoundly down-regulated during brain development (19). To investigate whether this ICE/CED-3-related protease (now designated caspase 3)‖ is involved in the apoptotic death of neurons in the developing brain we studied its expression in cultured cerebellar granule neurons (CGNs). CGNs are among the most abundant neuronal phenotype in the mammalian brain (20) and are readily maintained in primary culture in their fully differentiated state if depolarized with high concentrations of K+ (21). Cultured CGNs can be induced to apoptose if subsequently exposed to nondepolarizing culture conditions and the latter requires new RNA and protein synthesis (8, 22). Exposure of CGNs to nondepolarizing culture conditions results in overexpression of caspase 3 mRNA, which can be blocked by adding depolarizing concentrations of K+ before the commitment point for induction of apoptosis (19). These data suggest that caspase 3 may play a role in apoptosis of CGNs induced by K+ withdrawal.
Recently, excitotoxic death of CGNs induced by glutamate has been shown to occur via both necrosis and apoptosis, with apoptosis predominating after exposure to relatively low concentrations of glutamate and with a relatively delayed time course (10). In the present study we confirm that exposure of CGNs to low concentrations of glutamate induces a delayed apoptosis, but in contrast to K+ withdrawal-induced apoptosis does not require new RNA or protein synthesis. Rather, glutamate-induced apoptosis of CGNs is associated with a marked increase in cytosolic caspase 3 activity, cleavage of one of its known substrates, poly(ADP-ribose) polymerase (PARP), as well as proteolytic processing of pro-caspase 3 to its active p12 subunit. Both cell death and the increase in caspase 3 activity induced by glutamate are blocked by coincubation with the N-methyl-d-aspartate (NMDA) receptor antagonist dizocilpine or a cell permeable inhibitor of caspase 3. Our data strongly suggest that glutamate-induced apoptosis of CGNs is mediated by a posttranslational activation of the CED-3-related cysteine protease caspase 3.
MATERIALS AND METHODS
Neuronal Cell Cultures and Assessment of Neuronal Viability.
CGNs were prepared from 8-day-old Sprague–Dawley rat pups (Harlan, Indianapolis) as described (8, 23). Freshly dissected cerebella were dissociated and the cells seeded at a density of 1.2 to 1.5 × 106 cells/ml on poly-l-lysine coated dishes in basal medium Eagle supplemented with 10% FBS, 25 mM KCL, and 0.1 mg/ml gentamicin. Cytosine arabinoside (10 μM) was added to the culture medium 24 hr after initial plating. All experiments used neurons after 7–8 days in vitro. Viable granule neurons were quantified by counting fluorescein (green) positive cells, which result from the de-esterification of fluorescein diacetate (FDA) by living cells. Briefly, cultures were incubated with 10 μg/ml FDA for 5 min, examined, and photographed using UV light microscopy, and the number of neurons from representative low-power fields were counted as described (8, 24). Propidium iodide (PI), which interacts with nuclear DNA producing a red fluorescence, was used to identify dead neurons (24). For PI staining, cultures were incubated with PI (5 μg/ml), examined, and photographed using UV light microscopy as described (24).
Assessment of Apoptosis.
To quantify and assess nuclear morphology, CGNs were fixed for 10 min with 4% formaldehyde in PBS at 4°C, washed with distilled water, and stained for 5 min with 5 μg/ml Hoechst 33258 to reveal nuclear condensation/aggregation. Representative photomicrographs were taken with a Leica DM/LM microscope under UV illumination (8). Internucleosomal DNA cleavage was assessed by conventional gel electrophoresis after extraction of nuclear DNA (25). Briefly, CGNs (6 × 107 per treatment condition) were cultured in 100-mm dishes and after treatment were collected in cold PBS. After centrifugation (1,000 rpm, 5 min) CGNs were lysed in 100 μl buffer (50 mM Tris⋅HCl, pH 7.5/20 mM EDTA/1% Nonidet P-40). After centrifugation (10,000 × g, 5 min), supernatants were treated with 1% SDS and RNase A (5 μg/μl final concentration) for 2 hr at 37°C followed by proteinase K (2.5 μg/μl final concentration) overnight at 56°C. DNA was precipitated by the addition of 1:10 (vol/vol) of 3 M potassium acetate and 1 vol of isopropyl alcohol for 1 hr at 4°C, pelleted, and then redissolved in loading buffer. DNA samples were then size-fractionated on a 1.5% agarose gel (25).
Electron Microscopy.
Electron microscopy of CGNs was carried out to further assess and quantify features of apoptotic death. Neuronal cultures were collected by centrifugation, and the media decanted and replaced with a buffered fixative solution containing 2% formaldehyde and 2.5% glutaraldehyde. Further tissue processing consisted of a secondary fixative with 2% buffered osmium tetroxide. After rinsing with 0.12 M cacodylate buffer, samples were dehydrated with serially increasing concentrations of ethanol, and then embedded in epoxy resin. Areas for ultrastructural examination were chosen from toluidine blue stained 1-μm thick sections by examination using light microscopy. Ultrathin sections (80–100 nm) were mounted on copper grids, stained with uranyl acetate and lead citrate, and examined with a transmission electron microscope (Philips, 410LS) (26).
Protease Activity.
Treated cells were collected, washed three times with PBS, pH 7.2, and resuspended in precooled buffer (50 mM Tris⋅HCl, pH 7.4/1 mM EDTA/10 mM EGTA/1 mM DTT/0.1 mM phenylmethylsulfonyl fluoride/2 μg/μl aprotinin) (27). Cells were allowed to swell on ice for 20 min. After homogenization with 20 strokes of a B-type pestle, lysates were centrifuged at 15,000 rpm at 4°C for 20 min, and protein concentrations were determined (Pierce). Extracts either were used immediately or stored at −80°C. Aliquots of protein (30 μg) were incubated with 100 μM caspase 3 substrate (Ac-DEVD-p-nitroaniline; Calbiochem) in a total volume of 1.0 ml at 37°C. The colorimetric release of p-nitroaniline from the Ac-DEVD-pNA substrate was recorded every 10 min at 405 nm (28). One unit of enzyme is defined as the amount of enzyme required to release 0.22 pmol of p-nitroaniline at 37°C per min. Enzymatic activity for caspase 3 was linear over the range of protein concentrations used to calculate specific activity.
Western Blot Analysis.
Western blot analysis was performed on 50 μg whole-cell and 200 μg cytoplasmic extracts from treated and untreated cultures. Whole-cell extracts were prepared by lysing cells in a buffer containing 1% Nonidet P-40, 0.1% SDS, 50 mM Tris (pH 8.0), 50 mM NaC1, 0.05% deoxycholate, protease inhibitors (Boehringer Mannheim). Proteins were size-fractionated (SDS/PAGE) on a 10–20% polyacrylamide gradient gel and transferred onto nitrocellulose (Hybond N, Amersham). The blots then were probed with polyclonal antibodies specific for PARP (Enzyme Systems Products, Dublin, CA) or CPP32 (Upstate Biotechnology, Lake Placid, NY), followed by anti-rabbit IgG horseradish peroxidase-linked antibody (Jackson ImmunoResearch). Bound antibody was visualized using enhanced chemiluminescence (Amersham).
Whole-Cell Voltage-Clamp Recording and Analysis.
Recordings were conducted at room temperature (20°C–24°C) using cells from CGN cultures maintained 9–12 days in vitro (29). Immediately before a recording session the culture medium was replaced with an external solution consisting of 130 mM NaC1, 5.0 mM KC1, 2.0 mM CaC12, 1.0 mM MgC12, 5.6 mM glucose, and 5.0 mM Hepes (pH 7.4). Patch pipettes contained 140 mM KC1, 0.2 mM MgC12, 11.0 mM EGTA, 1.0 mM CaC12, 0.5 mM NaC1, and 10.0 mM Hepes (pH 7.2). Macroscopic currents elicited with 0.5–1 sec duration pulses of 30 μM glutamate using a Picospritzer II were recorded with a List EPC7 patch-clamp amplifier. Ac-DEVD-CHO, made up as a stock in dimethyl sulfoxide, was diluted to the experimental concentration (100 μM) in the external solution (0.1% final dimethyl sulfoxide concentration). Both Ac-DEVD-CHO and 1 μM dizocilpine were administered directly through the bath. Whole-cell currents assessed from a membrane holding potential of −70 mV were filtered, digitized and leak-and capacitative-subtracted for off-line analysis (pCLAMP).
RESULTS
Glutamate-Induced Apoptosis of CGNs Is Mediated by NMDA Receptors and Does Not Require New RNA and Protein Synthesis.
As has been reported previously (30), glutamate-induced cell death of CGNs observed over a broad range of glutamate concentrations was completely prevented by the noncompetitive NMDA receptor antagonist dizocilpine (MK-801) [(5R,10S)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine hydrogen maleate], but not by the non-NMDA receptor antagonists CNQX (6-cyano-7-nitroquinoxaline-2,3-dione) and DNQX (6,7-dinitroquinoxaline-2,3-dione) (31) (Fig. 1A). More than 70% of the nuclei in CGNs exposed to a relatively low concentration of glutamate (30 μM, 24 hr) displayed the morphological features of apoptosis (8, 10, 14) (Fig. 1 D and E), including pyknotic nuclei, condensation of nuclear chromatin, and heterochromatic clumping when assessed by transmission electron microscopy, compared with 5% of those from control (untreated) culture (>200 nuclei counted per treatment group; P < 0.001 by Student’s t test, data not shown). Internucleosomal DNA fragmentation, also a typical feature of apoptosis, readily was detected in CGNs exposed to glutamate (30 μM, 24 hr) (see Fig. 4B).
Figure 1.
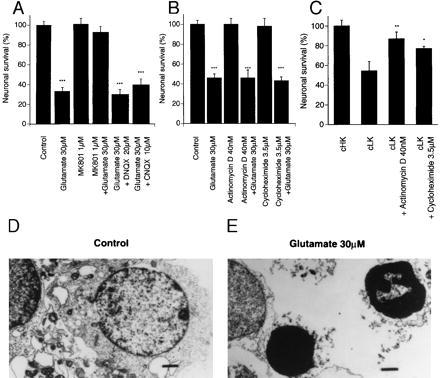
(A) Glutamate-induced neurotoxicity (excitotoxicity) is mediated by the NMDA receptor. Cultured CGNs were pretreated with 1 μM dizocilpine (MK-801), 10 μM CNQX (6-cyano-7-nitroquinoxaline-2,3-dione), or 20 μM DNQX (6,7-dinitroquinoxaline-2,3-dione) for 5 min, then exposed to 30 μM glutamate for 24 hr. (B) Glutamate-induced apoptosis is not dependent on de novo gene transcription or translation because neither 40 nM actinomycin D nor 3.5 μM cycloheximide attenuates glutamate-induced neurotoxicity (∗∗∗, P < 0.001 treated vs. control by Student’s t test). (C) Apoptosis of CGNs induced by switching from depolarizing (high K+, cHK) to nondepolarizing (low K+, cLK) media is greatly attenuated by 40 nM actinomycin D and 3.5 μM cycloheximide (cLK = 5 mM KCl, cHK = 20 mM KCl) (∗, P < 0.05, ∗∗, P < 0.01, cLK treated vs. cLK alone by Student’s t test). Neuronal viability was determined as described in Materials and Methods and calculated as a % of control (untreated cultures). Values are expressed as the mean ± standard error of triplicate determinations from at least three separate experiments (∗∗∗, P < 0.001 treated vs. control by Student’s t test). Ultrastructural analysis (×8,400) of CGNs after glutamate exposure reveals morphological features typical of apoptosis including condensed chromatin (bar = 1 μm) (E), as compared with control (D). Note markedly condensed chromatin and small pyknotic nuclei. Lower power electron microscopy photomicrographs (×4,400) were used to quantify number (%) apoptotic CGNs after glutamate exposure. Data is presented in text.
Figure 4.
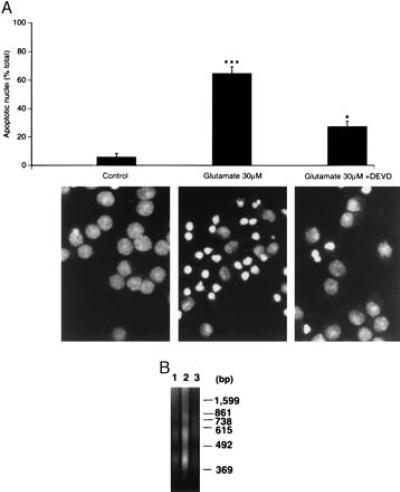
(A) The caspase 3 protease inhibitor Ac-DEVD-CHO attenuates the morphological features of apoptosis induced by glutamate in cultured CGNs. CGNs were treated with 30 μM glutamate for 24 hr in the presence or absence of 200 μM Ac-DEVD-CHO and then stained with 5 μg/ml Hoechst 33258. The number of apoptotic nuclei (small with condensed chromatin) were counted from representative photomicrographs (×1,250) and are represented as a % of the total number of nuclei counted (n = 400) (∗∗∗, P < 0.001 glutamate vs. control; ∗, P < 0.05 glutamate vs. glutamate + Ac-DEVD-CHO by Student’s t test). (B) Internucleosomal DNA fragmentation (DNA laddering) induced in CGNs after exposure to glutamate (30 μM, 24 hr) is greatly attenuated by 200 μM Ac-DEVD-CHO. CGNs (control, lane 1) were exposed to glutamate (30 μM, 24 hr) in the absence (lane 2) or presence of 200 μM Ac-DEVD-CHO (lane 3).
We then investigated whether glutamate-induced apoptosis (like apoptosis observed in many other cell types/tissues) is dependent on de novo transcription and translation. We were unable to block glutamate-induced apoptosis with the RNA and protein synthesis inhibitors, 40 nM actinomycin D, and 3.5 μM cycloheximide (Fig. 1B), respectively. By contrast, apoptosis of CGNs, which is reliably induced by nondepolarizing culture conditions, is almost completely blocked by the same concentrations of actinomycin D or cycloheximide, as we (8) and others (22) previously have reported (Fig. 1C).
Glutamate-Induced Apoptosis of CGNs Is Blocked by a Cell Permeable Inhibitor of Caspase 3, But Not Caspase 1.
Cultured CGNs were exposed to glutamate for 24 hr and subsequently examined for cell viability by double-staining with FDA/PI. Approximately 50–70% of CGNs died when they were exposed to a glutamate concentration of 30 μM. Neuronal killing was attenuated by 75% when cells were coincubated with glutamate (30 μM, 24 hr) and the specific caspase 3 inhibitor Ac-DEVD-CHO (200 μM). No protection was observed, however, when CGNs were coincubated with glutamate and the caspase 1 (ICE) inhibitor Ac-YVAD-CHO (Figs. 2A–H and 3A). To further confirm the protective effect of Ac-DEVD-CHO against glutamate-induced neuronal death, CGNs were exposed to a range of glutamate concentrations (3–1,000 μM) in the absence or presence of Ac-DEVD-CHO. The most robust neuroprotection was observed when 200 μM Ac-DEVD-CHO is coincubated with glutamate at concentrations of 30 μM or less (P < 0.001). When CGNs were exposed to higher concentrations of glutamate (>100 μM), Ac-DEVD-CHO still attenuated glutamate-induced neurotoxicity, but to a lesser degree. Moreover, Ac-DEVD-CHO failed to protect CGN when exposed to glutamate concentrations of 1 mM (Fig. 3B). The inhibition of glutamate-induced toxicity of CGNs by Ac-DEVD-CHO was concentration-dependent with an EC50 of approximately 50 μM (Fig. 3C), similar to the EC50 reported for inhibition of Fas-mediated apoptosis of various cell types (32, 33).
Figure 2.
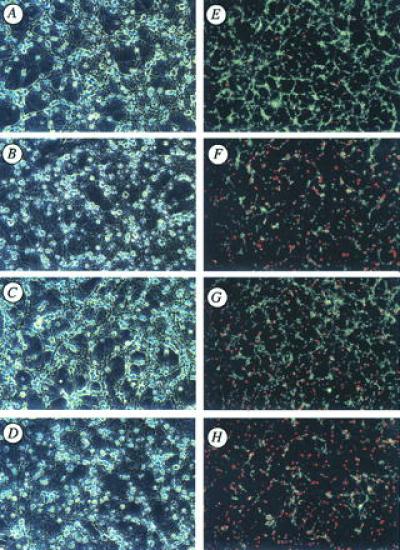
(A–H) Effects of the cell-permeable caspase 3 (CPP32) and caspase 1 (ICE) protease inhibitors, Ac-DEVD-CHO and Ac-YVAD-CHO, respectively, on glutamate-induced apoptosis of cultured CGNs. Representative fields of CGNs were photographed under phase contrast microscopy (A–D) and after double staining with FDA and PI (E–H) as described in Materials and Methods. Compare untreated control cultures (A and E) to glutamate (30 μm; 24 hr)-treated (B and F) vs. glutamate + Ac-DEVD-CHO (C and G) to glutamate + Ac-YVAD-CHO (D and H). Notice that the CPP32/apopain protease inhibitor, Ac-DEVD-CHO, but not the ICE-specific protease inhibitor Ac-YVAD-CHO, greatly attenuates glutamate-induced toxicity of CGN as measured by phase contrast microscopy or double staining with FDA and PI. (×100)
Figure 3.
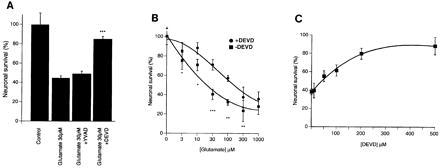
(A) Quantification of the effects of Ac-DEVD-CHO and Ac-YVAD-CHO on glutamate-treated (30 μm, 24 hr) CGNs also shows almost complete protection by Ac-DEVD-CHO but not by Ac-YVAD-CHO. (B) The CPP32 inhibitor Ac-DEVD-CHO significantly attenuates glutamate-induced apoptosis of CGNs (ANOVA, P < 0.001). CGNs were exposed to increasing concentrations of glutamate (3–1,000 μM) for 24 hr in the presence or absence of 200 μM Ac-DEVD-CHO (∗∗∗, P < 0.001, ∗, P < 0.05 by Student’s t test). (C) CGNs were exposed to 30 μM glutamate for 24 hr in the presence of increasing concentrations of the CPP32 protease inhibitor, Ac-DEVD-CHO. Neuronal viability was assessed by FDA and PI staining and visual counting of viable neurons from representative photomicrographs. Values in A, B, and C are expressed as a % of control cultures for each experiment, and the data represent the mean ± standard error of triplicate determinations from a representative experiment repeated at least three times with similar results.
Additionally, we investigated the effects of Ac-DEVD-CHO on glutamate-induced apoptosis of CGNs measured by staining with Hoechst 33258 and DNA fragmentation analysis. The number of apoptotic nuclei induced by glutamate treatment (30 μM, 24 hr) was reduced significantly by 200 μM Ac-DEVD-CHO from 70% to 30%, as measured by Hoechst 33258 staining (Fig. 4A). Similar results were observed by electron microscopy analysis (data not shown). Internucleosomal DNA fragmentation induced by glutamate was completely prevented in the presence of 200 μM Ac-DEVD-CHO, i.e., when nuclear DNA from CGNs exposed to glutamate with and without Ac-DEVD-CHO was fractionated by gel electrophoresis (Fig. 4B).
Glutamate-Induced Apoptosis of CGNs Is Associated with a Concentration and Time-Dependent Activation of Caspase 3.
To confirm that caspase 3 mediates glutamate-induced apoptosis of CGNs, we examined the effects of glutamate treatment of CGNs on caspase 3 protease activity as measured by monitoring the release of p-nitroaniline from the caspase 3-specific substrate Ac-DEVD-pNA (Fig. 5) (34). In all experiments cytosolic extracts of glutamate-treated (30 μM; 24 hr) CGNs were found to have specific caspase 3 protease activity that is almost 8-fold greater than extracts prepared from control (untreated) cultures (Fig. 5A). Importantly, the specific NMDA receptor antagonist dizocilpine (MK-801) (1 μM), which completely blocks glutamate-induced apoptosis of CGNs, also prevents the increase in caspase 3 protease activity induced by glutamate (Fig. 5A). Caspase 3 protease activity markedly increased 15 hr after 30 μM glutamate exposure, which roughly correlates with an increase in the number of apoptotic nuclei as measured by Hoechst 33258 staining (Fig. 5B). To further characterize this protease activity, we incubated cytosolic extracts from glutamate-treated (30 μM; 24 hr) CGNs with the caspase 3 tetrapeptide inhibitor Ac-DEVD-CHO or the caspase 1 (ICE) protease inhibitor Ac-YVAD-CHO. Caspase 3 protease activity in cytosolic extracts of glutamate-treated CGNs is inhibited specifically by Ac-DEVD-CHO (IC50 < 10nM), but not by Ac-YVAD-CHO (Fig. 5A, Inset). Consistent with the enzyme activity experiments, we confirmed activation of caspase 3 by demonstrating cleavage of PARP, a known caspase 3 substrate, and the proteolytically active p12 subunit from pro-caspase 3 in Western blots after exposure of CGNs to glutamate (30 μM; 24 hr) in the absence or presence of 40 nM actinomycin D (Fig. 5C) (35, 36).
Figure 5.
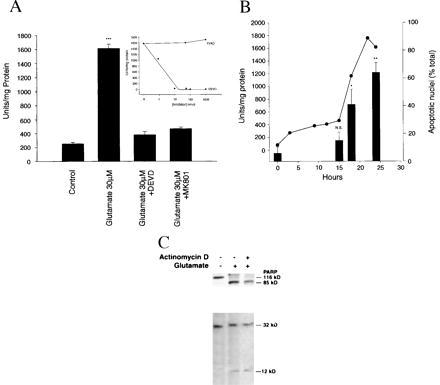
Caspase 3 protease activity in cultured cerebellar granule neurons is induced by glutamate. (A) The caspase 3-specific color substrate Ac-DEVD-pNA was incubated for 60 min at 37°C with 30 μg cytosolic protein collected from CGNs treated with glutamate (30 μM, 24 hr) in the presence or absence of Ac-DEVD-CHO (200 μM, 24 hr) or dizocilpine (MK-801), (1 μM, 24 h). Measurements were taken at 405 nm (see Materials and Methods for details). One unit of enzyme is defined as the amount of enzyme required to release 0.22 pmol of p-nitroaniline per min at 37°C. Note that both Ac-DEVD-CHO and dizocilpine (MK-801) completely inhibit glutamate-induced activation of caspase 3 activity. (B) Caspase 3 protease activity is induced significantly at 15 hr after glutamate exposure and increases 8-fold over the ensuing 9 hr (solid dark line), just before the measurement of apoptosis (bars). Note increase in caspase 3 activity that is temporally associated with an increase in the number (%) apoptotic nuclei measured by Hoechst 33258 staining (see Materials and Methods for details). Caspase 1 protease activity was not detected at any time point after glutamate exposure (30 μM, 24 hr). Protease activity was determined as described above. Glutamate-induced caspase 3 protease activity measured in cytosolic extracts from CGNs exposed to glutamate is potently inhibited in vitro by Ac-DEVD-CHO (IC50 < 10nM), but not by the caspase 1-specific inhibitor Ac-YVAD-CHO (A, Inset). (C) PARP and caspase 3 proenzyme from cell extracts of CGNs exposed to glutamate (30 μM, 24 hr) are proteolytically processed to their 85-kDa and 12-kDa subunits, respectively. Protein from CGNs exposed to glutamate (30 μM, 24 hr) were size-fractionated on a 10–20% gradient gel, transferred to nitrocellulose, and detected with polyclonal antibodies directed to PARP or the 12-kDa subunit of caspase 3 using enhanced chemiluminescence (see Materials and Methods for details).
Neuroprotection Afforded by Ac-DEVD-CHO Is Not Due to Channel Block of Ionotropic Glutamate Receptors.
We next examined the possibility that the excitoprotective effects of Ac-DEVD-CHO against glutamate-induced apoptosis of CGNs may have been due to a direct interaction of the peptide with ion channels known to be involved in glutamate toxicity. Individual CGNs were clamped in the whole-cell configuration (29) to assess changes in glutamate-evoked currents in the presence of an Ac-DEVD-CHO concentration that enhances neuronal survival and blocks apoptosis induced by glutamate. Macroscopic currents activated by 30 μM glutamate remained unchanged when simultaneously exposed to 100 μM Ac-DEVD-CHO (data not shown). Moreover, because coapplication of Ac-DEVD-CHO and 1 μM dizocilpine (MK-801) produced an almost complete (93 ± 0.7%) block of glutamate currents, the Ac-DEVD-CHO responses described here cannot be attributed to a direct block of NMDA or other ionotropic glutamate receptors.
DISCUSSION
This report demonstrates a critical role for an ICE/CED-3 related protease in the excitotoxic and apoptotic death of mammalian neurons induced by glutamate. Excitotoxicity of CGNs, which under our experimental conditions is mediated by NMDA receptors, occurs by either necrosis or apoptosis, depending on the degree of receptor stimulation (10). Indeed, our data strongly suggest that the specific CED-3-related cysteine protease CPP32 (now designated caspase 3) is, in fact, the specific protease mediating glutamate-induced apoptosis of CGNs. We caution, however, that although caspase 3 is activated by glutamate and with a time course that parallels the apoptotic death of CGNs, it is still conceivable that a closely related Caspase 3-like enzyme (rather than Caspase 3 itself) may be responsible. Moreover, the inhibition of glutamate-induced apoptosis of CGNs by Ac-DEVD-CHO suggests, but does not prove, that caspase 3 is involved. Experiments carried out in CGNs derived from the CPP32 knockout mouse (18) obviously could be useful in answering this question. Other reports have strongly implicated ICE/CED-3-related proteases in trophic/growth factor-deprived apoptosis of peripheral sympathetic neurons (37) or PC12 cells (38) or in apoptosis of CGNs induced by K+ withdrawal (19, 39, 40), but a specific protease(s) has not been identified. Finally, we show here that the activity of the CED-3-related cysteine protease caspase 3 is increased in CGNs by relatively low concentrations of glutamate, the principal excitatory neurotransmitter in brain.
Cultured cerebellar granule neurons have been used by several laboratories to study neuronal apoptosis (8, 22, 41, 42) and to characterize excitotoxicity induced by various glutamate receptor agonists (5, 8, 11, 30, 31, 40). In all of these studies CGNs have been maintained in primary culture by using culture conditions that effectively block their programmed death (via apoptosis); i.e., that which would occur normally in the absence of any specialized culture conditions. It is likely that the apoptotic death of CGNs induced by nondepolarizing culture conditions mimics the natural death of granule neurons, which occurs in the developing cerebellum (43) and appears to be critical for matching in rather precise stoichiometry the number of granule neurons and Purkinje cells observed in the adult cerebellum (44).
Apoptosis of CGNs can be induced in several ways, including switching CGNs from depolarizing (high K+) to nondepolarizing (low K+) media and (or) by serum withdrawal. Recently, Miller and Johnson (42) have characterized apoptosis of CGNs induced by either K+ or serum deprivation and have delineated both fast-dying (T½ = 4 hr) and slow-dying (T½ = 25 hr) populations. The latter population (which is induced to die by K+ withdrawal) accounts for approximately 80% of CGNs that apoptose after combined K+/serum (42). We previously have characterized apoptosis of CGNs induced by nondepolarizing culture conditions (switching to low K+ medium) and have found it to require new RNA and protein synthesis, but to be insensitive to ionotropic glutamate receptor antagonists (8). Similar findings recently have been reported by Schulz and colleagues (39). By contrast, Atabay and coworkers (41) have reported that glutamate receptor antagonists (of both the NMDA and AMPA/kainate receptor subtypes) can block a portion of serum withdrawal-induced apoptosis of CGNs. Moreover, we also have found that the wasp venom peptide mastoparan, which stimulates the G proteins Go/Gi, readily induces apoptosis of CGNs. The latter is attenuated by cholera toxin (an activator of Gs) and cycloheximide, but not by glutamate receptor antagonists (45). Taken together, these data suggest that apoptosis of CGNs can be induced by different stimuli and environmental conditions and that different intracellular mechanisms are likely to be involved.
Our data strongly suggest that the majority of NMDA receptor-mediated apoptosis of CGNs is mediated by posttranslational activation of caspase 3. Unlike Fas-mediated apoptosis of mouse lymphoma cells (32) caspase 1 (ICE) itself is not involved, as no evidence shows increased caspase 1 activity after glutamate exposure of CGNs, nor does a specific caspase 1 inhibitor affect glutamate-induced apoptosis. Moreover, we previously have failed to detect caspase 1 mRNA in CGNs under either depolarizing or nondepolarizing culture conditions (19). Our experiments provide no information, however, as to how caspase 3 is activated after NMDA receptor stimulation, nor can we exclude the involvement of other ICE-related proteases, either upstream or downstream of caspase 3. Conceivably, activation of an as-yet-unidentified Ca2+-dependent protease by the high levels of intracellular Ca2+ induced by NMDA receptor activation, and previously shown to initiate the excitotoxic death cascade (6), may induce processing and activation of the caspase 3 proenzyme. Nonetheless, it is clear from our work (8) and the work of others (11, 22, 31) that apoptosis of CGNs can proceed via mechanisms that require de novo gene transcription or, as reported here, by the activation of already translated protein(s).
Finally, if our findings can be extended to other populations of neurons (and/or) other glutamate receptor subtypes, brain-penetrable inhibitors of caspase 3 may prove to be of therapeutic value for a number of neurodegenerative disorders where overstimulation of glutamate receptors has been implicated (1, 3).
Acknowledgments
We thank Drs. Tom Bumol, Patrick May, Sheila Little, Elcira Villarreal, and H. Robert Horvitz for critical review and comments on our manuscript. We thank Theresa Moore and Pamela Edmonds for excellent editorial assistance.
ABBREVIATIONS
- ICE
interleukin 1β-converting enzyme
- CGN
cerebellar granule cell
- PARP
poly(ADP-ribose) polymerase
- NMDA
N-methyl-d-aspartate
- CED
Caenorhabditis elegans cell death
- dizocilpine (MK-801)
(5R,10S)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a, e]cyclohepten-5,10-imine hydrogen maleate
- FDA
fluorescein diacetate
- PI
propidium iodide
Footnotes
Alnemri and colleagues (13) recently have proposed a new nomenclature for the human ICE/CED-3 protease family. We have adopted this nomenclature and now refer to the CED-3-related cysteine protease CPP32/Yama/apopain as caspase 3.
References
- 1.Choi D W. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- 2.Meldrum B, Garthwaite J. Trends Pharmacol Sci. 1990;11:379–387. doi: 10.1016/0165-6147(90)90184-a. [DOI] [PubMed] [Google Scholar]
- 3.Coyle J T, Puttfarcken P. Science. 1993;262:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- 4.Lipton S A, Rosenberg P A. N Engl J Med. 1994;330:613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- 5.Manev H, Favaron A, Guidotti A, Costa E. Mol Pharmacol. 1989;36:106–112. [PubMed] [Google Scholar]
- 6.Choi D W. Trends Neurosci. 1995;18:58–60. [PubMed] [Google Scholar]
- 7.Dessi F, Charriaut-Marlangue C, Khrestchatisky M, Ben-Ari Y. J Neurochem. 1993;60:1953–1955. doi: 10.1111/j.1471-4159.1993.tb13427.x. [DOI] [PubMed] [Google Scholar]
- 8.Yan G M, Ni B, Weller M, Wood K A, Paul S M. Brain Res. 1994;656:43–51. doi: 10.1016/0006-8993(94)91364-1. [DOI] [PubMed] [Google Scholar]
- 9.Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton S A. Proc Natl Acad Sci USA. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ankarcrona M, Dypbukt J M, Orrenius E, Lipton S A, Nicotera P. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- 11.Simonian N A, Getz R L, Leveque J C, Konrad C, Coyle J T. Neuroscience. 1996;74:675–683. doi: 10.1016/0306-4522(96)00141-8. [DOI] [PubMed] [Google Scholar]
- 12.Ellis H M, Horvitz H R. Cell. 1986;44:817–829. doi: 10.1016/0092-8674(86)90004-8. [DOI] [PubMed] [Google Scholar]
- 13.Alnemri E S, Livingston D J, Nicholson D W, Salvesan G, Thornberry N A, Wong W W, Yuan J. Cell. 1996;87:171. doi: 10.1016/s0092-8674(00)81334-3. [DOI] [PubMed] [Google Scholar]
- 14.Martin S J, Green D R. Cell. 1995;82:349–352. doi: 10.1016/0092-8674(95)90422-0. [DOI] [PubMed] [Google Scholar]
- 15.Oppenheim R W. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- 16.Raff M C, Barres B A, Burne J F, Coles H S, Ishizaki Y, Jacobson M D. Science. 1993;262:695–700. doi: 10.1126/science.8235590. [DOI] [PubMed] [Google Scholar]
- 17.Bredesen D E. Ann Neurol. 1995;38:839–851. doi: 10.1002/ana.410380604. [DOI] [PubMed] [Google Scholar]
- 18.Kuida K, Zheng T S, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell R A. Nature (London) 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- 19.Ni B, Wu X, Du Y, Su Y, Hamilton-Byrd E, Rockey P K, Rosteck P, Poirier G G, Paul S M. J Neurosci. 1997;17:1561–1569. doi: 10.1523/JNEUROSCI.17-05-01561.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ito M. In: The Cerebellum and Neural Control. Ito M, editor; Ito M, editor. New York: Raven; 1984. pp. 74–85. [Google Scholar]
- 21.Gallo V, Kingsbury A, Balazs R, Jorgensen O S. J Neurosci. 1987;7:2203–2213. doi: 10.1523/JNEUROSCI.07-07-02203.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.D’Mello S R, Gall C, Ciotti T, Calissano P. Proc Natl Acad Sci USA. 1993;90:10989–10993. doi: 10.1073/pnas.90.23.10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gallo V, Ciotti M T, Coletti A, Aloisi F, Levi G. Proc Natl Acad Sci USA. 1982;79:7919–7923. doi: 10.1073/pnas.79.24.7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manev H, Favaron M, Vicini S, Guidotti A, Costa E. J Pharmacol Exp Ther. 1990;252:419–427. [PubMed] [Google Scholar]
- 25.Herrmann M, Lorenz H-M, Voll R, Grünke M, Woith W, Kalden J R. Nucleic Acids Res. 1994;22:5506–5507. doi: 10.1093/nar/22.24.5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Colacino J M, Horn J W, Horn D M, Richardson R C. Toxicol in Vitro. 1996;10:297–303. doi: 10.1016/0887-2333(96)00016-1. [DOI] [PubMed] [Google Scholar]
- 27.Liu X, Kim C N, Yang J, Jemmerson R, Wang X. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 28.Barrett A J. Methods Enzymol. 1981;80:561–565. doi: 10.1016/s0076-6879(81)80044-4. [DOI] [PubMed] [Google Scholar]
- 29.Hamill O P, Marty A, Neher E, Sakmann B, Sigworth F J. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 30.Marini A M, Paul S M. Proc Natl Acad Sci USA. 1992;89:6555–6559. doi: 10.1073/pnas.89.14.6555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chuang D M, Gao X M, Paul S M. Mol Pharmacol. 1992;42:210–216. [PubMed] [Google Scholar]
- 32.Enari M, Talanian R V, Wong W W, Nagata S. Nature (London) 1996;380:723–726. doi: 10.1038/380723a0. [DOI] [PubMed] [Google Scholar]
- 33.Hasegawa J I, Kamada S, Kamiike W, Shimizu A, Imazu T, Matsuda H, Tsujimoto Y. Cancer Res. 1996;56:1714–1718. [PubMed] [Google Scholar]
- 34.Lazebnik Y A, Kaufmann S H, Desnoyers S, Poirier G G, Earnshaw W C. Nature (London) 1994;371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- 35.Nicholson D W, Ali A, Thornberry N A, Vaillancourt J P, Ding C K, Gallant M, Gareau Y, Griffin P R, Labelle M, Lazebnik Y A, Munday N A, Raju S M, Smulson M E, Yamin T T, Yu V L, Miller D K. Nature (London) 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 36.Tewari M, Quan L T, O’Rourke H K, Desnoyers S, Zeng Z, Beidler D R, Poirier G G, Salvesen G S, Dixit V M. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- 37.Greenlund L J, Deckwerth T L, Johnson E M. Neuron. 1995;14:303–315. doi: 10.1016/0896-6273(95)90287-2. [DOI] [PubMed] [Google Scholar]
- 38.Troy C M, Stefanis L, Prochiantz A, Greene L A, Shelanski M L. Proc Natl Acad Sci USA. 1996;93:5635–5640. doi: 10.1073/pnas.93.11.5635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schulz J B, Weller M, Klockgether T. J Neurosci. 1996;16:4696–4706. doi: 10.1523/JNEUROSCI.16-15-04696.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Armstrong R C, Aja T J, Hoang K D, Gaur S, Bai X, Alnemri E S, Litwack G, Karanewsky D S, Fritz L C, Tomaselli K J. J Neurosci. 1997;17:553–562. doi: 10.1523/JNEUROSCI.17-02-00553.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Atabay C, Cagnoli C M, Kharlamov E, Ikonomovic M D, Manev H. J Neurosci. 1996;43:465–475. doi: 10.1002/(SICI)1097-4547(19960215)43:4<465::AID-JNR7>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 42.Miller T M, Johnson E M., Jr J Neurosci. 1996;16:7487–7495. doi: 10.1523/JNEUROSCI.16-23-07487.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wood K A, Dipasquale B, Youle R J. Neuron. 1993;11:621–632. doi: 10.1016/0896-6273(93)90074-2. [DOI] [PubMed] [Google Scholar]
- 44.Williams R W, Herrup K. Annu Rev Neurosci. 1988;11:423–453. doi: 10.1146/annurev.ne.11.030188.002231. [DOI] [PubMed] [Google Scholar]
- 45.Yan G M, Lin S Z, Irwin R P, Paul S M. J Neurochem. 1995;65:2425–2431. doi: 10.1046/j.1471-4159.1995.65062425.x. [DOI] [PubMed] [Google Scholar]