

Abstract
We explore charge migration in DNA, advancing two distinct mechanisms of charge separation in a donor (d)–bridge ({Bj})–acceptor (a) system, where {Bj} = B1,B2, … , BN are the N-specific adjacent bases of B-DNA: (i) two-center unistep superexchange induced charge transfer, d*{Bj}a → d∓{Bj}a±, and (ii) multistep charge transport involves charge injection from d* (or d+) to {Bj}, charge hopping within {Bj}, and charge trapping by a. For off-resonance coupling, mechanism i prevails with the charge separation rate and yield exhibiting an exponential dependence ∝ exp(−βR) on the d-a distance (R). Resonance coupling results in mechanism ii with the charge separation lifetime τ ∝ Nη and yield Y ≃ (1 + δ̄ Nη)−1 exhibiting a weak (algebraic) N and distance dependence. The power parameter η is determined by charge hopping random walk. Energetic control of the charge migration mechanism is exerted by the energetics of the ion pair state d∓B1±B2 … BNa relative to the electronically excited donor doorway state d*B1B2 … BNa. The realization of charge separation via superexchange or hopping is determined by the base sequence within the bridge. Our energetic–dynamic relations, in conjunction with the energetic data for d*/d− and for B/B+, determine the realization of the two distinct mechanisms in different hole donor systems, establishing the conditions for “chemistry at a distance” after charge transport in DNA. The energetic control of the charge migration mechanisms attained by the sequence specificity of the bridge is universal for large molecular-scale systems, for proteins, and for DNA.
In 1962, Eley and Spivey proposed (1) that π–π interactions between stacked base pairs in double-strand DNA could provide a pathway for rapid, one-dimensional charge separation. In spite of subsequent theoretical and experimental effort in this intriguing field (2–7), experimental evidence for such “molecular wire” type conduction in DNA remained elusive. The studies of Warman et al. (8) in 1996 of radiation-induced conductivity in hydrated DNA argued against one-dimensional conduction confined to the base pair core. Interest in this fascinating subject (9–31) was triggered recently by the studies of Barton and her colleagues (9–19), which seemed to indicate the occurrence of long-range, almost distance-independent charge separation in DNA, manifesting “chemistry at a distance” (17). The problem of charge separation in DNA (9–31) is pertinent for the realization of a particular DNA repair mechanism as an alternative to the DNA-photolyase (20–23), which rests on long-range charge transfer to the defect site, i.e., a thymine dimer followed by concurrent or sequential bond breaking. Moreover, a deeper understanding of charge migration processes and of the effects of electronic excess charges localized at specific nucleic bases has wide range implications for (i) protein binding to DNA. Because electrostatic interactions are primarily responsible for the association of proteins to nucleic bases, changes in the charge density at the DNA core induced by charge separation may affect the specificity of protein binding; (ii) DNA sequencing. The control of duplex formation via charge migration may be important for specific DNA sequencing; and (iii) DNA-based biosensors. The development of biosensors, which depend on specific long-range charge separation along duplex structures in solution and preferentially at electrodes, is of considerable potential.
The interpretation of the early experiments of Barton, Turro, and their colleagues (9–13) on charge separation between donor and acceptor complexes attached to DNA was fraught with some difficulties because of the possibility of aggregation effects (24). The recent data of Dandliker, Holmlin, and Barton (17–19) on hole migration between the electronically excited metal intercalator Rh(phi)2DMB+3 and the thymine dimer, both of which are specifically incorporated in a 16-bp DNA duplex, provide evidence for long-range hole separation (over a distance scale of r = 19–26 Å) with the yield being independent of donor–acceptor distance (R). These results (17–19) are in dramatic conflict with other experiments on charge separation in DNA (25–27), as well as with the standard electron transfer theory (32–40). For a donor (d)–bridge–acceptor (a) system, the theory (33–40) predicts an exponential (donor–acceptor) distance R dependence of the hole (or electron) transfer rate, k = (2π/η)V2F of the Marcus– Levich–Jortner equation (33–40):
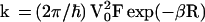 |
1 |
Here, F is the thermally averaged nuclear Franck–Condon density (involving both medium and intramolecular vibrational modes) whereas the electronic coupling V = V0 exp[−(β/2)R] is (approximately) characterized by the constant energy V0 and the exponential parameter β. Indeed, Beratan and colleagues (41) have applied electron transfer theory to the Rh(phi)2DMB+3–DNA–thymine dimer system (17, 18), predicting that the superexchange rate, Eq. 1, should have β = 1.2–1.6 Å−1. Recent experimental results (25–27) for hole transfer or migration to guanine (G), Eqs. 2a, 2c, and 2d, and for recombination, Eq. 2b, are well accounted for by conventional electron transfer theory, Eq. 1, i.e.,
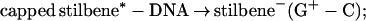 |
2a |
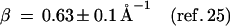 |
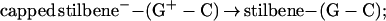 |
2b |
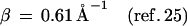 |
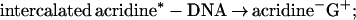 |
2c |
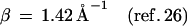 |
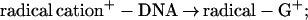 |
2d |
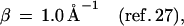 |
where * denotes an electronic singlet excited state of the hole donor. These charge separation/recombination/migration processes, Eqs. 2a–2d, provide an apparent contradiction to the results of Barton et al. (17, 18). However, it should be borne in mind that different charge donors, which are characterized by different energetics, may give rise to different mechanisms for charge separation in DNA. The perspectives of such energetic control raise the distinct possibility of an alternative mechanism of charge separation in DNA. We propose two mechanisms for charge separation in DNA: (i) The two-center unistep superexchange mediated electron/hole transfer described by Eq. 1; and (ii) electron/hole multistep transport via some bases of the DNA backbone superstructure. This charge transport mechanism was explored in molecular crystals (42–48) and in polymers (47, 48) and was advanced for DNA (40).
The open questions in the area of charge transfer and transport in DNA and related model systems are (i) under what conditions can long-range, efficient charge transport with a weak distance dependence prevail in DNA? (ii) When will the superexchange-mediated, unistep, exponentially distance-dependent charge separation occur in DNA? (iii) What are the structural and energetic attributes, e.g., base sequence specificity, of the donor–DNA–acceptor systems, which will exhibit charge transport or charge transfer in DNA? (iv) How can one induce the “transition” from electron transport to electron transfer in DNA by the modification of the donor or/and acceptor centers? And (v) are there basic differences between charge separation in DNA and in proteins?
Our starting point rests on the distinction between the charge transport and charge transfer mechanisms in DNA. We address the energetic–dynamic control of the mechanism of charge separation and the novel implications of the charge transport mechanism. These fascinating phenomena of charge migration in finite, large molecular-scale systems pertain to the novel areas of molecular electronics (49) and to the control of the primary, ultrafast, charge separation processes in photosynthesis (50), providing cornerstones for the relations between structure, energetics, dynamics and function in chemistry.
Mechanisms For Charge Separation in DNA.
The prevalence of the two distinct mechanisms of charge separation: (i) two-center donor–acceptor, unistep, superexchange-induced charge transfer, and (ii) multistep charge transport in DNA, are controlled by the energetics of the ion pair states of the system (Fig. 1). The relevant electronic states are
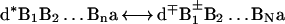 |
3 |
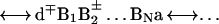 |
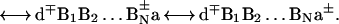 |
Here, d is the electron donor (capped or intercalated), a is the electron acceptor (which can constitute part of the DNA) while {Bj} ≡ B1,B2, … BN are the proper adjacent bases. In what follows, we consider the generic example of the separation of an electron–hole pair. The same approach will account for positive ion–hole charge migration from (d+q)*{Bj}a to d+q−1{Bj}a+. The two distinct charge separation mechanisms in system 3 will be addressed.
Figure 1.

Vibronic level scheme for the two distinct charge migration mechanisms in DNA. (A) Unistep charge transfer via superexchange. (B) Multistep charge transport via hopping and trapping.
Superexchange Charge Transfer.
Electron/hole transfer (mechanism i) will occur for off-resonance coupling between the electronic origin and low vibronic states (in the energy range ≈kBT) of the initial d*{Bj}a state with all of the d∓{Bj}±a vibronic manifolds, with a large energy gap δE ≫ 0 (Fig. 1A). Following the McConnell perturbation theory (51) and using the scattering matrix formalism (52), the electronic coupling for the superexchange interaction can be expressed in the form (52) V = (tdta/Δ1)(t/Δ)N−1, where the nearest-neighbor electronic matrix elements are
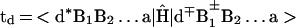 |
4a |
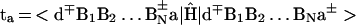 |
4b |
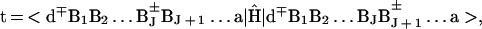 |
4c |
where |> denote the diabatic (valence bond) electronic states and Ĥ is the system’s electronic Hamiltonian. The vertical electronic energy gaps are Δ1 = δE + λ and Δ = λ, where δE is the electronic energy gap (Fig. 1A) while λ is the reorganization energy. For the sake of simplicity, we take the matrix elements t in the {BJ} chain to be site-independent. The superexchange electronic coupling assumed the form (52) V = (tdta/Δ1) exp[−Nln(Δ/t)]. The number, N, of units in the bridge is given by N = R/r0, where r0 is the nearest-neighbor B–B distance. Under these energetic conditions, superexchange-mediated transfer will occur with the rate
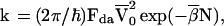 |
5 |
i.e., k ∝ exp(−β̄R/r0), where V̄0 = tdta/Δ1 and β̄ = 2ln(Δ/t). Fda is the Franck–Condon density between the donor |d*B1B2 … > vibronic doorway state and the |d∓B1B2 … a±> vibronic quasicontinuum (53). Eq. 5 manifests the exponential donor–acceptor distance and N dependence of the superexchange mediated rate and yields in accord with Eq. 1.
Multistep Charge Transport.
Electron/hole transport (mechanism ii) will prevail when three energetic conditions are satisfied simultaneously (Fig. 1B): (i) resonant coupling between the electronic origin and low vibronic levels of the initial d*{Bj}a state with the vibronic manifold of the primary ion pair d∓B1±B2 … BNa, i.e., δE < 0; (ii) near degeneracy of the origins of the vibronic manifolds of the ion pair states d∓B1±B2 … BNa, d∓B1B2± … BNa, and d∓B1B2 … BN±a; and (iii) degeneracy of the vibronic manifolds of the base ion pair state d∓B1B2 … BN± and the ion-pair state d∓B1B2 … BNa±. The resonant coupling (i) results in the injection of the hole/electron into the appropriate B1 base of DNA; the near degeneracy (ii) then will induce charge hopping between the {Bj} (n = 1, … , N) bases while the degeneracy (iii) will result in charge (hole/electron) trapping at the acceptor center.
We now address the microscopic description of charge injection, transport and trapping. We shall use an extension of the theoretical models for the dynamics in multiple Franck–Condon quasicontinua (53). The level structure corresponds to the initial doorway state |α> of the d*B1B2 … BNa vibronic state, intermediate vibronic manifolds, i.e., d∓B1±B2 … BNa{|β1>}, d∓B1B2± … BNa{|β2>} … d∓B1B2 … BN±a{|βN>} and the final d∓B1B2 … BNa±{|γ>} vibronic manifold (Fig. 1B). Resonance |α>–{|β1>} − {|β2>} … {|βN>} − {|γ>} coupling results in a reversible–sequential kinetic scheme for the populations of the doorway state
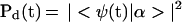 |
6a |
of the intermediate vibronic manifolds J ≡ {βJ>}
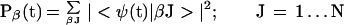 |
6b |
and of the final acceptor manifold
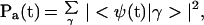 |
6c |
where ψ(t) is the wave function of the system, with the initial conditions ψ(t = 0) = |α>. The prevalence of reversible–sequential kinetics for the population probabilities manifests phase erosion caused by weakly correlated intercontinuum coupling in conjunction with vibrational relaxation and vibrational excitation within the individual vibronic manifolds (53). The kinetic scheme is
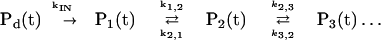 |
7 |
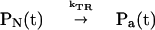 |
being characterized by the following microscopic rates.
(i) The charge injection rate is
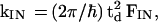 |
8 |
where the electronic coupling td is given by Eq. 4a. FIN is the thermally averaged nuclear Franck–Condon density for the |α>–{|β1>} vibronic coupling. Electron transfer theory gives the thermally averaged Franck–Condon density F, which incorporates both medium and intramolecular vibrational modes, by (34–36)
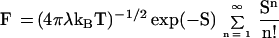 |
9 |
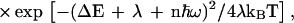 |
where λ is the medium reorganization energy involving the contribution of low frequency vibrational modes while the high frequency intramolecular vibrational modes are characterized by the (mean) vibrational frequency ω and coupling S, and ΔE is the (free) energy gap. For the charge injection process, Eq. 8, we use Eq. 9, i.e., FIN = F, taking λ = λIN, with λIN being the medium reorganization energy for injection and the energy gap ΔE = δE (Fig. 1B). The exoergic charge injection process (Eqs. 8 and 9 and Fig. 1B) can be normal, i.e., with a low activation energy (δE + λIN)2/4λIN, activationless, or inverted, with the latter exhibiting a weak temperature dependence caused by vibrational excitation induced by electron transfer (35, 36, 39, 40). A more detailed analysis also has to incorporate the recombination of the primary ion pair d∓B1± to the triplet state and to the ground electronic state of d.
(ii) The charge hopping rates are ki, i ± 1 = (2π/ℏ)t2Fi,i±1 for i = 1, … , N, with the intersite electronic coupling t (approximately site invariant) given by Eq. 4c. For bridge units separated by higher oxidation potential bases, t is determined by nearest-neighbor superexchange interactions. Fi, i ± 1 is the nuclear Franck–Condon density between the {|βi>} − {|βi ± 1>} vibronic manifolds. Assuming near degeneracy of the electronic origins of the ion pair states, charge hopping in the {βj} chain occurs between nearly isoenergetic ion pair states of adjacent bases. Under these circumstances, the hopping rates, Eq. 10, correspond to symmetric electron transfer (ΔG = 0) with the hopping rates being site-independent, i.e., ki, i ± 1 = kHOP (for all values of i) with
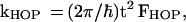 |
10 |
where the Franck–Condon density FHOP is given by Eq. 9, i.e., FHOP = F, with λ = λHOP being the medium (low frequency) reorganization energy for the hopping, with the process being symmetric, i.e., ΔE = 0. At sufficiently high temperatures, at which nuclear tunneling effects are negligible, the forward and backward hopping processes in the {Bj} chain are expected to be activated, with an activation energy of ≈λHOP/4.
(iii) The trapping rate is
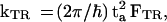 |
11 |
where the electronic coupling ta is given by Eq. 4b while F = FTR represents the thermally averaged nuclear Franck–Condon density between the {|βN>} − {|γ>} vibronic manifolds, given by Eq. 9, with ΔE = δETR being the (free) energy gap for trapping (Fig. 1B) and λ = λTR being the corresponding reorganization energy. The trapping process may be normal (activated), activationless, or inverted (with a weak temperature dependence) (35, 36, 39, 40), depending on the relation between δETR and λTR. In this analysis, we have disregarded the thermally activated detrapping process
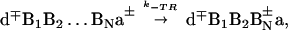 |
with the rate k−TR ≅ kTRexp(−δETR/ kBT), which readily can be incorporated in the kinetic scheme.
The specification of the individual microscopic rates for charge injection, hopping, and trapping provides an adequate description of the compound charge transport process. Multistep charge transport involves as a central ingredient charge hopping among (nearly) isoenergetic ion pair states of specific adjacent bases. This mechanism of stepwise charge transport involves an incoherent process with memory loss on each site being imposed by vibronic phase erosion and thermally induced vibrational excitation/relaxation processes (53). The charge transport process is analogous to Holstein’s incoherent small polaron motion (47) in (one-dimensional) solids. The multistep charge transport processes constitute a series of individual charge transfer steps between adjacent units in the d{Aj}a system. This description makes contact between multistep charge transport and intersite charge transfer.
A central conclusion emerging from the kinetic analysis of charge hopping transport in DNA is that this mechanism provides a weak donor–acceptor distance dependence for the rate (and yield) of the acceptor oxidation/reduction. A heuristic description of charge transport will describe this incoherent process in terms of a charge diffusion process among the appropriate bases. Many of the concepts developed in the area of charge transport (42–45, 47) and triplet excitation transport (54, 55) in pure and mixed molecular crystals are applicable to the DNA charge transport problem. A simple approximate way to describe hopping charge transport in the {Bj} one-dimensional chain rests on the diffusion model. A diffusion coefficient D for the hopping charge transport can be defined in analogy to triplet exciton transport (54, 55) in the form D ≃ kHOP(r0)2, where kHOP is given by Eq. 10. The time scale τ for charge diffusion over the donor-acceptor spatial distance R, as characterized by the diffusion process, is given by R = (2Dτ)1/2. Making contact between the time scale for diffusive hopping and the number of constituents in the chain N = R/r0, we obtain a weak distance (N) dependence of the charge separation, i.e., τ = N2(r0)2/2D = N2/2kHOP. This lifetime can be used for the analysis of time-resolved data for charge separation in DNA. Several experimental studies (17–19, 26, 27) provide information on the charge separation yield in DNA, which now will be addressed.
We introduce a characteristic lifetime τCOM for competitive charge depletion processes, which can occur from the donor site and from the bridge sites. A detailed analysis will require consideration of site-specific competitive charge depletion, but, for the present analysis, an average value of τCOM will be taken. The yield for charge transport Y = τ−1(τ−1 + τCOM−1)−1 then can be expressed in the form Y = (1 + δN2)−1, where δ = (2kHOPτCOM)−1. A more elaborate treatment of charge transport in the d{Bj}a system can be described in terms of mean residence times and first passage times in finite one-dimensional systems (56–62). These characteristic times give insight into the kinetics of trapping processes, with the mean first passage time providing a useful measure for the efficiency of the trapping. Symmetric random walks were explored (60–62) with nearest-neighbors forward hopping rates k+ and backward hopping rates k−. For unbiased processes with equal rates, i.e., k+ = k− = kHOP (where kHOP is given by Eq. 10), the mean first passage time, τMFPT, for a random walk initially injected to the first site to reach the trap is given by τMFPT = N(N+1)/2kHOP, with the trapping yield being given by Y = [1 + (τMFPT/τCOM)]−1. One obtains a weak, algebraic type N and a distance dependence of the charge separation lifetimes and yields of the form τ = (2kHOP)−1N(N+1) and Y ≃ [1+δN(N+1)]−1. The description of diffusive type unbiased charge transport has to be extended to incorporate biased charge hopping and static disorder effects.
Energetics of the B+ States of the DNA Bases Determine the Nature of the Bridge for Hole Transfer or Transport in DNA.
From the foregoing analysis, we infer that a proper energetic manipulation of the donor, with the modification of the energetics of the ion pairs, will modify the energy level structure from off-resonant (δE > 0) coupling to resonant (δE < 0) coupling, resulting in a transition from charge transfer to charge transport (Fig. 2). We shall limit our analysis to hole separation, with a hole donor attached (i.e., substituted, capped, or intercalated) to DNA being reduced while the acceptor is oxidized. The energetics of the initial d*{Bj}a state was estimated by us from the redox potentials E(d*/d+) of d*/d+ (25–27, 65) evaluated from the Rehm–Weller relation (63): E(d*/d+) = E(d/d+) + Es + C, where E(d/d+) is the ground state d/d+ redox potential, Es is the excited singlet state energy, and C is the solvent-dependent Coulomb attraction energy, which can presumably be neglected in a polar solvent such as water (64). For some hole donors already studied, Eqs. 2a, 2b, and 2d, the redox potentials of electronically excited donors in solution are given in Table 1. The oxidation potentials of the single DNA bases (66, 67) in solution on a relative scale (Table 1) are guanine (G) E = 0, adenine (A) E = 0.45 eV whereas for thymine (T) and cytosine (C) E = 0.6–0.7 eV. These single-base redox potentials presumably preserve the hierarchy of the relative energies in DNA. From the energetics of the different bases we draw conclusions regarding the following.
Figure 2.

Energetics of charge separation in DNA. (A)Unistep superexchange. The dashed lines represent off-resonance coupling. (B) Multistep hopping. The arrows represent individual charge transfer rates between adjacent units. Electronic origins of the doorway state, of the ion pair states, and of the oxidized donor state are marked by horizontal bars. The energetic data of the hole donors d* (or d+) control the dynamics. The dashed region marks energy uncertainty.
Table 1.
Redox potentials for elecronically excited donors E(d*/d+) and for DNA bases E(B/B+)
| d*/d+ | E(d*/d) | Ref. |
|---|---|---|
| stilbene*/stilbene− | 1.75 | 25 |
| Rh(phi)2phen+3*/Rh(phi)2phen+2 | 2.0 ± 0.15 | 65 |
| B/B† | E(B/B*) | |
| Guanine/Guanine+ | 1.45 ± 0.05 | 67 |
| Adenine/Adenine+ | 1.94 ± 0.05 | 67 |
| Thymine/Thymine+; Cytosine/Cytosine+ | 2.09–2.14 ± 0.05 | 67 |
All redox potentials are given in volt units vs. normal hydrogen electrode.
Hole acceptors in DNA.
The low oxidation potential of G makes this single base a hole acceptor in the d*-DNA system, in accord with the experimental observations (25–27). Other guanine-based hole acceptors, aG, can involve 8-oxo-guanine (29) or guanine triplets (GGG) (68), whose oxidation potential is lower than that of a single G.
Bridge bases.
Two generic classes of systems for the realization of charge separation in B-DNA and model systems can be considered: (i) d*{Aj}G and (ii) d*{Gj}aG. The nearest neighbor adenine bases {Aj} = A1,A2, … ,AN, whose oxidation potential is higher by 0.45 eV than that of G, constitute the components of the bridge in system i. The nearest-neighbor guanine bases {Gj} = G1,G2, … ,GN, whose oxidation potential is higher by ≈0.4 eV for 8-oxo-guanine (29), constitute the components of the bridge in system ii. These {Aj} and {Gj} bridges can mediate charge separation either via superexchange transfer or multistep transport.
Zigzagging between the two strands of B-DNA.
The {Aj} or {Gj} bridges of nearest-neighbor A or G bases can be located either on a single strand or on different strands of B-DNA (Fig. 3). The connectivity of the nearest neighbor {Aj} or {Gj} (j = 1, … , N) bases determines the length N of the bridge (Fig. 3).
Figure 3.

Zigzagging between the nearest-neighbor adenine or guanine bases on the two strands of DNA determines the length of the bridge for hole transport. (1) The adenines bridge in d*{Aj}G. (2) The guanines bridge in d*{Gj}aG (aG = 8-oxo-guanine or GGG). The base stacking distance (a) is in the range 2.5–4.4 Å while the average distances (b) are 3.5 Å for edge-to-edge and 7.1 Å for center-to center.
Energetic control of mechanism.
The d*{Aj}G and d*{Gj}aG systems can exhibit either hole superexchange transfer or multistep hole transport depending on the energetics of the d*A1A2 … −d−A1+A2 … system (Fig. 2) or of the d*G1-G2 … d−G1+G2 … system.
The role of T and C.
The high-energy ionic states of the d−T+ or d−C+ states are off-resonance for most hole donors (Table 1) and only can make a small additional superexchange contribution to hole migration.
Energetic Control of Hole Transfer and Transport in DNA.
Turning to the mechanisms of charge migration in DNA for the d{Aj}G systems previously studied (17, 25–29), we infer on the basis of the energetic data summarized in Fig. 2, together with our distinct mechanisms, that the hole donor stilbenedicarboxyamide (25) is characterized by low energies of d*A1 relative to d−A1+. Accordingly, off-resonance coupling with the {Aj} bridge (δE > 0) prevails in these systems, and hole migration will occur via unistep superexchange transfer, with an exponential distance dependence. A similar situation of off-resonance coupling and unistep superexchange hole transfer is expected to prevail for the hole donors 9-amino-6-chloro-2–methoxyacridine (26) and 4′-ribose radical cation (27). On the other hand, the energy of the hole donor Rh(phi)2phen+3* system (Table 1) is sufficiently high to warrant resonance coupling (δE < 0) to the {Aj} bridge. Accordingly, in the system studied by Barton et al. (17, 18), hole transport with a weak distance dependence can be realized. An analogous situation of resonance coupling resulting in hole transport prevails for DNA oxidation by excited anthraquinones (28, 29). The conceptual framework advanced by us, based on energetic-dynamic relations for hole migration, seems to remove apparent inconsistencies and accounts for the gross features of all of the available experimental data for charge migration in DNA (17–19, 25–30) in terms of unistep two-center hole transfer (25–27) or multistep hole transport (17–19, 28, 29).
Attainment of a Weak Distance Dependence of τ and Y in Multistep Charge Transport.
In the foregoing analysis of charge hopping transport, we alluded to diffusive type, unbiased random walk in the {Bj} bridge, which implies the degeneracy of the ion-pair states d−B1 … Bi+ … BNa for all i. The removal of the degeneracy of the electronic origins of the ion pair states can be induced by two types of electrostatic effects that result in level shifts: (i) a coulomb ladder. Here, the energies of the ion-pairs increase with increasing d− − Bi+ distance for the electron-hole charge separation and decrease with increasing the d+q-1 − Bi+ distance for the shift of the hole from a positive ion. The electrostatic interaction is diminished by solvent dielectric screening and ionic screening effects; (ii) electrostatic interactions with counterions. These can either stabilize or destabilize ion pairs with a large scale charge separation. Charge random walk biased toward the donor may arise from the coulomb ladder for electron-hole charge separation or from electrostatic destabilization. Random walk biased toward the acceptor can be induced from the coulomb ladder for a hole migration from a positive ion or from electrostatic stabilization. Biased charge hopping may occur in the {Bj} chain, being specified by the ratio of the rates K = k−/k+ (60–62). For donor direction-biased charge hopping, K > 1 while for acceptor direction-biased charge hopping, K < 1. The analysis (60–62) for biased random walks gives the lifetimes τ ≃ τMFPT = k+−1[α1N + α2(KN − 1)], where α1 = (1 − K)−1 and α2 = K(1 − K)−2, and yields Y = (1 + τ/τCOM)−1. For the relevant values of N and K, we infer that the lifetimes and yields for charge separation exhibit an algebraic N and distance dependence of the form
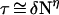 |
12 |
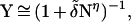 |
13 |
where the numerical constants are δ ≈ kHOP−1 and δ̄ ≈ (kHOPτCOM)−1. The power parameter η is, (i) for unbiased diffusive hopping, η = 2; (ii) for acceptor direction-biased random walk, (K < 1) 1 ≤ η ≤ 2. τ assumes a linear N dependence (60–62) for K ≪ 1; and (iii) for donor direction-biased random walk (K > 1) η ≥ 2 for moderate K values. τ assumes a fast exponential N dependence ∝ exp(NlnK) (60–62) only for large values of K and N. Our analysis establishes the prevalence of a weak N and distance dependence of the dynamic observables for charge separation when charge transport can be described in terms of unbiased/biased random walk. A scrutiny of additional effects of static disorder on charge transport is relevant and interesting. Diagonal disorder may originate from the inhomogeneous broadening of the sites of distinct ion pairs with different energies. Furthermore, the effects of off-diagonal disorder, i.e., variations of nearest neighbor electron coupling terms caused by zigzagging between bridge units across the two strands of DNA, have to be taken into account. The effects of static disorder will modify the details of the charge-hopping process but will not affect our general conclusion regarding the weak distance dependence of the dynamic observables. The energetic control of the charge separation mechanism in DNA is attained by the base sequence specificity of the {Bj} bridge.
Epilogue.
We explored the two mechanisms of unistep hole transfer and multistep hole transport in DNA and in model finite systems, establishing the energetic control of the mechanism of charge migration in DNA. On the basis of the energetics of the ion pair/donor, i.e., d−B1+B2 …/d*B1B2 in a finite d*{Bj}a system and in similar systems involving hole injection to DNA, we provided criteria for the realization of the two distinct mechanisms of charge separation in different donor-bridge systems. The energetic control of charge separation in DNA is determined by the specificity of the base sequence of the {Bj} bridge. Of considerable interest is the transition between the two limiting situations of charge transfer and transport in DNA (69), which will be exhibited for off-resonance coupling with a small energy gap (69), i.e., δE ≈ kBT. A parallel superexchange-sequential charge separation mechanism then will prevail, including both unistep transfer and multistep transport processes, which will be induced from different vibronic levels of d*{Bj}a. Interesting disorder effects and temperature effects are expected in the transition region. The energetic control of the charge separation mechanism in large molecular-scale systems is general. The two distinct charge separation mechanisms in DNA bear a close analogy to the two mechanisms of the primary charge separation between cofactors in bacterial photosynthesis, i.e., unistep superexchange and sequential mediated electron transfer (50). The prevalence of each of these mechanisms is controlled by the energetics of the ion-pair states P+B− ≡ (bacteriochlorophyll dimer)+(bacteriochlorophyll)− relative to P*B, with the sequential mechanism (induced by resonance P+B− − P*B coupling) involving a single intermediate P+B− state (50), providing another cardinal example for structure-energetics-dynamic-function relations in chemistry (32).
Multistep charge transport under the resonance donor coupling condition exhibits a weak (algebraic) N and distance dependence of the lifetime and yield for charge separation in the form given by Eqs. 12 and 13, allowing for the realization of chemistry at a distance in DNA and in any large molecular-scale system. The basic principles, which determine the energetic control attained by the sequence specificity of a bridge for short-range charge transfer or for long-range charge hopping, are universal, pertaining to both polynucleic acid structures and to proteins. Accordingly, no fundamental differences have to be envisioned between the two most important biostructures with respect to the mechanisms of charge transfer and transport.
Acknowledgments
We are indebted to Professor Joseph Klafter for stimulating discussions and for prepublication information and to Professor Richard Bersohn and Professor Bernd Giese for their perceptive comments on the manuscript. This research was supported at The Technical University of Munich and the Tel Aviv University by the Volkswagen Foundation.
References
- 1. Eley D D, Spivey D I. Trans Faraday Soc. 1962;58:411–415. [Google Scholar]
- 2.Hoffmann T A, Ladik J. Adv Chem Phys. 1964;7:84–158. [Google Scholar]
- 3.Ladik J, Biczo G. J Chem Phys. 1965;42:1658–1668. [Google Scholar]
- 4.Ladik J. Int J Quantum Chem. 1971;3S:307–330. [Google Scholar]
- 5.Suhai S. J Chem Phys. 1972;57:5599–5603. [Google Scholar]
- 6.Dee D, Bauer M E. J Chem Phys. 1974;60:541–560. [Google Scholar]
- 7.Eley D D. Mol Cryst Liq Cryst. 1989;171:1–21. [Google Scholar]
- 8.Warman J M, De Haas M P, Rupprecht A. Chem Phys Lett. 1996;249:319–322. [Google Scholar]
- 9.Murphy C J, Arkin M R, Jenkins Y, Ghattia N D, Bossmann S H, Turro N J, Barton J K. Science. 1993;262:1025–1029. doi: 10.1126/science.7802858. [DOI] [PubMed] [Google Scholar]
- 10.Murphy C J, Arkin M R, Ghattia N D, Bossmann S H, Turro N J, Barton J K. Proc Natl Acad Sci USA. 1994;91:5315–5319. doi: 10.1073/pnas.91.12.5315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stemp E D A, Arkin M R, Barton J K. J Am Chem Soc. 1995;117:2375–2376. [Google Scholar]
- 12.Holmlin R E, Stemp E D A, Barton J K. J Am Chem Soc. 1996;118:5236–5244. [Google Scholar]
- 13.Arkin M R, Stemp E D A, Holmlin R E, Barton J K, Hörmann A, Olson E J C, Barbara P F. Science. 1996;273:475–480. doi: 10.1126/science.273.5274.475. [DOI] [PubMed] [Google Scholar]
- 14.Stemp E D A, Arkin M R, Barton J K. J Am Chem Soc. 1997;119:2921–2925. [Google Scholar]
- 15.Arkin M R, Stemp E D A, Pulver S C, Barton J K. Chem Biol. 1997;4:389–400. doi: 10.1016/s1074-5521(97)90129-0. [DOI] [PubMed] [Google Scholar]
- 16.Hall D B, Holmlin R E, Barton J K. Nature (London) 1996;382:731–735. doi: 10.1038/382731a0. [DOI] [PubMed] [Google Scholar]
- 17.Dandliker P J, Holmlin R E, Barton J K. Science. 1997;275:1465–1468. doi: 10.1126/science.275.5305.1465. [DOI] [PubMed] [Google Scholar]
- 18.Holmlin R E, Dandliker P J, Barton J K. Angew Chem Int Ed Engl. 1997;36:2715–2730. [Google Scholar]
- 19.Dandliker P J, Nunez M E, Barton J K. Biochemistry. 1998;37:6491–6502. doi: 10.1021/bi980041w. [DOI] [PubMed] [Google Scholar]
- 20.Sancar A. Biochemistry. 1994;33:2–9. doi: 10.1021/bi00167a001. [DOI] [PubMed] [Google Scholar]
- 21.Heelis, P. F., Hartman, R. F. & Rose, S. D. (1995) Chem. Soc. Rev. 289–297.
- 22.Langenbacher T, Zhao X, Bieser G, Heelis P F, Sancar A, Michel-Beyerle M E. J Am Chem Soc. 1997;118:10532–10536. [Google Scholar]
- 23.Park W H, Kim S T, Sancar A, Deisenhofer J. Science. 1995;268:1866–1872. doi: 10.1126/science.7604260. [DOI] [PubMed] [Google Scholar]
- 24.Olson E J C, Hu D M, Hormann A, Barbara P F. J Phys Chem. 1997;101:299–303. [Google Scholar]
- 25.Lewis F D, Wu T, Zhang Y, Letsinger R L, Greenfield S R, Wasielewski M R. Science. 1997;277:673–676. doi: 10.1126/science.277.5326.673. [DOI] [PubMed] [Google Scholar]
- 26.Fukui K, Tanaka K. Angew Chem Int Ed Engl. 1998;37:158–161. [Google Scholar]
- 27.Meggers E, Kusch D, Spichty M, Wille U, Giesse B. Angew Chem Int Ed Engl. 1998;37:460–462. doi: 10.1002/(SICI)1521-3773(19980302)37:4<460::AID-ANIE460>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 28.Breslin D T, Schuster G B. J Am Chem Soc. 1996;118:2311–2319. [Google Scholar]
- 29.Gasper S M, Schuster G B. J Am Chem Soc. 1997;119:12762–12771. [Google Scholar]
- 30.Brun A M, Harriman A. J Am Chem Soc. 1992;114:3656–3660. [Google Scholar]
- 31.Cowan, J. A., ed. (1998) J. Biol. Inorg. Chem.3, 195–225.
- 32.Jortner J, Bixon M. In: Femtochemistry and Femtobiology: Ultrafast Dynamics at Atomic Scale Resolution. Sundström V, editor. London: Imperial College Press; 1997. pp. 349–385. [Google Scholar]
- 33.Marcus R A. Annu Rev Phys Chem. 1964;15:155–196. [Google Scholar]
- 34.Kestner N R, Logan J, Jortner J. J Phys Chem. 1974;78:2148–2166. [Google Scholar]
- 35.Efrima S, Bixon M. Chem Phys. 1976;13:447–460. [Google Scholar]
- 36.Ulstrup J, Jortner J. J Chem Phys. 1976;63:4358–4368. [Google Scholar]
- 37.Newton M D, Sutin N. Annu Rev Phys Chem. 1984;35:437–480. [Google Scholar]
- 38.Marcus R A, Sutin N. Biochim Biophys Acta. 1985;811:265–320. [Google Scholar]
- 39.Jortner J. J Chem Phys. 1976;64:4860–4867. [Google Scholar]
- 40.Bixon, M. & Jortner, J. (1998) Adv. Chem. Phys. 106, in press.
- 41.Priyadanshy S, Risser S M, Beratan D N. J Phys Chem. 1996;100:17678–17682. [Google Scholar]
- 42.Rice S A, Jortner J. In: Physics of Solids at HPLCs. Tomizuka C T, Emrick R M, editors. New York: Academic; 1965. pp. 63–170. [Google Scholar]
- 43.Katz J L, Rice S A, Choi S I, Jortner J. J Chem Phys. 1963;39:1683–1697. [Google Scholar]
- 44.Silbey R, Jortner J, Rice S A, Vala M T. J Chem Phys. 1965;42:733–737. [Google Scholar]
- 45.Silbey R, Jortner J, Rice S A, Vala M T. J Chem Phys. 1965;43:2925–2926. [Google Scholar]
- 46.Plato M, Möbius K, Michel-Beyerle M E, Bixon M, Jortner J. J Am Chem Soc. 1988;110:9279–9285. [Google Scholar]
- 47.Pope M, Swenberg C E. Electronic Processes in Organic Crystals. Oxford: Clarendon; 1982. [Google Scholar]
- 48.Simon J, André J-J. Molecular Semiconductors. Berlin: Springer; 1985. [Google Scholar]
- 49.Jortner J, Ratner M A, editors. Molecular Electronics. Oxford: Blackwell; 1997. [Google Scholar]
- 50.Bixon M, Jortner J, Michel-Beyerle M E. Chem Phys. 1995;197:389–404. [Google Scholar]
- 51.McConnell H M. J Chem Phys. 1961;35:508–518. [Google Scholar]
- 52.Jortner J, Bixon M. In: Protein Structure: Molecular and Electronic Reactivity. Austin R, Buhks E, Chance B, DeVault D, Dutton P L, Frauenfelder H, Goldanskii V I, editors. New York: Springer; 1985. pp. 277–308. [Google Scholar]
- 53.Bixon M, Jortner J. J Chem Phys. 1997;107:5154–5170. [Google Scholar]
- 54.Sommer B, Jortner J. J Chem Phys. 1968;49:3919–3928. doi: 10.1063/1.1670701. [DOI] [PubMed] [Google Scholar]
- 55.Levine M, Jortner J, Szoke A. J Chem Phys. 1966;45:1591–1604. [Google Scholar]
- 56.Weiss G H. Adv Chem Phys. 1967;13:1–18. [Google Scholar]
- 57.Van Kampen N G. Stochastic Processes in Physics and Chemistry. Amsterdam: North–Holland; 1981. [Google Scholar]
- 58.Procaccia L, Mukamel S, Ross J. J Chem Phys. 1978;68:3244–3253. [Google Scholar]
- 59.Seshadri V, West B J, Lindenberg K. J Chem Phys. 1980;72:1145–1150. [Google Scholar]
- 60.Bar-Haim A, Klafter J. J Phys Chem. 1998;102:1662–1664. [Google Scholar]
- 61.Bar-Haim A, Klafter J. J Lumin. 1998;76:197–200. [Google Scholar]
- 62.Bar-Haim, A. & Klafter, J. (1998) J. Chem. Phys., in press.
- 63.Rehm D, Weller A. Isr J Chem. 1970;8:259–271. [Google Scholar]
- 64.Weller A. Z Phys Chem (Wiesbaden) 1982;133:93–98. [Google Scholar]
- 65.Turro C, Evenzahav A, Bossmann S H, Barton J K, Turro N J. Inorg Chim Acta. 1996;243:101–108. [Google Scholar]
- 66.Steenken S, Jovanovic S V. J Am Chem Soc. 1997;119:617–618. [Google Scholar]
- 67.Seidel C A M, Schulz A, Sauer M H M. J Phys Chem. 1996;100:5541–5553. [Google Scholar]
- 68.Prat F, Houk K N, Foote C S. J Am Chem Soc. 1998;120:845–846. [Google Scholar]
- 69.Okada A, Chernyak V, Mukamel S. J Phys Chem A. 1998;102:1241–1251. [Google Scholar]