

Abstract
Atherosclerotic lesions are characterized by a profound alteration in the architecture of the arterial intima, with a marked increase of fibronectin (FN) and the appearance of the alternatively spliced FN variant containing the Extra Domain A (EDA). To analyze the role of FN isoforms in atherosclerotic lesion formation we utilized mouse strains devoid of EDA-exon regulated splicing, which constitutively include (EDA+/+) or exclude (EDA−/−) the exon. Both mutant mice had a 40% reduction in atherosclerotic lesions after the atherogenic-diet treatment (Mean±SE, μm2; 22969±2185; 13660±1533; 14260±2501 for EDAwt/wt, EDA+/+ and EDA−/−, respectively; p≤0.01 ANOVA test) associated to a lower capacity of macrophages to uptake modified LDL and undergo foam-cell formation. Lesions in control mice were more numerous and bigger, with augmented and deeper macrophage infiltration, and increased FN expression in the sub-endothelial area. Previous experiments have shown that apoE−/−EDA−/− mice have a decreased number and size of atherosclerotic lesions and, on this basis, it has been proposed that the EDA domain has a pro-atherogenic role. Our data with the EDA+/+ mice rules out this hypothesis and suggest that regulated splicing of the EDA exon of the FN gene is involved in progression of atherosclerosis, highlighting the importance of alternative splicing in regulating cellular processes.
Keywords: atherosclerosis, alternative splicing, fibronectin isoforms, macrophage, cholesterol uptake
Introduction
The highly organized architecture of the vascular wall and the spatial and functional relationships between different cell types are perturbed in pathological conditions such as atherosclerosis. Activation of the endothelium is postulated to play a key role in initiation and progression of the atherosclerotic process, followed by changes in the extracellular matrix (ECM) composition of the arterial intima [1]. Under normal conditions, the main components of the ECM are basement membrane proteins, collagen type IV, laminin, and fibronectin (FN). FN isoforms, generated by alternative splicing at three sites -the Extra Domain B (EDB), Extra Domain A (EDA), and the Type III Homologies Connecting Segment (IIICS) [2-4]-, have been associated with a variety of cellular processes such as embryogenesis, malignancy, hemostasis, wound healing and maintenance of tissue integrity [3, 5]. In the last few years, the development of mouse models produced conclusive evidence of the roles of FN and its isoforms in some of the processes mentioned above [6-13].
In the normal artery wall, significant amounts of fibronectin (FN) are present, strictly devoid of the EDA and EDB domains. In atherosclerotic lesions and experimentally induced thickening of rat aorta, there is a marked increase in FN and FN containing the EDA domain [14, 15]. However, the precise role of EDA+FN in the development and progression of atherosclerotic lesions remains unclear.
In the present work we have studied the role of fibronectin isoforms in atherosclerosis taking advantage of two mouse strains unable to undergo alternative splicing of the fibronectin EDA exon [9]. One strain contains optimized spliced sites at both splicing junctions of the EDA exon and constitutively includes the exon (EDA+/+), whereas the other strain contains an EDA-null allele of the EDA exon (EDA−/−) [9]. Surprisingly, we found that mice having constitutive inclusion or exclusion of the EDA exon have a similar phenotype, with a 40% reduction in the number and area of atherosclerotic lesions after the atherogenic-diet treatment, associated to a lower efficiency of macrophages to take up modified LDL and undergo foam-cell transformation. Lesions in control mice were more numerous and bigger, had increased macrophage cellularity and FN expression in the sub-endothelial lesion when compared with those of both types of mutant animals. Our data suggest that regulated splicing of the EDA exon of the FN gene is involved in progression of atherosclerosis.
Experimental Methods
Mice and diets
We have previously described the mouse strains devoid of regulated splicing at the EDA exon [9]. Mice were backcrossed into the C57Bl/6 genetic background for at least six generations (98.44% or higher C57Bl/6 background [13]) and were 16 months old at the beginning of the atherogenic-diet treatment. Female mice were fed for the indicated time with D12109-diet from Research Diets, NJ, containing 1.25% cholesterol, 17.2% cocoa butter and 0.5% Sodium Cholate. Control mice were treated for the same time with D12102-control diet, containing 0% cholesterol, 1.9% cocoa butter and 0% Sodium Cholate. Sodium cholate was used to generate moderate and consistent atherosclerotic lesions in mice where atherosclerosis is only diet induced, and not genetically induced [16]. Serum lipids, analysis of aortic lesion and modified LDL uptake were performed as described [17].
Serum Lipids
Mice were fasted for 4 hours, and the serum total cholesterol and triglycerides were determined as described [17].
Analysis of Atherosclerotic Lesions
The aorta was flushed through the left ventricle and the heart with aorta was embedded in O.C.T. Compound (Sakura Finetek USA, Torrance, CA). Cryosections of 10-μm thickness were taken from the region of the proximal aorta starting from the end of the aortic sinus as described [18]. The sections (15 sections per mouse) were stained with oil red O and Mayer's hematoxylin (Sigma), and then analyzed using an Imaging system KS 300 (Kontron Electronik GmbH) as described [19].
Immunocytochemistry
Serial 5-μm cryosections of the proximal aorta were incubated with monoclonal rat antibodies to macrophages, MOMA-2 (Accurate Chemical & Scientific Corp., Westbury, NY). The sections were treated with goat biotinylated antibodies to rat IgG (PharMingen, San Diego, CA), incubated with avidin-biotin complex labeled with alkaline phosphatase (Vector Lab, Burlingame, CA.) and visualized with Fast Red TR/Naphthol AS-NX substrate (Sigma). A non-immune rat serum was used as a negative control.
Modified LDL Uptake
Thioglycolate-elicited peritoneal macrophages were cultured in DMEM with 10% fetal bovine serum for 2 days as described[17]. Then cells were incubated with DiI-labeled human acetylated LDL (AcLDL) or oxidized LDL (Intracel Corp, Rockville, Md) at 37°C for the indicated time and analyzed under a fluorescent microscope or by fluorescence-activated cell sorter (FACSCalibur, Becton Dickinson) flow cytometry as described [17]. For modified-LDL binding experiments, macrophages were incubated with DiI-labeled human AcLDL or oxLDL at 4°C for 60 min.
RNA preparation and RT-PCR
Total RNA was prepared from macrophages and freshly extracted aorta (2-3 mice per genotype, 2-3 month-old mice) [20]. The radioactive RT-PCR reactions were performed and quantified as previously described [21]. Briefly, FN cDNAs were generated from total RNA using the MMLV- Reverse Transcriptase (InVitrogen), and the EDA inclusion vs. EDA exclusion ratio was quantified via RT-PCR by using primers in the exons flanking the EDA exon, using α32P-dCTP in the PCR mix. RT-PCR products (28–30 PCR cycles) were then quantified by using a Cyclone (Storage Phosphor System, Canberra Packard, USA) and corrected by size and G+C content. Quantification analysis was performed always within the different RT-PCR amplified products derived from the alternatively spliced exons of the same RNA sample.
Statistical analysis
Quantitative data are shown as the mean±SD. Comparisons between genotypes were done by using unpaired ANOVA test, followed by post-hoc test when a significant general effect was observed. A P value of 0.05 was chosen as the limit of statistical significance.
Experimental Results
FN distribution in aorta sections
First, we performed immunohistochemical analysis of cross-sections in the proximal aorta and observed the presence of FN in the intima and media, with all three genotypes displaying a similar pattern of FN distribution (Figure 1A). RT-PCR analysis of RNA prepared from total aorta showed that most of the FN mRNA in the untreated EDAwt/wt mice did not contain the EDA exon (Figure 1B) in the absence of atherosclerotic changes. The aortas of EDA+/+ and EDA−/− mice, as already observed for other tissues [9, 21], showed complete inclusion and complete exclusion of the EDA exon, respectively.
Figure 1. Panel A. Immunohistochemical analysis of FN distribution in mouse aorta.

Sections from the proximal aorta of EDAwt/wt (A and D), EDA+/+ (B and E) and EDA−/− mice (C and F) were incubated with anti-total FN rabbit polyclonal antibody (A, B, and C) or control antibody (D, E and F). Original magnification 650x; Panel B. RT-PCR analysis of FN-EDA expression in aorta samples. RT-PCR analysis of RNA prepared from total aorta of EDAwt/wt untreated 2-3 month old mice showed that FN is mainly of the EDA-minus form (Lanes 2-3). The EDA+/+ and EDA−/− mice showed complete inclusion and complete exclusion of the EDA exon, respectively (lanes 4-5 and 6-7, respectively). Cerebellum EDAwt/wt (Cer) and liver EDA+/+ are shown as controls (lanes 8 and 9, respectively). Lane 10 corresponds to PCR control without cDNA. “L” indicates vessel lumen.
Levels of cholesterol and triglycerides in mutant mice
To analyze in vivo the specific effects of the alteration of alternative splicing pattern of the EDA exon of fibronectin in atherosclerosis, EDAwt/wt, EDA+/+ and EDA−/− mice were backcrossed with C57Bl/6 mice. To increase the size and progression of intimal lesions we used aged mice (16 months old at the beginning of the experiment) fed with atherogenic-diet for 14 or 18 weeks.
Serum samples were analyzed at baseline and at 8, 14 and 18 weeks after starting the diets. The basal levels of cholesterol in the EDA−/− mice were lower than those observed in the EDAwt/wt animals (Table 1). Triglyceride levels were higher in both the EDA+/+ and EDA−/− animals. However, these differences observed at basal status disappeared after initiation of the treatment as mice showed similarly elevated levels of serum cholesterol and reduced levels of triglycerides at 8, 14 or 18 weeks of atherogenic-diet (Table 2).
Table 1.
Serum total cholesterol and triglycerides levels in EDAwt/wt, EDA+/+ and EDA−/− mice at basal levels and after 8, 14 and 18 weeks of the atherogenic diet.
| Type of animals |
Serum Lipids | Basal levels |
8 weeks of the diet |
14 weeks of the diet |
18 weeks of the diet |
|---|---|---|---|---|---|
| EDAwt/wt mice |
Cholesterol Triglycerides |
2.48±0.85 0.93±0.20 |
3.06±0.24 0.94±0.14 |
3.72±0.90 0.77±0.14 |
4.08±0.47 0.77±0.12 |
| EDA+/+ mice |
Cholesterol Triglycerides |
2.25±0.42 1.11±0.22* |
2.97±0.32 0.97±0.16 |
3.22±1.25 0.79±0.12 |
3.94±0.56 0.69±0.09 |
|
EDA−/− mice |
Cholesterol Triglycerides |
2.07±0.34# 1.03±0.17+ |
2.99±0.31 1.04±0.16 |
3.18±0.79 0.80±0.09 |
4.08±1.35 0.65±0.15 |
Values are in mmol/L (Mean ± SEM). The number of animals (n) in each group is varied (n=34-39 at the basal level; n=22-25 after 8 weeks, n=17-24 after 14 weeks and n=11-17 after 18 weeks of the atherogenic diet). Data were analyzed by ANOVA test with Bonferroni's corrections:
p<0.05
p<0.01
p<0.001.
Atherosclerosis Studies
EDA+/+ and EDA−/− mice showed a reduction in the aorta atherosclerotic lesions of 31% and 59%, respectively, after 14 weeks of diet (Figure 2A). At 18 weeks the reduction was ∼40% for both mutant genotypes (ANOVA, p≤0.01, Bonferroni's comparison test, EDAwt/wt vs. EDA+/+ and EDAwt/wt vs. EDA−/−, p≤0.05).
Figure 2. Panel A. EDA+/+ and EDA−/− mice are protected against atherosclerosis after atherogenic-diet.
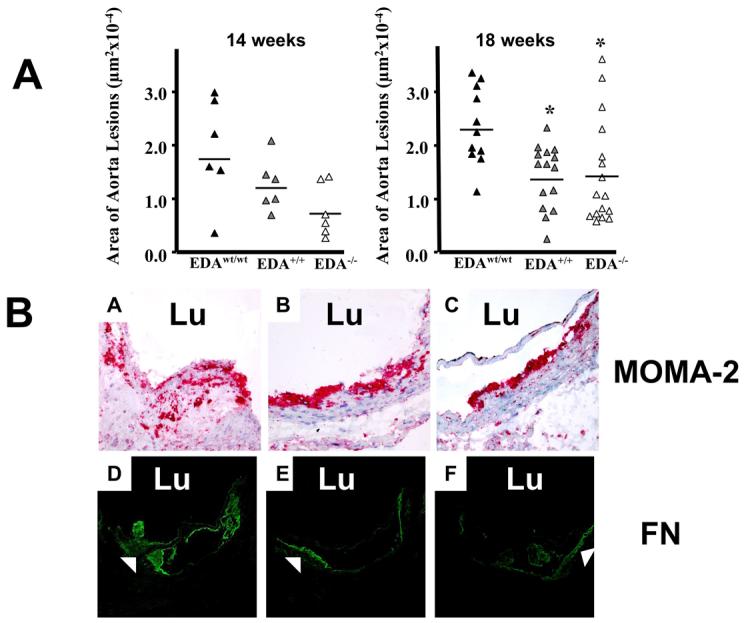
The atherosclerotic lesion area in the proximal aorta region was analyzed at 14 and 18 weeks after the initiation of the diet. Each triangle corresponds to the mean values of 15 sections from a single mouse. Compared to the EDAwt/wt mice, the EDA+/+ and EDA−/− mice had lower atherosclerotic lesion area at both 14 and 18 weeks of diet (Mean±SE, μm2; 14 weeks: 17482±3716; 12028±2011; 7216±2036; 18 weeks: 22969±2185; 13660±1533; 14260±2501 for EDAwt/wt, EDA+/+ and EDA−/−, respectively; 14 weeks, n=6 mice per genotype; 18 weeks, n=11, 15 and 16 mice for EDAwt/wt, EDA+/+ and EDA−/− , respectively). Data was analyzed by ANOVA test (14 weeks, NS; 18 weeks, p≤0.01) with the Bonferroni's multiple comparison Test (18 weeks, EDAwt/wt vs. EDA+/+ p≤0.05; EDAwt/wt vs. EDA−/−, p≤0.05). Panel B. Detection of macrophages and the FN distribution in atherosclerotic lesions of the proximal aorta. Sections from the proximal aorta of EDAwt/wt (A and D), EDA+/+ (B and E) and EDA−/− mice (C and F) fed with atherogenic-diet for 18 weeks were stained with the MOMA-2 monoclonal antibody (A, B and C) or with the anti-FN polyclonal antibody (D, E and F). (Original magnification, 400x and 200x for MOMA-2 and FN antibodies, respectively). “Lu” indicates vessel lumen.
A more detailed histological analysis of the lesions revealed larger and more numerous lesions in the EDAwt/wt compared to the mutant mice. In addition, MOMA2 staining showed that macrophages extended deeper in the subendothelial area in EDAwt/wt mice than in either mutant strain (Figure 2B, Panels A-C).
Analysis of control and mutant mice tissue sections containing lesions of different size suggested that the observed differences in FN distribution depended primarily on the size of the lesion (Figure 2B, Panels D-F).
Macrophages from EDA+/+ and EDA−/− mice show reduced uptake of modified LDL
The process of macrophage-derived foam cell formation was analyzed by incubating macrophages with acetylated and oxidized DiL labeled LDL. Lipid uptake was very efficient and reached a plateau at 4 h (Figure 3A). Consequently, we performed uptake experiments at 30 min of incubation, in the linear range of the curve. Microscopic analysis showed that wild type and mutant macrophages were able to take up both types of modified LDL efficiently (Figure 3B). Cell sorting analyses showed that both mutant genotypes had a significant decrease in AcLDL and OxLDL uptake at 30 min (Figure 3C). We observed no differences in the binding capacity of macrophages for modified LDL particles (Figure 3D) suggesting similar expression levels of scavenger receptors in macrophages from all three strains.
Figure 3. Panel A. Time course of the modified LDL uptake by EDAwt/wt peritoneal macrophages.

Peritoneal macrophages were incubated with 15 μg/ml of human DiL-oxidized for the indicated times and the uptake was examined by flow cytometry. The data are presented in relative units as an average of triplicate repeats. Panel B. Visualization of the modified-LDL uptake by peritoneal macrophages from EDAwt/wt, EDA+/+ and EDA−/− mice. Peritoneal macrophages from EDAwt/wt (A), EDA+/+ (B) and EDA−/− mice (C) were incubated with 15 μg of Ac-DiL LDL for 30 min. and visualized under UV light. Original magnification, 400x; Panels C and D. FACS analysis of the modified-LDL uptake (C) and binding (D) by peritoneal macrophages from EDAwt/wt, EDA+/+ and EDA−/− mice. Peritoneal macrophages were incubated for 30 min at 37°C (uptake) or 60 min at 4°C (binding) with Ac-DiL LDL or Ox-DiL LDL and uptake was examined by flow cytometry. The data of the uptake are presented in relative units as an average of three independent experiments (each experiment having three mice per genotype, each mouse having three repeats) ± Standard Deviation. The data of the binding experiment are presented in relative units as an average of three mice per genotype, each mouse having three repeats (a, ANOVA p=0.0023; b, ANOVA, p=0.0002; Bonferroni's Correction: *, p≤0.05; **, p≤0.01; ***, p≤0.001). wt, +/+ and −/− indicate EDAwt/wt, EDA+/+ and EDA−/− samples, respectively.
Given the pivotal role of macrophages in atherosclerotic lesions, the observed decrease in modified LDL uptake may account for the observed protection of the EDA+/+ and EDA−/− animals after the atherogenic-diet treatment.
Therefore, to determine whether EDA splicing in macrophages is induced by exposure to AcLDL or OxLDL we performed RT-PCR analysis on mRNA isolated from stimulated and non-stimulated macrophages. We observed that inclusion of the EDA exon increased very rapidly when non-stimulated macrophages from EDAwt/wt mice were placed in culture (Supplementary Figure 1), arriving to a plateau after 30 h. When EDAwt/wt macrophages were incubated with modified-LDL we observed no differences in the amount of EDA exon inclusion after incubation of macrophages with Ox-LDL for 2 and 20 h (Figure 4A and B) indicating that EDA+FN is not induced by the treatment.
Figure 4. RT-PCR analysis of the FN-EDA splicing of peritoneal macrophages from EDAwt/wt after incubation with modified Ox-DiL LDL.

(A) Macrophages from three EDAwt/wt animals (numbered 1 to 3) were plated, washed after 40 minutes (t=0), total RNA was prepared at different times (t=4, 24, 30 and 48 hs) and analyzed by radioactive RT-PCR. Total RNA from EDA+/+ cerebellum was run as control. (B) The PCR products from Panel A were quantified using a Phosphor Imager. (C) Similarly, EDAwt/wt, EDA+/+ and EDA−/− samples were plated for 0 and 48 hs, the RNA prepared and analyzed by radioactive RT-PCR. (D) Macrophages from EDAwt/wt mice were prepared, plated and after 48 hs were incubated with Ox-DiL LDL for 2 and 20 additional hs. (E) The radioactivity was quantified. Each bar corresponds to macrophages prepared from separate EDAwt/wt mice (numbered 1 to 3). The data was analyzed by One Way ANOVA and is presented as the mean ± standard deviation. No differences were observed.
Discussion
Alternative splicing of the EDA exon is a very tightly controlled event suggesting important and specific roles for the EDA segment [3, 4]. Here we present new evidence on the role of the alternatively spliced FN isoforms in atherosclerosis. Mice having constitutive inclusion or constitutive exclusion of the EDA exon of the fibronectin gene [9] showed decreased development of atherosclerotic lesions after an atherogenic-diet treatment, highlighting the importance of regulated splicing in complex cellular processes.
In a recent attempt to study the role of the EDA domain in atherosclerosis, double ApoE−/− and EDA−/− mutant mice showed reduced atherosclerosis after an atherogenic-diet treatment [12]. Our results obtained with the EDA−/− mice confirmed the decrease in atherosclerotic lesions and the defect in modified LDL uptake by macrophages observed by Tan et al [12], and extended their observation to a background where the hyperlipidemia is not genetic but diet-induced. However, the unexpected and paradoxical reduction in lesion size and LDL uptake by macrophages in the EDA+/+ mice seems to rule out the proposed pro-atherogenic role for the EDA domain in the pathophysiology of the disease [12].
The decreased efficiency of LDL uptake by macrophages could act in synergism with other potentially altered atherogenic events in mutant mice, such as macrophage rolling, adhesion and migration to the lesion area (and/or migration of SMC), or formation of foam cells, leading to a reduction in the formation of atherosclerotic plaques. Macrophages display the integrin α4β1 or VLA-4 which recognizes the EDA exon [22], in addition to the CS1 cell-binding site present in the IIICS region of FN. It plays an important role in monocyte rolling and adhesion, regulating leukocyte entry into early [23] and advanced lesions [1], and in the initial phase of atherosclerotic lesion formation and lipid accumulation [1]. Being the EDA domain a ligand for this integrin it is possible to hypothesize that EDA-FN isoforms could also have a role in any of those processes, as already observed for other cell adhesion and ECM proteins such as P-selectins, VCAM-1 and von Willebrand factor [24, 25].
As the migratory capacity of cells also depends in the composition of the ECM [26], migration of SMC and/or macrophages to the site of the lesion might also be altered. Preliminary experiments showed that EDA+/+ and EDA−/− MEFs migrated less than EDAwt/wt cells (having about 50% of EDA exon inclusion), while an EDA+/+ and EDA−/− mixture of MEFs recovered the migratory capacity (AFM, unpublished).
Therefore, the “block” in EDA splicing might affect any of the processes mentioned above. Additionally, strong interactions between monocytes and ECM are necessary for the formation of “foam-cells”, as lipid does not accumulate in monocytes that do not form a stable interaction with tissues, suggesting specific ECM-dependent signaling [27]. Therefore, as both mutant mice are unable to modulate EDA expression after initial injury, it is possible to suppose that macrophage signaling from the ECM is affected, leading to less active macrophages in both mutant mice. These results suggest that the main event leading to a reduction in plaque formation might not be the constitutive presence or absence of the EDA domain but the absence of modulation of the EDA domain levels by alternative splicing.
Alternatively, constitutive absence or inclusion of the EDA exon might produce similar atherogenic phenotypes by different mechanisms. We have looked at different parameters associated to the development of atherosclerotic lesions such as foam cell formation, levels of serum triglycerides and cholesterol in non-treated and treated mice, distribution and amount of macrophages and FN in the lesion. Most of them were different in the mutants to those of EDAwt/wt mice, but were similar between EDA+/+ and EDA−/− mice supporting the idea of a common mechanism.
Our EDA−/− mice also showed decreased serum cholesterol levels confirming the previous results [12] although we observed the reduction in basal levels but not in the treated mice, in addition to an increase in basal triglyceride levels in both EDA+/+ and EDA−/− mice. Additionally, the stimulation of EDAwt/wt macrophages with modified LDL produced no effect in EDA splicing, differing from previous data [12] but we observed a time-dependent increase in the EDA+/EDA− ratio of non-stimulated macrophages after plating of the cells. These variations could be due to differences in our experimental protocol, which differed from those of Tan et al [12] in at least the following aspects: a) We exclusively analyzed the role of the FN-EDA isoforms, as mice did not have the atherosclerosis prone apoE null background, using an atherogenic-diet containing sodium cholate [16, 28]; b) In addition to the EDA− FN isoform, we analyzed the role of the FN isoforms having constitutive inclusion of the EDA domain (EDA+ FN); and c) We used aged, 16 month-old mice to increase the incidence of intimal lesions.
In conclusion, we have shown that mice having constitutive absence or inclusion of the EDA-FN exon have decreased levels of atherosclerosis. The lesions in aorta from both EDA+/+ and EDA−/− mutant animals are smaller and have less infiltration of macrophages in the sub-endothelial area, with a reduced expression and distribution of FN. The cause of the reduced lesions in mutant mice may be related to a decreased uptake of modified LDL by macrophages from both mutant mice. We propose that the regulation of the EDA domain by alternative splicing has an important role in the progression of the atherogenic process.
Acknowledgments
The authors are thankful to Youmin Zhang and Lei Ding for excellent technical expertise, and to Mauro Sturnega and Giancarlo Lunazzi for help in animal handling. The study was supported by the Lipid, Lipoprotein and Atherosclerosis Core of the Vanderbilt Mouse Metabolic Phenotyping Centers (NIH DK59637).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Osterud B, Bjorklid E. Role of monocytes in atherogenesis. Physiol Rev. 2003;83:1069–112. doi: 10.1152/physrev.00005.2003. [DOI] [PubMed] [Google Scholar]
- 2.ffrench-Constant C. Alternative splicing of fibronectin--many different proteins but few different functions. Exp Cell Res. 1995;221:261–71. doi: 10.1006/excr.1995.1374. [DOI] [PubMed] [Google Scholar]
- 3.Hynes RO. Fibronectins. Springer-Verlag; New York: 1990. p. 544. [Google Scholar]
- 4.Kornblihtt AR, Pesce CG, Alonso CR, Cramer P, Srebrow A, Werbajh S, Muro AF. The fibronectin gene as a model for splicing and transcription studies. Faseb J. 1996;10:248–57. doi: 10.1096/fasebj.10.2.8641558. [DOI] [PubMed] [Google Scholar]
- 5.Mosher DF. Fibronectin. Academic Press; New york: 1989. p. 474. [Google Scholar]
- 6.George EL, Georges-Labouesse EN, Patel-King RS, Rayburn H, Hynes RO. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development. 1993;119:1079–91. doi: 10.1242/dev.119.4.1079. [DOI] [PubMed] [Google Scholar]
- 7.George EL, Baldwin HS, Hynes RO. Fibronectins are essential for heart and blood vessel morphogenesis but are dispensable for initial specification of precursor cells. Blood. 1997;90:3073–81. [PubMed] [Google Scholar]
- 8.Fukuda T, Yoshida N, Kataoka Y, Manabe R, Mizuno-Horikawa Y, Sato M, Kuriyama K, Yasui N, Sekiguchi K. Mice Lacking the EDB Segment of Fibronectin Develop Normally but Exhibit Reduced Cell Growth and Fibronectin Matrix Assembly in Vitro. Cancer Res. 2002;62:5603–10. [PubMed] [Google Scholar]
- 9.Muro AF, Chauhan AK, Gajovic S, Iaconcig A, Porro F, Stanta G, Baralle FE. Regulated splicing of the fibronectin EDA exon is essential for proper skin wound healing and normal lifespan. J Cell Biol. 2003;162:149–60. doi: 10.1083/jcb.200212079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ni H, Yuen PS, Papalia JM, Trevithick JE, Sakai T, Fassler R, Hynes RO, Wagner DD. Plasma fibronectin promotes thrombus growth and stability in injured arterioles. Proc Natl Acad Sci U S A. 2003;100:2415–9. doi: 10.1073/pnas.2628067100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakai T, Johnson KJ, Murozono M, Sakai K, Magnuson MA, Wieloch T, Cronberg T, Isshiki A, Erickson HP, Fassler R. Plasma fibronectin supports neuronal survival and reduces brain injury following transient focal cerebral ischemia but is not essential for skin-wound healing and hemostasis. Nat Med. 2001;7:324–30. doi: 10.1038/85471. [DOI] [PubMed] [Google Scholar]
- 12.Tan MH, Sun Z, Opitz SL, Schmidt TE, Peters JH, George EL. Deletion of the alternatively spliced fibronectin EIIIA domain in mice reduces atherosclerosis. Blood. 2004;104:11–8. doi: 10.1182/blood-2003-09-3363. [DOI] [PubMed] [Google Scholar]
- 13.Chauhan AK, Moretti FA, Iaconcig A, Baralle FE, Muro AF. Impaired Motor coordination in mice lacking the EDA exon of the fibronectin gene. Behav Brain Res. 2005 doi: 10.1016/j.bbr.2005.02.020. In Press. [DOI] [PubMed] [Google Scholar]
- 14.Glukhova MA, Frid MG, Shekhonin BV, Vasilevskaya TD, Grunwald J, Saginati M, Koteliansky VE. Expression of extra domain A fibronectin sequence in vascular smooth muscle cells is phenotype dependent. J Cell Biol. 1989;109:357–66. doi: 10.1083/jcb.109.1.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Magnusson MK, Mosher DF. Fibronectin: structure, assembly, and cardiovascular implications. Arterioscler Thromb Vasc Biol. 1998;18:1363–70. doi: 10.1161/01.atv.18.9.1363. [DOI] [PubMed] [Google Scholar]
- 16.Nishina PM, Verstuyft J, Paigen B. Synthetic low and high fat diets for the study of atherosclerosis in the mouse. J Lipid Res. 1990;31:859–69. [PubMed] [Google Scholar]
- 17.Babaev VR, Yancey PG, Ryzhov SV, Kon V, Breyer MD, Magnuson MA, Fazio S, Linton MF. Conditional knockout of macrophage PPARgamma increases atherosclerosis in C57BL/6 and low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2005;25:1647–53. doi: 10.1161/01.ATV.0000173413.31789.1a. [DOI] [PubMed] [Google Scholar]
- 18.Paigen B, Morrow A, Holmes PA, Mitchell D, Williams RA. Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis. 1987;68:231–40. doi: 10.1016/0021-9150(87)90202-4. [DOI] [PubMed] [Google Scholar]
- 19.Fazio S, Babaev VR, Murray AB, Hasty AH, Carter KJ, Gleaves LA, Atkinson JB, Linton MF. Increased atherosclerosis in mice reconstituted with apolipoprotein E null macrophages. Proc Natl Acad Sci U S A. 1997;94:4647–52. doi: 10.1073/pnas.94.9.4647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate- phenol-chloroform extraction. Anal Biochem. 1987;162:156–9. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 21.Chauhan AK, Iaconcig A, Baralle FE, Muro AF. Alternative splicing of fibronectin: a mouse model demonstrates the identity of in vitro and in vivo systems and the processing autonomy of regulated exons in adult mice. Gene. 2004;324:55–63. doi: 10.1016/j.gene.2003.09.026. [DOI] [PubMed] [Google Scholar]
- 22.Liao YF, Gotwals PJ, Koteliansky VE, Sheppard D, Van De Water L. The EIIIA segment of fibronectin is a ligand for integrins alpha 9beta 1 and alpha 4beta 1 providing a novel mechanism for regulating cell adhesion by alternative splicing. J Biol Chem. 2002;11:11. doi: 10.1074/jbc.M201100200. [DOI] [PubMed] [Google Scholar]
- 23.Huo Y, Hafezi-Moghadam A, Ley K. Role of vascular cell adhesion molecule-1 and fibronectin connecting segment-1 in monocyte rolling and adhesion on early atherosclerotic lesions. Circ Res. 2000;87:153–9. doi: 10.1161/01.res.87.2.153. [DOI] [PubMed] [Google Scholar]
- 24.Methia N, Andre P, Denis CV, Economopoulos M, Wagner DD. Localized reduction of atherosclerosis in von Willebrand factor-deficient mice. Blood. 2001;98:1424–8. doi: 10.1182/blood.v98.5.1424. [DOI] [PubMed] [Google Scholar]
- 25.Ramos CL, Huo Y, Jung U, Ghosh S, Manka DR, Sarembock IJ, Ley K. Direct demonstration of P-selectin- and VCAM-1-dependent mononuclear cell rolling in early atherosclerotic lesions of apolipoprotein E-deficient mice. Circ Res. 1999;84:1237–44. doi: 10.1161/01.res.84.11.1237. [DOI] [PubMed] [Google Scholar]
- 26.Giancotti FG, Tarone G. Positional control of cell fate through joint integrin/receptor protein kinase signaling. Annu Rev Cell Dev Biol. 2003;19:173–206. doi: 10.1146/annurev.cellbio.19.031103.133334. [DOI] [PubMed] [Google Scholar]
- 27.Wesley RB, 2nd, Meng X, Godin D, Galis ZS. Extracellular matrix modulates macrophage functions characteristic to atheroma: collagen type I enhances acquisition of resident macrophage traits by human peripheral blood monocytes in vitro. Arterioscler Thromb Vasc Biol. 1998;18:432–40. doi: 10.1161/01.atv.18.3.432. [DOI] [PubMed] [Google Scholar]
- 28.Vergnes L, Phan J, Strauss M, Tafuri S, Reue K. Cholesterol and cholate components of an atherogenic diet induce distinct stages of hepatic inflammatory gene expression. J Biol Chem. 2003;278:42774–84. doi: 10.1074/jbc.M306022200. [DOI] [PubMed] [Google Scholar]