

Abstract
Tissue-targeted metabonomics provides tissue specific metabolic information while still retaining the profiling approach of traditional metabonomics. Microdialysis sampling is used to generate site-specific samples of endogenous metabolites. The dialysate samples are subjected to proton NMR analysis with data analysis by principal components analysis and partial least squares regression. In this study, sample and data pretreatment methods were examined for their impact on the quality of the data analysis. Specifically, the effects of speedvacuuming, sample solubility, sample pH stability, and sample storage stability were examined. Data pretreatment methods examined included the effects of standardization and normalization to internal standards. In addition, the ability of tissue-targeted metabonomics to generate time trend data was explored and more fully characterized using principal components analysis and partial least squares regression.
Keywords: Metabonomics, microdialysis, NMR
Introduction
Metabonomics is an in vivo profiling technique that simultaneously monitors changes in the levels of endogenous metabolites in response to a metabolic stressor such as the administration of a drug or the onset of a disease [1,2]. From an analytical standpoint, metabonomics experiments are comprised of three primary steps: sampling, detection, and data analysis.
Metabonomics experiments typically employ biofluid samples, such as urine or plasma, which can be obtained fairly noninvasively and in sufficient quantity and concentration for typical metabonomics analytical techniques. The detection schemes used in metabonomics studies aim for nonselectivity in order to detect a large number of endogenous metabolites in a single analysis. Most popular for metabonomics experiments are proton NMR and LC/MS. Proton NMR is nonselective, quantitative, nondestructive to the sample and spectra are amenable to multivariate statistical analysis. However, NMR suffers from both poor sensitivity and limits of detection. Also, in complex biofluids like plasma, macromolecules can give rise to broad resonances that can overshadow those from small molecules, making detection and quantitation difficult. LC/MS, on the other hand, has better sensitivity and detection limits than NMR but does not offer the nonselectivity of NMR. Quantitation is also much more difficult due to fluctuations in ionization efficiencies. Also, the sample is destroyed during analysis. Other techniques like GC/MS [3-6], LC/UV [7], CE/UV [8-10], CE/MS [10-16], CE/EC [17] and LC-coulometric array [18,19] are also being explored.
Once the data has been collected, patterns in the metabonomics data are then described using multivariate statistical analyses, which reduce the dimensionality of the data set to a more manageable size. Unsupervised methods like principal components analysis allow the researcher to observe patterns in the data and identify molecules responsible for the patterns. Other supervised statistical approaches, such as partial least squares analysis, can correlate variables like time with the data.
Sampling biofluids like urine and plasma does not provide spatial information about the metabolic changes. Many metabonomics studies have made use of tissue biopsies to obtain localized metabolic information, which has limitations including the termination of the experiment after a biopsy is taken, utilizing large numbers of animals to characterize metabolic differences and increasing experimental error through animal-to-animal variability. A viable alternative to tissue biopsy is microdialysis sampling of extracellular fluid [20-23].
Microdialysis is a sampling technique in which a probe with a semipermeable membrane component is implanted in a tissue [24]. An isotonic solution is perfused through the probe, and molecules in the surrounding extracellular fluid diffuse into the membrane lumen along their concentration gradient. Samples of these molecules are collected at the probe outlet. Although previous reports describe some tissue damage and stimulation of an immune response as a result of probe implantation [25,26], the impact of implantation on the tissue metabolic profile has not been studied. Although work is currently in progress to evaluate whether probe implantation measurably affects the metabolic profile, such results are beyond the scope of this study. Microdialysis offers an inherent advantage for site-specific metabonomics sampling. Careful selection of the molecular weight cutoff of the membrane can selectively exclude macromolecules. Thus, samples are much cleaner and do not require additional cleanup steps that reduce throughput and increase method error. Microdialysis samples are typically small in volume (flow rates of ∼ 1 μl/min). While not typically a problem for metabolic profiling with mass spectrometry, this does require some special consideration when conducting NMR analysis, such as using tube inserts or low volume probes.
NMR has the advantage of being inherently quantitative in nature – that is, the intensity of the signal is directly proportional to the number of nuclei giving rise to the signal. The addition of a single standard compound of known concentration is the only additional sample preparation step that is required for quantitation. Proton NMR analysis of biofluids requires some special attention to sample preparation and experimental setup including the addition of a deuterated solvent to provide a lock signal and the use of pulse sequences that selectively suppress the water resonance.
For microdialysis studies, additional sample preparation is required. Dialysate samples are very low in volume and many components border on the detection limits of the NMR instrument. Thus, any dilution becomes problematic. In many experiments, dialysate samples are taken to dryness and reconstituted in a deuterated solution to achieve lock signal and avoid any dilution effects. There are two primary options for low volume NMR analysis: capillary tube inserts and capillary probes or probes with reduced detection volumes. Capillary tube inserts are made to sit inside a typical 5 mm NMR tube with the aid of a plastic adapter. The capillary is submerged in sample-matching solvent (usually D2O) and then the sample is centered in the active region of the magnet for analysis. Although tube inserts do not provide an efficient filling factor, they allow the analysis of small volume samples without specialized NMR probes. The low volume detection cells of specialized capillary probes and nanoprobes can accommodate smaller sample volumes and typically provide better mass sensitivity than capillary inserts.
Data generated by metabonomics analysis is complex, as the number of variables is much higher than typical single biomarker experiments. One of the primary objectives of metabonomics data analysis is to reduce the dimensionality of the data set, thus improving its accessibility and streamlining data interpretation. For NMR spectra, integrals are taken over portions of the NMR spectrum or a set of individual data points [27-29]. Each integral region corresponds to a variable in the data matrix. After integration, normalization and standardization of the data it is extremely important to remove any extraneous variation due to experimental error and to reduce domination of higher intensity variables in the statistical analysis, respectively.
The purpose of this study was to characterize experimental parameters and establish analytical methodology pertinent to metabonomics studies of microdialysis samples. These included microdialysis probe calibration, sample preparation, and data pretreatment for multivariate statistical analysis.
Methods
Reagents and Solutions
All deuterated solvents were purchased from Cambridge Isotope Laboratories (Andover, MA). Salts for Ringer's solution and other reagents were purchased from Sigma Chemical Corporation (St. Louis, MO). Ringer's solution was 145 mM NaCl, 2.8 mM KCl, 1.2 mM CaCl2, and 1.2 mM MgCl2. Solutions were prepared in nanopure water (Labconco, Kansas City, KS) and filtered through a 0.22 μm pore filter prior to use.
Microdialysis Methods
Microdialysis Probe Preparation and Calibration
All probes were prepared in-house, using polyacrylonitrile (PAN) membrane (Hospal, Lakewood, CO) of dimensions 350 μm O.D. by 250 μm I.D. and polyimide tubing (MicroLumen, Tampa, FL) of dimensions 170 μm O.D. by 120 μm I.D. All probes used Tygon tubing attached at the inlet as a syringe adapter. Vascular probes were prepared with a 10 mm membrane length. Liver probes were linear and also had a 10 mm membrane length. The outlet of the probe had a 7 cm piece of fused silica (360 μm O.D. by 200 μm I.D.) attached to the polyimide to use as a needle guide. Heart probes were linear and were approximately 2 mm in membrane length. A 1 mm long Tygon tubing “stop” was secured directly behind the active window of the membrane to aid in implantation of the probe. The outlet of the probe had a 27 gauge stainless steel needle (bent to 45°) attached to aid in implantation. All probes were perfused with Ringer's solution at a rate of 1 μl/min. Probes were flushed for 30 minutes prior to the start of sampling.
Probes were calibrated continuously through the experiment by retrodialysis of 7 μM antipyrine in Ringer's solution [30,31]. Antipyrine is an established marker of microdialysis membrane extraction efficiency [32] and will account for global changes in probe performance. Antipyrine was detected using LC/UV at 254 nm (Shimadzu, Columbia, MD). Samples were injected onto a Phenomenex Synergi Polar-RP column 4 μm, 2 mm × 150 mm. The mobile phase was acetonitrile-sodium phosphate (pH 2.5, 25 mM) (17:83, v/v), and was set to a flow rate of 0.3 ml/min.
Animal Setup
All experiments utilized male Sprague-Dawley rats (Sasco, Wilmington, MA), weight range 350−450 g. Rats were pre-anesthetized with isoflurane and then fully anesthetized with a ketamine (67.5 mg/kg)/xylazine (3.4 mg/kg)/acepromazine (0.67 mg/kg) cocktail, administered subcutaneously. For nonsurvival surgeries, rats were maintained under anesthesia for the duration of all experiments, using ketamine booster doses administered intramuscularly at one quarter of the initial dose. Rats undergoing survival procedures were only given the initial anesthesia cocktail, which maintained anesthesia for the duration of probe implantation. Rat body temperature was maintained using an electric heating pad.
For survival surgeries, all procedures were performed aseptically. Rats were given 3 ml saline subcutaneously after the surgery to facilitate recovery and were housed in a Raturn© system (BAS, West Lafayette, IN). All animal experiments were performed under approval of the local Institutional Animal Care and Use (IACUC) committee.
Jugular Probe Implantation
The jugular vein was exposed through a small midline incision in the neck skin tissue. The vein was isolated by separating the vessel from fine connective tissue using blunt dissection and cotton swabs. A flat metal spatula was placed under the vein to externalize it, and a small incision made in the vessel using a pair of fine spring scissors. A vascular probe with an introducer was inserted into the vein toward the heart. Silk sutures were used to secure the probe in the vessel, and the introducer was then removed.
Heart Probe Implantation
A tracheotomy was performed by externalizing the trachea through an incision on the midline of the neck. The trachea was isolated using a spatula and any fluid around the trachea was removed with cotton swabs. A small nick was made in between the rings of the trachea and an intubation tube was immediately inserted and secured with sutures. The rat was attached to a Digi-Med Sinus Rhythm Analyzer (Louisville, KY) to monitor heart rate and sinus rhythm. Immediately prior to opening the chest cavity, the intubation tube was connected to a respirator that supplied artificial ventilation for the rat. A constant-volume respirator using room air (Model 683 Rodent Respirator, Harvard Apparatus, Holliston, MA) was used. To expose the heart, a thoracotomy was performed on the left side of the animal by making an incision between and parallel to the fifth and sixth ribs approximately 1.5 to 2 cm in length, and the pericardium was opened, allowing a linear microdialysis probe (prepared in-house) to be inserted into the myocardium. Specifically, a bent 27 gauge needle attached to the probe was used to implant the probe into the apex of the beating heart muscle, near the left descending coronary artery along the longitudinal axis of the heart. The lungs were then fully expanded.
Liver Probe Implantation
A 2 cm incision was made along the midline of the abdomen directly below the diaphragm, and the abdominal cavity was opened. The incision was spread with Bowman retractors and tissue forceps were used to expose the median liver lobe from beneath the ribs. Two microdialysis probes were implanted using the fused silica as a guide needle. The probes were implanted 1 cm apart, which has been shown by previous studies to be a sufficient distance to prevent cross-talk between the probes [33]. The tissue entrance and exit points for each probe were secured with a small amount of tissue glue. After probe implantation, the incision was closed with sutures, with the probe inlets and outlets externalized through the incision. For survival surgeries, the probe tubing was tunneled underneath the skin and externalized through a small incision in the back of the neck.
Sample Preparation
All dialysate samples (50 μl original volume) were speedvacuumed to dryness at room temperature after collection and stored at −20°C in the dark until analysis. Immediately prior to NMR analysis, the sample was thawed to ambient temperature and reconstituted in 25 μl of 25 mM deuterated acetate solution in D2O, pD = 7.8, with 5 mM TSP as an internal standard. Samples were vortexed for 30 seconds and then transferred to a linear capillary tube insert (Wilmad-Labglass, Buena, NJ) using a 10 μl syringe with a fused silica needle. The insert was submerged using a PTFE adapter in 600 μl D2O in a 5 mm NMR tube.
NMR Spectroscopy
1H-NMR analysis was performed on a Varian INOVA 600 MHz spectrometer equipped with VnmrJ software (Varian Instruments Inc., Palo Alto, CA) at 25°C. Samples were analyzed on a 5 mm Varian triple-axis gradient probe. All samples were analyzed with the WET pulse sequence. A tip angle of 90° was utilized, with an acquisition time of 1.8 sec and 25200 points. Spectra were coadded for 3000 transients for a total acquisition time of 95 minutes, zero-filled to 65536 points, and a line broadening of 1 Hz was applied. All spectra were referenced to TSP at 0.00 ppm. Resonances were assigned by comparison to standard spectra and literature chemical shift values.
Data Analysis
NMR spectra were exported to ACD/Labs software (Toronto, Ontario) and spectral processing (zero-filling, line-broadening, baseline correction, and referencing) were performed as described above. Spectra were manually integrated over 40 defined integral regions between 0.8 and 4.5 ppm, and integral tables were exported as a spreadsheet to Excel for data manipulation. (For sample stability analyses, spectra were manually integrated over 57 defined integral regions.) Integral values were normalized to the TSP integral and concentration, as well as the % delivery of the corresponding microdialysis probe. Analysis by principal components, partial least squares and univariate methods were performed in Minitab® Statistical Software (Release 14.20, Minitab, Inc., State College, PA). Principal components analysis was performed on a correlation matrix (data matrix was centered and standardized), with five principal components calculated. Partial least squares regression between time and metabolic profiles was performed on a correlation matrix (data matrix was centered and standardized), with a maximum of five components calculated. The PLS model was cross-validated through jackknifing.
Results
Comparison of 1H-NMR Spectra of Plasma and Plasma Dialysate
To illustrate the differences between conventional metabonomics data and tissue-targeted metabonomics data, a comparison of 1H-NMR spectra of rat plasma and plasma dialysate is shown in Figure 1. D2O was added to the plasma to a final concentration of 10% to provide a lock signal. The plasma, as expected, showed underlying broad signals throughout the spectrum due to proteins and lipids. Very few small molecule resonances are detectable above this background. The visible sharper resonances in the region from 3.1 to 3.9 ppm correspond to glucose and the sharp singlet at 3.0 ppm is due to creatine. Other small molecule resonances may be present, but are not sufficiently above the background for positive identification. In contrast, the plasma dialysate was entirely devoid of any macromolecule signal. Glucose and creatine resonances are present at the frequencies indicated above. Lactate, alanine, lysine, valine, leucine, isoleucine, citrate, tyrosine, and many other small molecules are detectable. Spectral resolution is high, indicating that the use of inserts for NMR analysis of small volume dialysate samples does not compromise the quality of the NMR spectrum.
Figure 1.

Comparison of the 1H-NMR spectra of (A) plasma and (B) plasma dialysate.
Sample Preparation Effects and Stability
Speedvacuuming Effects
The effect of speedvacuuming on the composition of heart and plasma dialysate samples was determined. Samples were split into two fractions, with one frozen at −20°C and the other speedvacuumed to dryness before storage at −20 °C. All sample pairs were analyzed by NMR on the same day. Immediately prior to NMR analysis, frozen samples were diluted to a final TSP concentration of 5 mM and deuterated acetate concentration of 25 mM. Speedvacuumed samples were reconstituted in 25 mM deuterated acetate solution in D2O with 5 mM TSP to the same final volume as the frozen samples.
Visual inspection of the spectra revealed few differences between the sample preparation methods. For heart and plasma dialysate, the aliphatic regions (< 3 ppm) and aromatic regions (> 5 ppm) showed no detectable differences in both resonance intensity and resonances present. For both dialysates, small differences were noted in the 3 − 4.5 ppm region, primarily containing glucose resonances.
Principal components analysis (Figure 2) showed a clear segregation between the plasma dialysate and heart dialysate samples (with one set of plasma dialysate samples appearing as an outlier). No segregation was observed between dialysate samples that were directly frozen and those that were speedvacuumed before freezing.
Figure 2.

Score plot comparing sample preparation methods for dialysate samples. ◆ represents samples speedvacuumed to dryness and reconstituted in deuterated solution immediately prior to NMR analysis. ▽ represents samples frozen prior to NMR analysis and then diluted to 10% D2O. Each point is labeled with its individual sample number.
The normalized integrals from each sample were averaged for the frozen samples and for the speedvacuumed samples. A student's t-test was performed on the two means for each integral region. Only one out of 40 integral regions showed significant differences between the speedvacuumed and frozen heart dialysate samples at 95% confidence: 3.22−3.29 ppm. For the plasma dialysate samples, one region was significant at 95% confidence: 3.75−3.77 ppm.
Recovery by Dissolution
The recovery of plasma dialysate components by reconstitution in Ringer's solution was examined. Plasma dialysate was collected and speedvacuumed to dryness. The plasma was reconstituted in deuterated acetate solution in D2O (pD=7.8) and analyzed by NMR. Any solution remaining in the sample vial was removed and the same volume of methanol-d4 added to solubilize any residual metabolites that were not dissolved when the plasma dialysate was initially reconstituted. The NMR spectrum measured for this residual metabolite solution was compared to a control methanol-d4 spectrum and to the original plasma dialysate spectrum. Only solvent resonances were detected in the residual metabolite solution, indicating that no detectable levels of metabolites were left undissolved by the reconstitution buffer. No solubility concerns were noted with the dialysate sample preparation procedure.
Sample Storage Stability
The appropriate storage conditions of the speedvacuumed samples were assessed. Plasma dialysate samples were split into equal fractions and all samples were speedvacuumed to dryness. Half of the samples were stored at −20°C and the other half were stored at 4°C. All samples were reconstituted in equal volumes of 25 mM deuterated acetate solution in D2O with 5 mM TSP immediately prior to NMR analysis. Samples from each storage condition were analyzed on day 0 (immediately after speedvacuuming to determine the initial sample composition), 1, 2, and 7.
Results showed a clear demonstration of sample instability for some components at 4°C and sample stability at −20°C. Aliphatic resonances showed substantial changes seven days after storage at 4°C. Although some resonances (such as alanine) showed a decline in relative intensity, many new resonances appeared. Minor changes in the frozen samples were noted, although these were smaller relative to changes observed in the refrigerated samples. Specifically, six out of 57 integral regions were considered significantly different at 95% confidence when comparing the frozen samples to the original sample integral values. In contrast, 18 of 57 integral regions were considered significantly different at 95% confidence when comparing the refrigerated samples to the original sample integral values. Most of the changes in sample composition for both frozen and refrigerated samples were observed in the 3.0 − 4.0 ppm region, which primarily contains glucose resonances. Upon inspection of the NMR spectra, the levels of glucose stayed constant. The differences observed are due to unidentified resonances growing into the spectra as time elapsed. Also, when comparing the integral values for frozen and refrigerated samples, 23 integral regions were considered significantly different at 95% confidence. The significant integral regions were also primarily found in the 3.0 − 4.0 ppm region.
Data Analysis
Data Pretreatment
Several data treatment methods can be used prior to the application of principal components analysis. This pretreatment can strongly affect the statistical analysis and the interpretation of the results. Thus, data pretreatment steps must be chosen that will give results that accurately reflect trends in the data, rather than bringing irrelevant artifacts to light. With this in mind, several data pretreatment steps were examined for their effects on the PCA results.
First, the method of integration was considered. Many metabonomics studies use bucketing, an automated approach to integration in which the NMR spectrum is divided somewhat arbitrarily into integral regions of designated ppm width. Another approach is to use manual integration, whereby the researcher examines the spectra and determines the divisions between integral regions. In this comparison, a sample set of basal heart and liver dialysate was integrated in two ways. First, the spectra were bucketed from 4.5−0.8 ppm after spectral processing in ACD/Labs software with settings of 0.04 ppm bucket width and 10% looseness. (The % looseness setting determines the flexibility that the program is allowed in dividing spectra, so as to avoid splitting a resonance into two integral regions as much as possible.) Second, the same set of spectra was manually integrated from 4.5−0.8 ppm. Both sets of integrals were normalized to TSP intensity and to the average % delivery of the microdialysis probe. PCA was performed on a correlation matrix. The resulting scores plots were similar, with clusters for the liver dialysate samples observed in roughly the same coordinates in each plot. The heart dialysate shows about the same amount of spread with each integration mode. No differences were noted between integration modes.
Data normalization steps were also examined. Manual integrals from the basal heart and liver dialysate data set were again utilized. Three principal components analyses were performed: on the raw data set, the data set normalized to TSP, and the data set normalized to both TSP and the average percent delivery for the microdialysis probe. Figure 3 shows the three scores plots generated. Without normalization the data shows scatter, with no clustering or segregation between heart and liver dialysate. Data normalized to TSP intensity showed segregation between dialysate types, but with no real clustering. Finally, data normalized to both TSP and % delivery of the microdialysis probes showed a tight cluster of liver dialysate samples that were segregated from the more scattered heart dialysate samples. Although the liver dialysate data clustered tightly when normalized to both TSP and % delivery, the heart dialysate data showed less variance when normalized only to TSP.
Figure 3.
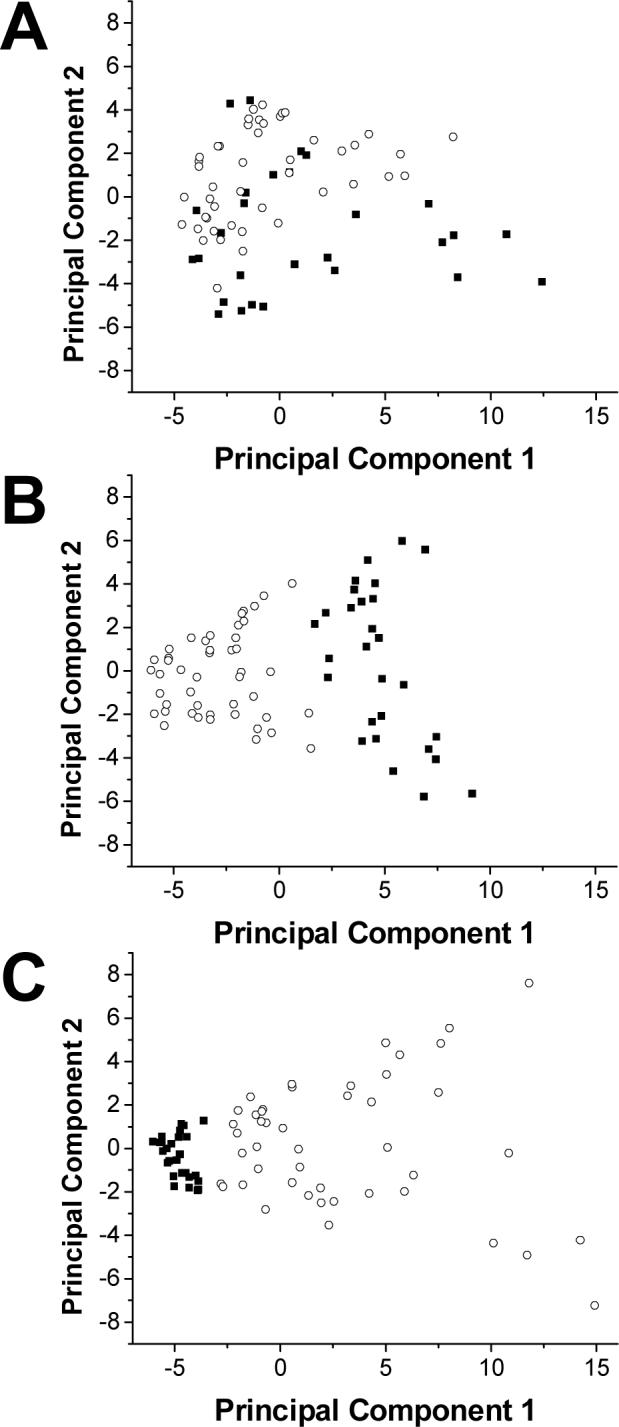
Effects of normalization on principal components analysis. A shows a score plot of basal heart and liver dialysate using unnormalized integrals. B shows a score plot of the same data normalized to TSP, the internal standard. C shows a score plot of the same data normalized to both TSP and the % delivery of the microdialysis probe. ■ represents basal liver dialysate samples. ○ represents basal heart dialysate samples.
Analysis of Tissue-Targeted Metabonomics Data: Perspectives and Time Trends
Metabonomics data sets can be analyzed on three primary levels: multivariate, univariate, and by spectral examination. For tissue-targeted metabonomics in particular, the description of time trends is key to data interpretation. These data analysis strategies were applied to a data set comparing basal liver dialysate from both anesthetized and awake rats. For the anesthetized rats, sampling began 30 minutes after probe implantation and continued for 12 hours. For the awake rats, sampling began 20 hours after probe implantation to allow the animals sufficient time to recover from the anesthesia and continued for 96 hours. Comparisons were made between tissue metabolic states (awake, anesthetized, and an awake rat that developed an infection post-surgery). Additionally, time trends were analyzed by both principal components analysis and partial least squares regression.
First, general groupings in the data were examined. Principal components analysis of the data, shown in Figure 4, shows three distinct groupings of data points along principal components 1 and 3. These three groupings correspond to previously noted metabolic states of the animal: awake healthy, anesthetized, and awake infected. Principal component 1 separates the healthy awake samples from the anesthetized and awake infected samples. Principal component 3 separates the awake infected samples from the anesthetized samples. The loadings indicate that principal component 1 is a contrast axis between time after probe implantation and all integral regions except those corresponding to lactate, meaning that the time variable has a negative loading while all loadings for the integral regions are positive in magnitude. Loadings for principal component 3 show lactate, valine, and resonances in the 3.51−3.63 ppm region (including some glucose resonances) and the 2.46−2.60 ppm region as important in segregating the samples from anesthetized and awake infected animals.
Figure 4.

Comparison of liver metabolic states by principal components analysis. ○ represents samples of basal liver dialysate from an awake rat with sepsis. ■ represents samples of basal liver dialysate taken from healthy awake rats. ◆ represents samples of basal liver dialysate taken from anesthetized healthy rats.
The data was also analyzed in a univariate manner by plotting time after probe implantation against the normalized integrals from each integral region of the NMR spectrum (40 in total). This allowed both time trends and segregation of groups within a single variable to be visualized. Examination of the plots showed discrepancies in regions containing glucose resonances. As shown in Figure 5, although figures A and B both contain glucose resonances, the trends observed in the awake infected samples differ remarkably between these regions, while the trends observed in the awake healthy and anesthetized samples remain the same. The 3.51−3.63 ppm region was also shown to be important in principal component 3 for segregating the anesthetized samples from the awake infected samples. Spectral examination of the data confirmed the findings from the PCA and univariate data analysis.
Figure 5.
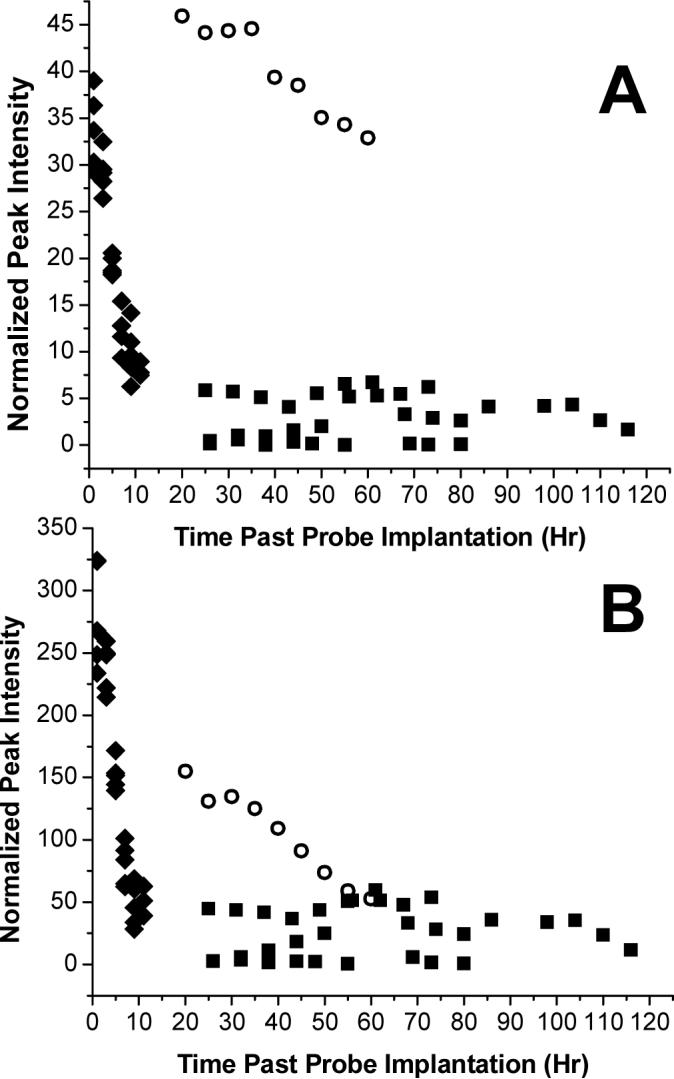
Comparison of liver glucose levels over time. ○ represents samples of basal liver dialysate from an awake rat with sepsis. ■ represents samples of basal liver dialysate taken from healthy awake rats (n=3). ◆ represents samples of basal liver dialysate taken from anesthetized healthy rats (n=4). A shows changes in the 3.51−3.52 ppm region and B shows changes in the 3.36−3.46 ppm region.
PCA (described above) showed a time trend along principal component 2, as illustrated in Figure 6. This time trend was unidirectional and confined to the samples from anesthetized animals. The samples from awake (both healthy and infected) animals did not show a definitive time trend although a trend was observed in the univariate time plots in Figure 5. Loadings for principal component 2 (not shown) indicate glucose, creatine and lysine as being most important in defining the time trends for the anesthetized samples.
Figure 6.

Time trends observed in basal liver dialysate using principal components analysis. ○ represents samples of basal liver dialysate from an awake rat with sepsis. ■ represents samples of basal liver dialysate taken from healthy awake rats. ◆ represents samples of basal liver dialysate taken from anesthetized healthy rats. The arrow shows the direction of the time trend observed in the anesthetized rats.
PLS regression showed a slightly different time trend. PLS was performed separately on correlation matrices for samples from anesthetized and awake animals, correlating the NMR integral data with sampling time. The anesthetized sample set showed a good correlation between the predictors (NMR integrals) and responses (sampling time), with an R2 of 0.916 for 2 components and an R2 predicted of 0.874 for 2 components, as determined through cross-validation. The standardized coefficients generated, which are similar to PCA loadings, are shown in Figure 7A. Again, there is a contrast between glucose and creatine, but glucose is now also contrasted with lactate and citrate. These resonances were not significant in the PCA loadings, likely because the samples from awake animals were also included in the analysis. Figure 7B shows the coefficients for the PLS regression correlation between the NMR integral data for the awake samples and the sampling time. Here, the correlation is not as strong as the observed for the anesthetized samples, with an R2 of 0.488 and an R2 predicted of 0.152, as determined by cross-validation. A similar trend was also observed in the PCA analysis. However, a loose correlation was still defined by the analysis largely by a contrast between the lysine, isoleucine, and valine and citrate, creatine, alanine, and β-hydroxybutyrate resonances.
Figure 7.
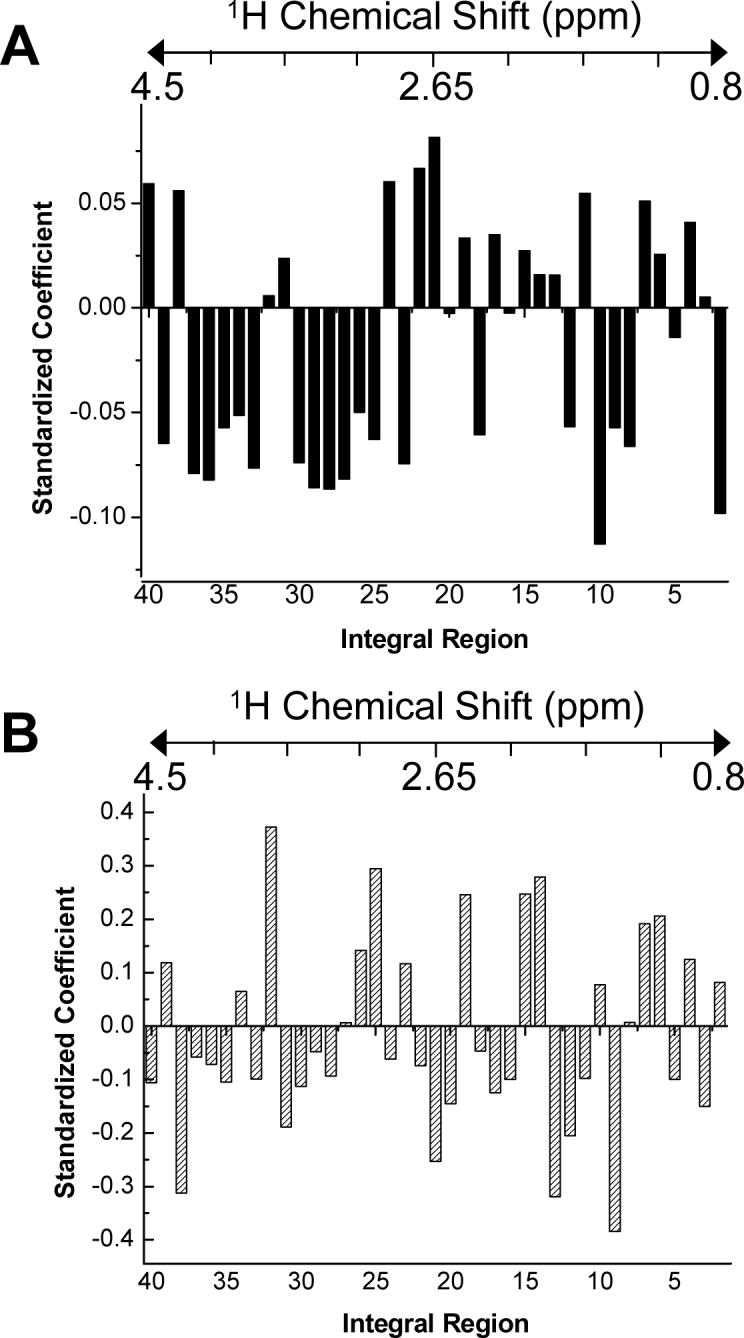
Standardized coefficients for PLS regression analysis. A shows the coefficients from PLS analysis of basal liver dialysate from anesthetized rats. Model statistics showed an R2 of 0.916 and an R2 predicted of 0.874. B shows the coefficients from PLS analysis of basal liver dialysate from awake rats. Model statistics showed an R2 of 0.488 and an R2 predicted of 0.152. The approximate correspondence of the integral regions to chemical shift is shown by the chemical shift scale above each graph.
Discussion
Tissue-targeted metabonomics shares many of the same goals as “traditional” metabonomics studies, the primary goal being the identification of new biomarkers without the prejudice of the more common single biomarker assays. Theoretically, by expanding the scope of metabolites studied, the probability of uncovering new biomarkers increases. However, tissue-targeted metabonomics differs from traditional metabonomics in several ways. First, of course, is the site specificity of the sampling approach. Second, microdialysis sampling does not require biopsy of the tissue of interest and the tissue remains intact. Third, because many samples can be obtained from the same animal over the time course of the experiment, tissue-targeted metabonomics better lends itself to the examination of time trends in metabolic data, rather than simpler group classification experiments. This is not to imply that traditional metabonomics cannot study time trends (or that tissue-targeted metabonomics cannot study group classification experiments) – in fact, many do [34,35]. However, the vast majority of metabonomics studies using urine or plasma sampling report group classifications as the end result of data interpretation. Finally, an additional benefit of the time trend approach of tissue-targeted metabonomics is the use of an animal as its own control, as is also done in some conventional metabonomics studies. By characterizing the basal metabolic patterns of an animal prior to perturbing its metabolism, corrections for animal-to-animal variation can be applied, potentially improving data interpretation.
Microdialysis Sampling Considerations
Basal metabolic profiles generated prior to the metabolic stressor event allow for normalization of the data, thus reducing animal-to-animal variability and perhaps increasing the prominence of pertinent metabolic trends. The picture generated by tissue-targeted metabonomics has the potential to be more complete than in metabolic profiling studies using conventional sampling techniques, in terms of sampling frequency. Urine sampling is limited by the excretion rate of the animal, and plasma samples are limited by the volume that can be drawn from the animal in a given period of time. Microdialysis sampling frequency, however, is limited only by the volume needed for analysis. Additionally, because many time points can be obtained from a single animal using microdialysis sampling, the number of animals required for experimentation is greatly reduced.
Calibration of the microdialysis probes is an important point in metabolic profiling experiments. Typical microdialysis experiments only examine one or two analytes. Thus, it is fairly straightforward to calibrate for the analytes of interest using the analytes themselves, allowing for quantitative studies. However, for metabonomics studies, the number of analytes could reach into the hundreds. Not only does this far exceed the capacity of the researcher to calibrate for each compound individually, the actual compounds of interest may be unknown at the beginning of the experiment due to the “fishing expedition” nature of profiling studies. Thus, exact quantitation of all analytes is not feasible for tissue-targeted metabonomics studies. However, exact quantitation is not necessarily the goal of these studies - rather, it is the relative change from baseline levels. This makes probe calibration by retrodialysis a good option for metabonomics studies. In retrodialysis, the delivery of a standard compound (antipyrine is commonly used) through the microdialysis probe to the tissue has been shown to be equivalent to the recovery of the same compound from the tissue to the probe lumen. Thus, the use of a single marker compound to account for changes in extraction efficiency of many endogenous small molecules is a simple and efficient way to account for variability in the data set due to changes in probe recovery. Normalizing the NMR integrals to the average probe delivery of antipyrine removed much of the random scatter in the liver dialysate data set as shown in Figure 3.
Sample Preparation Effects and Stability
Speedvacuuming
Dialysate samples are primarily collected into an isotonic Ringer's solution. Although it is possible to effectively suppress the water resonance in solutions that are predominately (i.e., 90%) protic water, large regions of the spectrum containing the analytes of interest are also suppressed. In addition, the residual water resonance may distort the spectral baseline, affecting analyte quantitation. For dilute solutions, the residual water signal may dominate the spectrum, limiting the dynamic range of the measurement. Therefore, we chose to reduce the water resonance by drying the dialysate sample and reconstituting in a deuterated solution. This approach is also advantageous because the sample could be concentrated, improving sample throughput by reducing the time required for signal-averaging.
However, it is possible that speedvacuuming could alter sample composition. The scores plot in Figure 2 suggests that speedvacuuming did not significantly alter the sample composition in either plasma or heart dialysate. Although segregation was observed between heart and plasma dialysate as expected (with one outlying sample), no segregation between sample preparation methods was observed within clusters of dialysate types. It should be noted that sample composition is not exactly the same, as corresponding frozen and speedvacuumed sample points do not overlap in the score plot. Thus, speedvacuuming alters dialysate composition slightly, but not in a large and systematic manner as would be indicated by clustering in the scores plot. Univariate analysis and spectral examination confirm these conclusions. Although it is apparent that speedvacuuming does alter dialysate composition slightly due to loss of volatile components (primarily acetate) or possible degradation of unidentified unstable compounds, the changes it causes are confined to a small region of the NMR spectrum. Speedvacuuming dialysate samples also improves sample stability (as discussed below) and spectral quality by improving water suppression. Although reconstitution in D2O can cause loss of resonances from exchangeable protons, in general for small organic molecules there is less information lost by this route than by suppression of the solvent resonance in 90% H2O.
Sample Recovery
Biofluid composition is complex, especially plasma, whose components have varying degrees of solubility in aqueous solution. Additionally, many of the dialysate samples collected for this project were speedvacuumed to dryness and reconstituted in half the original volume, potentially exacerbating any solubility issues. Solvent recovery experiments demonstrated that detectable plasma dialysate components had no solubility problems in deuterated solution.
Sample Storage Stability
The storage conditions of the speedvacuumed samples was important in that the need to for long NMR acquisition times to allow for signal averaging combined with the limitations on instrument access limit the number of samples that can be analyzed in a single day, requiring that samples from the same set be stored and analyzed on different days. Comparative studies between paired samples stored at 4°C and −20°C showed instability for some components at 4°C, but good stability at −20°C. Thus, speedvacuumed samples were frozen until NMR analysis.
Data Analysis
Data Pretreatment
Interpretation of large metabonomics data sets can be cumbersome when considered in a univariate manner. Multivariate statistical methods such as principal components analysis have been a tremendous tool for metabonomics researchers in the evaluation of their data. However, the quality of data input for principal components analysis determines the quality of the results obtained. One purpose of this study was to evaluate the effects of data pretreatment on principal components analysis of tissue-targeted metabonomics data.
Tissue-targeted metabonomics data is distinct from typical metabonomics data. The disparity between metabolite levels in an NMR spectrum seems to be exacerbated because most of the molecules of interest in the NMR spectrum are present at concentrations at least ten times lower than the most abundant molecule, glucose. However, the patterns in their changes are as important, if likely not more important, than that of glucose. Thus, all PCA analyses were performed using correlation matrices to equalize variable contribution to the statistical analysis [28].
Another consideration for tissue-targeted metabonomics data pretreatment was normalization of the data to correct for variation from NMR data acquisition and microdialysis probe performance. Figure 3 shows the progression of effects of normalization. It is clear that the variation in the data due to these two factors is greater than the variation between liver and heart dialysate composition. Therefore, the initial PCA was dominated by experimental (not metabolic) variance and underlying patterns could not be resolved. For the liver dialysate data, the more variation corrected for prior to the PCA, the tighter the clustering. This was not observed with the heart dialysate data, as more scatter was observed after normalization to probe delivery. This is likely due to two key differences in heart and liver microdialysis probe performance, as shown in Figure 8. First, due to spatial constraints in probe implantation in the heart apex, the membrane length is 8 mm shorter than the liver microdialysis probe. Thus, the extraction efficiency is approximately 30% less in the heart than in the liver. Second, the heart is a continuously moving organ, unlike the liver. This leads to greater variation in probe extraction efficiency. When this is considered with the smaller overall extraction efficiency of the heart microdialysis probes, it is likely that probe performance does not effectively remove variation in the heart dialysate data set. These results are consistent with what would be expected both experimentally and biologically.
Figure 8.

Comparison of microdialysis probe performance for heart and liver tissues as gauged by in vivo delivery of 7 μM antipyrine. Liver probes (n=5) showed an average % delivery of 41.8% ± 2.3%. Heart probes (n=4) showed an average % delivery of 6.58% ± 3.64%.
Analysis of Tissue-Targeted Metabonomics Data: Perspectives and Time Trends
The complexity of metabonomics data sets is a significant impairment to the throughput of the technique. Balancing a thorough investigation of the data with time constraints can be difficult. Using principal components analysis to identify the pertinent trends in the data, while efficient, could easily overlook important metabolic trends, particularly from lower abundance metabolites, which tend to become overshadowed in principal components analysis even with unit variance scaling.
This tension between high and low abundance metabolites is a significant barrier to metabolic profiling experiments in general, both for detection schemes and data analysis. The instrumental dynamic range required to effectively characterize the complete metabolome is immense. Thus, it should be considered that while the data generated by metabonomics analysis is extensive, it is not comprehensive. Many metabolites of key significance to the metabolic perturbation being studied but lower than the detections limits of the instrument may be excluded. A related problem is the difficulty of detecting metabolites present in low abundance due to overlap with the more intense resonances of the major components, an inherent consequence of performing the analysis on the intact mixture without a separation. This problem can be partially alleviated by using a higher field magnet since dispersion increases linearly with magnetic field. Still, there are practical limits such as the instrumentation available to the investigator, and even at the highest field available many important low abundance metabolites will be missed due to resonance overlap. Dynamic range is also a problem in metabonomics data analysis. A balance must be established between the dominance of high abundance metabolites in the multivariate analysis and the inflation of regions of noise when trying to increase the contribution of low abundance metabolites in the multivariate analysis.
This paper examines a three-tiered approach for the analysis of a metabonomics data set. Principal components analysis provided a good overall description of the trends in the data set. Groupings in the data were identified on the scores plot with principal components 1 and 3, while time trends were identified on the scores plot with principal components 1 and 2. While these trends were confirmed by examining the data from a univariate and spectral perspective, no significant trends were overlooked by the PCA. Analysis on the univariate and spectral levels, however, is still important to more fully describe trends identified in the multivariate analysis. The multivariate analysis can streamline the more time consuming univariate and spectral analysis steps by focusing attention on the most relevant integral regions, both increasing throughput and ensuring an in-depth analysis of the data set.
Because a time course of microdialysis samples are collected from a single animal, the data generated by tissue-targeted metabonomics naturally lends itself to analysis of time trends as opposed to the typical group classification. Time trends can be observed with principal components analysis and then more accurately described using partial least squares regression. PCA was useful in identifying data sets with strong time trends, but was unable to identify weaker time trends. The principal components loadings correctly identified most of the key resonances responsible for defining strong time trends, as they corresponded to the coefficients calculated from PLS regression. However compared with PCA, PLS identified more resonances as significant. Additionally, PLS regression could identify and describe both strong and weak time trends, as illustrated by the samples from anesthetized and awake animals.
Conclusions
Tissue-targeted metabonomics focuses on biomarker discovery for localized metabolic perturbations, as observed through time trends in the metabolic profiles. These time trends point to potential biomarkers for the condition, leading to more targeted single biomarker diagnostic assays. Perhaps more importantly, the improved time resolution with reduced animal-to-animal variability enables the researcher to observe the development or recession of a physiological condition, as well as to identify the key metabolic pathways involved throughout the onset or retraction of a metabolic perturbation. These perturbations may be identifiable well before any changes are observed in urine or plasma. Thus, the use of tissue-targeted metabonomics may lead to tests that enable the detection of site-specific diseases or drug toxicity much faster than conventional assays.
Acknowledgements
Funding from NIH grant HL069014 is gratefully acknowledged. K.E.P. gratefully recognizes the Madison and Lila Self Fellowship for financial support and Sara R. Logan for assistance with some of the animal surgeries.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- 1.Lindon JC, Holmes E, Nicholson JK. Pharm. Res. 2006;23:1075–1088. doi: 10.1007/s11095-006-0025-z. [DOI] [PubMed] [Google Scholar]
- 2.Nicholson JK, Lindon JC, Holmes E. Xenobiotica. 1999;29:1181–1189. doi: 10.1080/004982599238047. [DOI] [PubMed] [Google Scholar]
- 3.Trygg JA,J, Gullberg J, Johansson AI, Jonsson P, Antti H, Marklund SL, Moritz T. Anal. Chem. 2005;77:8086–8094. doi: 10.1021/ac051211v. [DOI] [PubMed] [Google Scholar]
- 4.Fancy S-A, Beckonert O, Darbon G, Yabsley W, Walley R, Baker D, Perkins GL, Pullen FS, Rumpel K. Rapid Commun. Mass Spectrom. 2006;20:2271–2280. doi: 10.1002/rcm.2583. [DOI] [PubMed] [Google Scholar]
- 5.O'Hagan S, Dunn WB, Brown M, Knowles JD, Kell DB. Anal. Chem. 2005;77:290–303. doi: 10.1021/ac049146x. [DOI] [PubMed] [Google Scholar]
- 6.Pohjanen E, Thysell E, Jonsson P, Eklund C, Silfver A, Carlsson I-B, Lundgren K, Moritz T, Svensson MB, Antti H. J. Proteom. Res. 2007;6:2113–2120. doi: 10.1021/pr070007g. [DOI] [PubMed] [Google Scholar]
- 7.Pham-Tuan H, Kaskavelis L, Daykin CA, Janssen H-G. J. Chromatogr. B. 2003;789:283–301. doi: 10.1016/s1570-0232(03)00077-1. [DOI] [PubMed] [Google Scholar]
- 8.García A, Barbas C, Aguilar R, Castro M. Clin. Chem. 1998;44:1905–1911. [PubMed] [Google Scholar]
- 9.Zomer S, Guillo C, Brereton RG, Hanna-Brown M. Anal. Bioanal. Chem. 2003;378:2008–2020. doi: 10.1007/s00216-004-2518-0. [DOI] [PubMed] [Google Scholar]
- 10.Sato S, Soga T, Nishioka T, Tomita M. Plant J. 2004;40 doi: 10.1111/j.1365-313X.2004.02187.x. [DOI] [PubMed] [Google Scholar]
- 11.Soga T, Ohashi Y, Ueno Y, Naraoka H, Tomita M, Nishioka T. J. Proteom. Res. 2003;2:488–494. doi: 10.1021/pr034020m. [DOI] [PubMed] [Google Scholar]
- 12.Soga T, Ueno Y, Naraoka H, Ohashi Y, Tomita M, Nishioka T. Anal. Chem. 2002;74:2233–2239. doi: 10.1021/ac020064n. [DOI] [PubMed] [Google Scholar]
- 13.Ullsten S, Danielsson R, Bäckström D, Sjöberg P, Bergquist J. J. Chromatogr. A. 2006;1117:87–93. doi: 10.1016/j.chroma.2006.03.048. [DOI] [PubMed] [Google Scholar]
- 14.Soga T, Baran R, Suematsu M, Ueno Y, Ikeda S, Sakurakawa T, Kakazu Y, Ishikawa T, Robert M, Nishioka T, Tomita M. J. Biol. Chem. 2006;281:16768–16776. doi: 10.1074/jbc.M601876200. [DOI] [PubMed] [Google Scholar]
- 15.McNally DJ, Hui JPM, Aubry AJ, Mui KKK, Guerry P, Brisson J-R, Logan SM, Soo EC. J. Biol. Chem. 2006;281:18489–18498. doi: 10.1074/jbc.M603777200. [DOI] [PubMed] [Google Scholar]
- 16.Lee R, Ptolemy AS, Niewczas L, Britz-McKibbin P. Anal. Chem. 2007;79:403–415. doi: 10.1021/ac061780i. [DOI] [PubMed] [Google Scholar]
- 17.Hong J, Baldwin RP. J. Capillary Electrophor. 1997;4:65–71. [PubMed] [Google Scholar]
- 18.Vigneau-Callahan K, Shestopalov AI, Milbury PE, Matson WR, Kristal BS. J. Nutr. 2001;131:9245–9325. doi: 10.1093/jn/131.3.924S. [DOI] [PubMed] [Google Scholar]
- 19.Gamache PH, Meyer DF, Granger MC, Acworth IN. J. Am. Soc. Mass Spectrom. 2004;15:1717–1726. doi: 10.1016/j.jasms.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 20.Khandelwal P, Beyer CE, Lin Q, McGonigle P, Schechter LE, Bach AC., II J. Neurosci. Methods. 2004;133:181–189. doi: 10.1016/j.jneumeth.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 21.Khandelwal P, Beyer CE, Lin Q, Schechter LE, Bach AC., II Anal. Chem. 2004;76:4123–4127. doi: 10.1021/ac049812u. [DOI] [PubMed] [Google Scholar]
- 22.Price KE, Vandaveer SS, Lunte CE, Larive CK. J. Pharm. Biomed. Anal. 2005;38:904–909. doi: 10.1016/j.jpba.2005.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bergström SK, Goiny M, Danielsson R, Ungerstedt U, Andersson M, Markides KE. J. Chromatogr. A. 2006;1120:21–26. doi: 10.1016/j.chroma.2006.01.110. [DOI] [PubMed] [Google Scholar]
- 24.Hansen DK, Davies MI, Lunte SM, Lunte CE. J. Pharm. Sci. 1999;88:14–27. doi: 10.1021/js9801485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davies MI, Lunte CE. Drug Metab. Disp. 1995;23:1072–1079. [PubMed] [Google Scholar]
- 26.Schiffer WK, Mirrione MM, Biegon A, Alexoff DL, Patel V, Dewey SL. J. Neurosci. Meth. 2006;155:272–284. doi: 10.1016/j.jneumeth.2006.01.027. [DOI] [PubMed] [Google Scholar]
- 27.Cloarec O, Dumas ME, Trygg J, Craig A, Barton RH, Lindon JC, Nicholson JK, Holmes E. Anal. Chem. 2005;77:517–526. doi: 10.1021/ac048803i. [DOI] [PubMed] [Google Scholar]
- 28.Craig A, Cloarec O, Holmes E, Nicholson JK, Lindon JC. Anal. Chem. 2006;78:2262–2267. doi: 10.1021/ac0519312. [DOI] [PubMed] [Google Scholar]
- 29.Forshed J, Torgrip RJO, Åberg KM, Karlberg B, Lindberg J, Jacobsson SP. J. Pharm. Biomed. Anal. 2005;38:824–832. doi: 10.1016/j.jpba.2005.01.042. [DOI] [PubMed] [Google Scholar]
- 30.Wang Y, Wong SL, Sawchuk RJ. Pharm. Res. 1993;10:1411–1419. doi: 10.1023/a:1018906821725. [DOI] [PubMed] [Google Scholar]
- 31.Yokel RA, Allen DD, Burgio DE, McNamara PJ. J. Pharmacol. Toxicol. Methods. 1992;27:135–142. doi: 10.1016/1056-8719(92)90034-x. [DOI] [PubMed] [Google Scholar]
- 32.Yokel RA, Allen DD, Burgio DE, McNamara PJ. J. Pharmacol. Toxicol. Meth. 1992;27:135–142. doi: 10.1016/1056-8719(92)90034-x. [DOI] [PubMed] [Google Scholar]
- 33.Davies MI, Lunte CE. Drug. Metab. Dispos. 1995;23:1072–1079. [PubMed] [Google Scholar]
- 34.Waters NJ, Holmes E, Williams A, Waterfield CJ, Farrant RD, Nicholson JK. Chem. Res. Toxicol. 2001;14:1401–1421. doi: 10.1021/tx010067f. [DOI] [PubMed] [Google Scholar]
- 35.Yap IKS, Clayton TA, Tang H, Everett JR, Hanton G, Provost J-P, Le Net J-L, Charuel C, Lindon JC, Nicholson JK. J. Proteom. Res. 2006;5:2675–2684. doi: 10.1021/pr0601584. [DOI] [PubMed] [Google Scholar]