

Abstract
Sgs1, the RecQ helicase homolog, and Top3, the type-IA topoisomerase, physically interact and are required for genomic stability in budding yeast. Similarly, topoisomerase III genes physically pair with homologs of SGS1 in humans that are involved in the cancer predisposition and premature aging diseases Bloom, Werner, and Rothmund-Thompson syndromes. In the absence of Top1 activity, sgs1 mutants are severely growth impaired. Here, we investigate the role of Sgs1 helicase activity and its N-terminal Top3 interaction domain by using an allele replacement technique to integrate mutant alleles at the native SGS1 genomic locus. We compare the phenotype of helicase-defective (sgs1-hd) and N-terminal deletion (sgs1-NΔ) strains to wild-type and sgs1 null strains. Like the sgs1 null, sgs1-hd mutations suppress top3 slow growth, cause a growth defect in the absence of Srs2 helicase, and impair meiosis. However, for recombination and the synthetic interaction with top1Δ mutations, loss of helicase activity exhibits a less severe phenotype than the null. Interestingly, deletion of the Top3 interaction domain of Sgs1 causes a top3-like phenotype, and furthermore, this effect is dependent on helicase activity. These results suggest that the protein-protein interaction between these two DNA-metabolism enzymes, even in the absence of helicase activity, is important for their function in catalyzing specific changes in DNA topology.
Keywords: Sgs1, Top3, helicase, topoisomerase, recombination, Saccharomyces cerevisiae
1. Introduction
The S. cerevisiae SGS1 gene belongs to a family of DNA helicases first defined by the E. coli RecQ helicase [1]. All RecQ homologs studied to date are important for genomic integrity (reviewed in [2, 3]). In humans, mutations in three of the five known RecQ/Sgs1 homologs are involved in tumor suppression and disease syndromes: Bloom syndrome (BS) and Werner Syndrome (WS), caused by mutations in the BLM and WRN genes, respectively, and three syndromes - Rothmund Thompson (RTS), RAPADALINO, and Baller-Gerold (BGS), caused by mutations in the RECQ4 gene [4-8]. These syndromes variously display features of premature aging, cancer predisposition, developmental abnormalities, and genomic instability (for recent review see [9])
The Sgs1 helicase was discovered by both genetic and physical interactions with Top3, a prokaryotic-like type-I topoisomerase [10]. Strains mutant for top3 have a pleiotropic phenotype including a severe growth defect caused by a cell-cycle delay in late S/G2, hyper-recombination at multiple loci, increased chromosome nondisjunction and sensitivity to the DNA damaging agents MMS and HU, meiotic defects, and an impaired intra S-phase checkpoint [10-15]. The phenotype of sgs1 mutants resembles that of top3 mutants but in each case the sgs1 defects are less severe and, for the most part, sgs1 is epistatic to top3 [10, 16, 17]. For example, mutation of sgs1 suppresses the slow growth of top3 mutants to the rate of an sgs1 mutant.
Recently, Rmi1, a third member of the Sgs1-Top3 complex, was discovered [18, 19]. Biochemical studies suggest that the Sgs1-Top3-Rmi1 complex plays a role in processing HR intermediates, restarting failed replication forks, and activating S-phase checkpoint arrest [17-25]. Rmi1 may promote binding specifically to branched DNA structures and/or stimulate Top3 strand passage [28,29]. In humans, a conserved complex of the homologous BLM-hTOPO IIIα-BLAP75/RMI1 functions in similar processes [26-30].
These functional roles for Sgs1-Top3-Rmi1 in yeast are evidenced by synthetic sickness or lethality with other genes involved in replication and recombination, such as srs2, rrm3, slx1, slx4, mus81, mms4 and top1 [12, 19, 31-36, 40]. These synthetic interactions, along with many of the defects in sgs1 and top3 mutant cells are likely a result of toxic or unresolved HR events, since they can be suppressed by mutation of genes involved in the early steps of HR such as RAD51, RAD52, RAD54, RAD55, and RAD57 [18, 19, 21, 37-39].
The RecQ, Blm, Wrn, and Sgs1 proteins all possess enzymatic helicase activity with a 3′ to 5′ polarity [1, 40, 42, 43]. Yet the signature helicase domain of the eukaryotic genes only covers about a third of the length of the respective proteins, approximately 400 amino acids (AA) (Fig. 1A). For example, helicase activity has been demonstrated for Sgs1 in a fragment from AA 400-1268, indicating that much of the protein is dispensable for helicase function [44]. Furthermore, loss of the enzymatic helicase activity of Sgs1 is responsible for some, but not all aspects of the sgs1 mutant phenotype including chromosome loss and missegregation as well as synthetic lethality with srs2Δ [47].
Fig. 1.

(A) Diagram of Sgs1 protein showing regions of interaction with other proteins as well as functional domains. The N-terminal 82-aa deletion is indicated in black and the Walker-A box invariant lysine at residue 706 is shown in red. Other features of the Sgs1 protein are illustrated as follows: the two acidic regions (“1” and “2”) are in yellow, the helicase domain is gray, the RecQ C-terminal homology region is in dark gray, and the Helicase and RNaseD C-terminal region (HRDC) is in green. (B) Protein blot of triple-HA-tagged alleles of Sgs1. Alleles are indicated at the top. Left arrow indicates full-sized 1447aa Sgs1 protein, right arrow indicates mobility shift of N-terminal deletions at 1365aa; size marker is indicated at right. Blot was probed with 1:10,000 dilution of HA antibody (Santa Cruz Biotechnology) and visualized using ECL Plus (Amersham Pharmacia Biotech) detection reagents according to product protocol (see Material and Methods for details).
Sgs1 interacts physically with Top3, Top2, Rad16, Rad51 and Rmi1 as well as other proteins [10, 13, 16, 18, 19, 48]. Genetic and physical evidence suggest that the interaction between eukaryotic RecQ and topoisomerase III homologs is of central importance for the function of both proteins in DNA metabolism. In humans, the BLM interacts with hTOPO IIIα and stimulates its strand-passage activity, and, together with RMI1, this complex can function as a double Holliday junction (dHJ) dissolvase [28, 29, 49, 50]. In yeast, fusion of Sgs1 and Top3 into a single peptide complements several aspects of the sgs1 phenotype [51]. The Sgs1-Top3 interaction is important in the absence of srs2 and top1, as well as for complementation of MMS sensitivity and suppression of HR [46, 52].
Here we investigate the roles of Sgs1 helicase activity and the Sgs1 interaction with Top3 by analyzing mutant alleles that disrupt these functions. In contrast to many previous studies, we only studied alleles integrated at their native genomic loci [53-55]. This permits an assessment of the true mutant phenotype and eliminates problems associated with plasmid-based complementation studies as well as position effects that may occur as a result of integration at non-native sites. We find that point mutations that inactivate the Sgs1 helicase result in many, but not all of the defects seen in sgs1 null mutants. For example, helicase defective mutants and the null mutant all exhibit the same top3 slow growth suppression as well as synthetic interaction with srs2. However, helicase mutants do not parallel the null allele for their effects on recombination at the SUP4-o locus or their growth defects in combination with top1 mutations. To investigate what other function of the Sgs1 protein may be responsible for these differences, we focused on the Sgs1 interaction with Top3 by deleting a region of the Sgs1 N terminus necessary for physical interaction with Top3. The phenotype of this deletion mimics the loss of Top3 when helicase activity is intact. However, when both the Top3 interaction domain and helicase activity are eliminated, an sgs1 null phenotype results. These data add support to the notion that Sgs1 acts upstream of Top3 in DNA metabolism, and its helicase activity creates an intermediate DNA structure that requires the recruitment of Top3 for efficient resolution. Furthermore, these observations suggest that the physical interaction between the two proteins is required to coordinate their activities.
2. Materials and Methods
2.1 S. cerevisiae strains and genetic methods
The W1588 segregants of W303-1A (MATa ade2-1 can1-100 his3-11,15 leu2-3,112 ura3-1 trp1-1 RAD5) and their derivatives used are listed in Table 1 [56, 57]. The crosses, growth, and transformation of strains were performed by standard methods [58]. Media were prepared as described [59], except twice the amount of leucine was used. Sporulation medium was prepared as described [60]. Standard procedures were used for mating, sporulation, and dissection [59]. Cells were grown at 30°C.
Table 1.
Strains used in this study
| Strain | Genotype |
|---|---|
| W1588-4C | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 RAD5 |
| W1588-4A | MATα ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 RAD5 |
| J276 | W1588-4A sgs1-K706R |
| J730 | W1588-4C sgs1-K706A |
| J734 | W1588-4A sgs1-NΔ82 |
| J735 | W1588-4A sgs1-NΔ82,K706R |
| J739 | W1588-4A SGS1-3HA |
| J740 | W1588-4A sgs1-K706R-3xHA |
| J741 | W1588-4A sgs1-K706A-3xHA |
| J742 | W1588-4A sgs1-NΔ82-3xHA |
| J743 | W1588-4A sgs1-NΔ82,K706R-3xHA |
| W1874 | Diploid wild-type (W1588-4C/W1588-4A) |
| W1956 | Diploid sgs1::URA3/SGS1 top1::HIS3/TOP1 |
| W1958 | Diploid sgs1::HIS3/SGS1 top3::TRP1/TOP3 |
| W1857 | Diploid sgs1::URA3/SGS1 srs2::HIS3/SRS2 |
| W1914 | Diploid sgs1-K706R/SGS1 srs2::HIS3/SRS2 |
| W2008 | Diploid sgs1-K706A/SGS1 srs2::HIS3/SRS2 |
| W2087 | Diploid sgs1-NΔ82/SGS1 srs2::HIS3/SRS2 |
| W2088 | Diploid sgs1-NΔ82,K706R/SGS1 srs2::HIS3/SRS2 |
| W1875 | Diploid sgs1::HIS3/sgs1::HIS3 |
| W1949 | Diploid top3::TRP1/top3::TRP1 |
| W1950 | Diploid sgs1-K706R/sgs1-K706R |
| W2034 | Diploid sgs1-K706A/sgs1-K706A |
| W1911 | Diploid sgs1-K706R/SGS1 |
| W2036 | Diploid sgs1-K706A/SGS1 |
| W2069 | Diploid sgs1-NΔ82/SGS1 |
| W2075 | Diploid sgs1-NΔ82,K706R/SGS1 |
| W2065 | Diploid sgs1-NΔ82/SGS1top1::HIS3/TOP1 |
| W2071 | Diploid sgs1-NΔ82,K706R/SGS1top1::HIS3/TOP1 |
| W2066 | Diploid sgs1-NΔ82/SGS1top3::TRP1/TOP3 |
| W2072 | Diploid sgs1-NΔ82,K706R/SGS1top3::TRP1/TOP3 |
| W2068 | Diploid sgs1-NΔ82/SGS1 srs2::HIS3/SRS2 |
| W2074 | Diploid sgs1-NΔ82,K706R/SGS1 srs2::HIS3/SRS2 |
| W1959 | Diploid sgs1::HIS3/SGS1 top3::TRP1/TOP3 SUP4-o::URA3 |
| W1934 | Diploid sgs1-K706R/SGS1 top3::TRP1/TOP3 SUP4-o::URA3 |
| W2082 | Diploid sgs1-NΔ82/SGS1 SUP4-o::URA3 |
| W2083 | Diploid sgs1-NΔ82,K706R/SGS1 SUP4-o::URA3 |
| W2089 | Diploid sgs1-NΔ82/SGS1 mec1::TRP1/MEC1 sml1-1/SML1 |
| W2090 | Diploid sgs1-NΔ82,K706R/SGS1 mec1::TRP1/MEC1 sml1-1/SML1 |
| W2094 | Diploid sml1::HIS3/SML1 mec1::TRP1/MEC1 sgs1::URA3/SGS1 top3::LEU2/TOP3 |
| W2706 | Diploid sgs1::URA3/SGS1 srs2::HIS3/SRS2 rad55Δ/RAD55 |
| W2722 | Diploid top3::TRP1/TOP3 srs2::HIS3/SRS2 rad55Δ/RAD55 |
| W2725 | Diploid sgs1-NΔ82/SGS1 srs2::HIS3/SRS2 rad55Δ/RAD55 |
| W2726 | Diploid sgs1-NΔ82,K706R/SGS1 srs2::HIS3/SRS2 rad55Δ/RAD55 |
| W3058 | Diploid sgs1-K706R/SGS1 srs2::HIS3/SRS2 rad51::LEU2/RAD51 |
The introduction of the K706R mutation into SGS1 was as described previously [53]. The plasmid-based sgs1-K706A allele (pJL37) was a gift from S. Brill [40]. A 4.1kb fragment (DrdI to SacII) containing the sgs1-K706A allele was used to replace the genomic sgs1ΔURA disruption in W1956-1D. 5-Fluoro-orotic acid (5-FOA) resistant colonies from this transformation were confirmed to contain the entire sgs1-K706A ORF by PCR and restriction digestion analysis. Segregation of the alleles after genetic crosses were scored by colony PCR of individual spores using primers SGS1aa541-F and SGS1aa890-R followed by restriction digest (using BglII for sgs1-K706R and NheI for sgs1-K706A).
The sgs1-NΔ82 allele was created by PCR amplification using Expand hi-fidelity polymerase (Roche Diagnostics) of two overlapping DNA fragments that were then fused by a third PCR reaction. Primers used in this study are listed in Table 2. One fragment contained 503bp directly upstream of SGS1 ending at the ATG (primers -500-F and SGS1aa1-R). The second fragment overlapped the 3′ 20 bases of the first fragment and contained 1145bp of the SGS1 ORF with the first 82 residues deleted (primers PROMOTER/aa83-F and SGS1-376-R). The 5′ primer contained this deletion by fusing 20 bases of the promoter to the ATG previously at residue 83. Fusion of the 503bp and 1145bp fragments by amplification with the outside primers (-500-F and SGS1-376R) yields a 1382bp fragment beginning 500bp upstream of the ATG and ending at amino-acid 376 of (wild-type) SGS1 that lacks the first 82 residues. This fusion fragment was digested with XhoI and AgeI and subcloned into the same sites of pWJ692 (pRS424-SGS1) to yield pWJ912 (pRS424-sgs1-NΔ82). The fragment was also digested with XhoI and AatII and subcloned into the same sites of pWJ795 (pRS415-sgs1-K706R) to create pWJ913 (pRS415-sgs1-NΔ82,K706R). A 4.5 kb XhoI/BstBI fragment containing the entire sgs1 ORF in addition to 206bp upstream and 202bp downstream was then used to replace the genomic sgs1ΔURA3 allele of W1956-1D by cotransformation followed by replica plating to 5-FOA. Colonies that grew on 5-FOA were confirmed to contain sgs1-NΔ82 (strain J734) and sgs1-NΔ82,K706R (strain J735) by PCR, restriction digests, and DNA sequencing. Sequence analysis of the entire region that had been amplified by PCR (up to bp1128 of wt SGS1) determined that no changes to the amino-acid sequence of Sgs1-NΔ and Sgs1-NΔ,K706R were introduced by PCR mutations.
Table 2.
Primers used in this study
| Name | Sequence |
|---|---|
| SGS1-PROMOTER/aa83-F | CACGTACACACAAGGCGGTAATGCAAACTTTGTCGAAC |
| SGS1us500-F | GGCTCAAACTGATCAGCGTTCGG |
| SGSaa1-R | CATTACCGCCTTGTGTGTACG |
| SGS1aa376-R | GACTGGTTCTCTTGAACGC |
| SGS1us116-F | GCTGATATACGGATCAATAGAG |
| SGS1aa119-R | CGAGCCATTGGGCGTCCTCGGG |
| SGS1aa376-R | GACTGGTTCTCTTGAACGC |
| GAD-SGS1.1-5′ | TAATATCCCCCGGGATGGTGACGAAGCCGTC |
| GAD-SGS1.83-5′ | TAATATCCCCCGGGATGCAAACTTTGTCGAACG |
| GAD-SGS1.795-3′ | AGGAAGATCTTCCTCTACTGATAGCTCTCTTGC |
| SGS1aa541-F | GCCGTCAACACATGCCCATGTC |
| SGS1aa890-R | GATTTAACCGCATCACAG |
| D/ds56-31r | GTGCTGCAGGTGTAAACTTGGAATGCTTGGCGAATGGTGTCG |
| E/1418f | GATACATCTACAACCGGCACCAAGGTCCATCAGTAAGTCGTCC |
| SGS1ct/NheI-F | TCAAAGGGCTAGCTTAGAGTAGAAAAATAAATAGTGTTAC |
| SGS1ct/NheI-R | TCAAAGGGCTAGCTCCTTTCTTCCTCTGTAGTGACCTCGG |
| SGSds252R | GACGCATTTATGCACATATGTAG |
| 3HA/NheR | CGAAGCGCTAGCTATCTTTTACCCATACGATG |
| 3HA/NheF | CGAAGCGCTAGCCTGGCCAGCGTAATCTGGAACG |
| SGS1HAcheck-R | GCCCGCATAGTCAGGAACATCG |
| Top3/BamH1f | TCGCGGATCCTAATGAAAGTGCTATGTGTCGCAGAG |
| Top3/BamH1r | TCGCGGATCCTTACATGGATGCCTTGACACGGTC |
We estimated Sgs1 protein levels in the various constructs used in this study by fusing each to a triple-HA epitope (see below). The introduction of the HA-tag did not significantly alter the function of the proteins as measured by testing each construct for growth in combination with top1 and top3 mutants and by comparing their sensitivities to HU and MMS (as singles or double mutants or both, data not shown). These results indicate that the tagged alleles are representative of the untagged version. However, at the same time, all experiments described in this study were performed with untagged alleles to avoid unforeseen phenotypic complications that might be caused by the presence of the HA-epitope.
Strains J739, J740, J741, J742 and J743, which contain fusions of a triple-HA tag to the C terminus of wild-type SGS1, sgs1-K706R, sgs1-K706A, sgs1-NΔ82, and sgs1-NΔ82,K706R, respectively, were created by the allele-replacement method as described previously [53]. Briefly, an in-frame fusion of a triple-HA repeat to the C terminus of SGS1 on plasmid pWJ691 was used as template for PCR amplification of a fragment of the C terminus, (bp 4110 of the SGS1 ORF through 187 bp downstream). This HA-containing fragment was then fused by PCR to two overlapping fragments of the K. lactis URA3 gene. Cotransformation of the two fragments and selection on medium lacking uracil yielded integrants containing the reconstructed K. lactis URA3 gene surrounded by a direct repeat of the triple-HA epitope at the genomic locus of SGS1 (or sgs1-K706R, etc.) Direct-repeat recombination events that excised the URA3 gene were selected on 5-FOA, leaving a single copy of the triple-HA fusion. The HA-tagged alleles were verified by PCR analysis, phenotype and protein blots (see Fig. 1B).
2.2 Protein blot analysis
Strains bearing the HA-tagged alleles (J739, J740, J741, J742 and J743) were grown overnight in YPD. Protein extraction was carried out at 4°C using chilled solutions. Approximately 5 × 108 cells were collected and washed once in 1ml PBS, followed by resuspension in 500μl 25% TCA. Cells were pelleted at 4000 RPM and washed 2 times in 90% acetone before resuspension in 100μl 1% SDS with 1X protease inhibitor cocktail (Boehringer Mannheim). Cell pellets were lysed by vortexing with glass beads before addition of 5X loading buffer. Samples were then boiled for 3 minutes followed by a brief centrifugation and electrophoresis on 5% Ready Gels (BioRad). Proteins were then electroblotted to Immobilion-P membranes (Millipore) followed by blocking with 5% nonfat dry milk and blots were probed with 1:10,000 dilution of anti-HA antibody (Santa Cruz Biotechnology). ECL Plus (Amersham Pharmacia Biotech) detection reagents were used according to product protocols. Densitometry of bands from scanned films was performed using ImageQuant (Molecular Dynamics). As shown in Fig. 1B, the mutant Sgs1 proteins were present at the same levels as wild-type Sgs1, indicating no significant change in the steady state levels of the mutant proteins.
2.3 Recombination assays
A replica-plating assay for detection of recombination events causing the deletion of the SUP4-o gene was performed as previously described [11, 61]. Briefly, various sgs1 alleles were introduced into the W1868 background (MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 SUP4-o::URA3 RAD5) by standard genetic crosses. At least 3 independent segregants of each genotype were assayed for recombination events of the δ-repeats surrounding the SUP4-o locus. Deletions of SUP4-o result in red (ade2-) colonies that require uracil for growth and are canavanine resistant.
2.4 Growth rates
Cultures were grown overnight in 5ml YPD cultures, from which between 300μl and 1ml were used to inoculate 50ml YPD cultures to similar OD600densities as measured by spectrophotometer. To avoid spontaneous suppressors, at least 3 independent segregants of each genotype were assayed after fresh dissection of heterozygous diploids. The OD600 of each culture was measured at hourly intervals for a total of 8 to 12 hours. The negative log of these numbers was then plotted. The growth rates were calculated from determining the slope of the straight-line portion of each graph, which defines the period of logarithmic growth after recovery from lag phase of the overnight culture before the cells reach stationary phase again.
2.5 HU and MMS sensitivities
Cells in mid-log phase cultures were counted and 10-fold dilutions from 105 to 10 cells per 10μl were made in YPD. 5μl of each was spotted on YPD plates and YPD plates containing HU or MMS. The plates were incubated at 30°C for 3-4 days. At least 2 segregants of each genotype were assayed twice and photographed.
2.6 Two-hybrid assays
The 2-hybrid strains used were PJ69 MATa and MATα and the plasmids used were pGBD-C2 and pGAD-C2 [62]. pGBD and pGAD fusions with TOP3 were constructed by amplification of the TOP3 ORF with primers Top3/BamH1f and Top3/BamH1r followed by digestion with BamHI and cloning into the BamHI site of pGBD-C2. Also tested (but not shown) was the pGBT9-TOP3 plasmid described in [10]. Both pGBD-Top3 fusions gave the same results. pWJ982 (pGAD-sgs1 AA 1 to 795) and pWJ983 (pGAD-sgs1 AA 83 to 795) were made by cloning a PCR fragment of SGS1 into the SmaI and BglII sites of pGAD. The primers used are GAD-SGS1.1-5′ (pWJ982) or GAD-SGS1.83-5′ (pWJ983) and GAD-SGS1.795-R. The presence of fusion peptides was confirmed by protein blot analysis of the two hybrid strains (diploids listed below) using antibodies specific to the GAL-4 DNA binding domain and activation domain (Santa Cruz Biotechnology, data not shown.)
2.7 Cell cycle distribution
4′,6-Diamidino-2-phenylindole (DAPI) staining was performed to visualize DNA as described in [63]. YPD liquid cultures of the different strains were grown to mid-log phase before fixing in 70% ethanol and washing in water. At least 500 cells of each genotype were counted and divided into categories by morphology, as indicated in Fig. 3B.
Fig. 3.

(A) Cell cycle distribution of different mutant strains. (A) Mid log-phase cultures of WT, sgs1Δ, sgs1-NΔ and top3 mutant strains were fixed in EtOH and stained with DAPI for visualization of DNA and photographed. (B) For the four strains shown in (A) as well as for sgs1-NΔ and sgs1-hd, at least 500 cells of each genotype were observed and classed according to morphology as shown. These classes correspond roughly to cell cycle phases G1, S, G2/M, M/G1. The percentages of each class are indicated.
3. Results
3.1 Mutant sgs1 alleles integrated at the genomic locus
A number of studies report the effect of various sgs1 mutations on different aspects of its phenotype. In most of these reports, alleles expressed from plasmids or integrated at non-native loci were used to study complementation [40, 41, 45, 46, 52, 64-68]. To avoid the ambiguities inherent in interpreting growth or lack of growth in cells transformed with a plasmid as well as to avoid potential position effects for loci integrated elsewhere, all mutant alleles used in this study were integrated at the SGS1 genomic locus (Table 1) [53, 54]. As described in the Materials and Methods, all alleles created for this study were confirmed by restriction digestion, sequence analysis and protein blot (Fig. 1B).
We analyzed the role of two major functions of the SGS1 gene in the sgs1 mutant phenotype--its helicase activity and its Top3 interaction. To understand the importance of the helicase activity, we studied sgs1 alleles that contain mutations of the invariant lysine in the ATPase domain known to abrogate biochemical helicase activity (sgs1-K706R and sgs1-K706A [40, 53]). In all assays described below, both sgs1-K706R and sgs1-K706A alleles behaved identically and we refer to them together as sgs1-hd, for helicase-defective. Biochemical and 2-hybrid data identified the Top3 interaction region of Sgs1 to N-terminal residues within the first 100 amino acids of Sgs1 [10, 41, 46, 52, 64]. We created an sgs1 allele encoding an N-terminal deletion protein by fusing the Sgs1 promoter to the ATG at residue 83 (sgs1-NΔ82). This 82 amino acid deletion eliminates the protein interaction with Top3 in 2-hybrid experiments (data not shown). This truncation was also combined with helicase-mutant alleles of SGS1.
3.2 sgs1-hd alleles exhibit synthetic interactions with mutations in other DNA metabolism genes
To compare the phenotype of sgs1-hd and sgs1 null alleles in combination with other genes, single and double mutant strains were constructed as described below. Since mutations in SGS1 were first discovered as suppressors of top3Δ slow growth, we analyzed suppression of the growth defect by measuring the doubling times of at least 3 independent spores of various mutant genotypes. Both sgs1-hd and sgs1Δ suppress top3Δ slow growth to the same extent (Fig. 2A). The slow growth of cells lacking top3 is largely the result of a delay in the late S/G2 phase of the cell cycle, and top3Δ cells are greatly enlarged with a majority of mid-log-phase cells as large budded cells with a single nucleus in the bud neck [10, 12, 69] (Fig. 3A). We examined cell morphology and cell cycle distribution of the mutant strains by staining mid-log phase cells with DAPI, and similar to that observed for growth suppression, helicase mutations suppress the enlarged morphology and cell-cycle delay of top3Δ cells to the same degree as the null allele (Fig. 3B and data not shown).
Fig. 2.
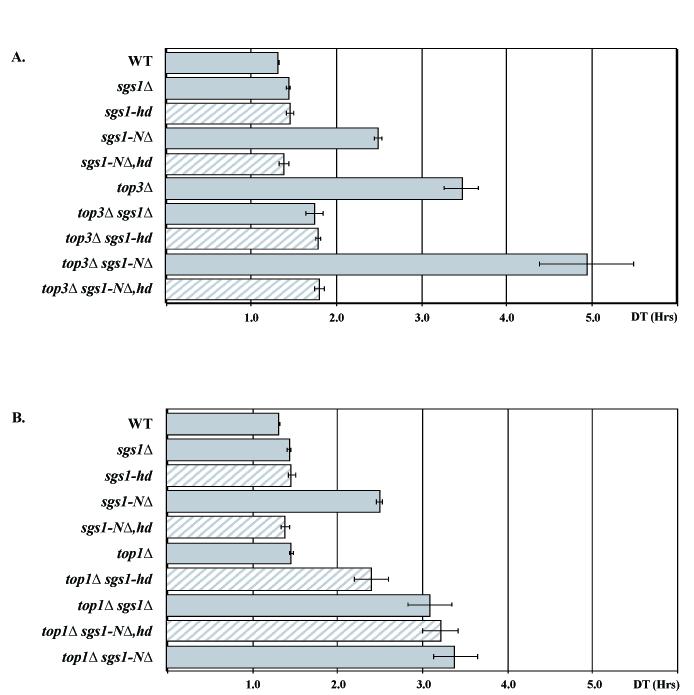
Bar graphs of doubling times (DT) of various mutant strains. Combinations of mutant strains are indicated on the left. (A) Strains mutant for sgs1 and top3. (B) Strains mutant for sgs1 and top1. The growth rate of logarithmic cells from each culture was calculated and is displayed. Growth rates were measured 3 times for each strain.
We next examined the synthetic growth interaction between sgs1 mutants and top1Δ. Fig. 2B illustrates the synergistic growth defect seen in sgs1Δ top1Δ double mutants [40]. The sgs1-hd top1Δ strains exhibit an intermediate doubling time between SGS1 top1Δ and sgs1Δ top1Δ strains. This difference suggests that loss of helicase activity is only partly responsible for the synergistic slow growth and that an additional non-helicase function of Sgs1 plays a role in growth in the absence of top1.
Loss of sgs1 function also causes a severe synthetic growth defect that often results in inviability with mutations in srs2 (Fig. 4A and [31]). This defect is alleviated by mutation of genes required for efficient HR such as RAD51, RAD52, RAD55, or RAD57 [21, 37, 70]. We confirmed that both sgs1-Δ srs2Δ and sgs1-hd srs2Δ double mutant segregants are inviable or form microcolonies (see tetrad dissections in Fig. 4A and B) [47]. Furthermore, homologous recombination also contributes to this phenotype, since the sgs1-hd srs2Δ growth defect is suppressed by mutation of rad51, 55 or 57 (Fig. 4C and data not shown).
Fig. 4.

Representative tetrads to illustrate genetic interactions between sgs1 and srs2. Tetrads are oriented horizontally, i.e., the third spore of the first tetrad in A. has not produced a colony. Genotypes of haploid parents are indicated in figure, and are: A. Diploid W1857, heterozygous for sgs1::URA3 and srs2::HIS3. B. Diploid 1914, heterozygous for sgs1-K706R and srs2::HIS3. C. W3058, heterozygous for sgs1-K706R, srs2::HIS3, and rad51::LEU2. D. W2068, heterozygous for sgs1-NΔ82 and srs2::HIS3. E. W2074, heterozygous for sgs1-NΔ,hd and srs2::HIS3. F. W2725, heterozygous for sgs1-NΔ82, srs2::HIS3, and rad55Δ. In A., B., D., and E., the colonies that are missing or are present as small colonies represent double mutants. In C. and F., triple mutants are indicated by squares.
3.3 Mutation of the Sgs1 helicase impairs recombination and meiosis
Mutations in sgs1 increase mitotic recombination at numerous genetic loci including the SUP4-o locus surrounded by δ-repeats [10, 12, 16, 69, 74]. Deletion of SUP4-o occurs by recombination events between these repeats. To examine the importance of the helicase function in the suppression of mitotic recombination, we measured marker loss of SUP4-o. As shown in Table 3, deletion of sgs1 results in a 39-fold increase in recombination. Mutation of the sgs1 helicase, however, increases recombination frequency to only half that of the null (19-fold).
Table 3.
Recombination frequency at SUP4-o
| Strain | Frequency (× 10-4) | Fold increase |
|---|---|---|
| SGS1 | 1.7 ± 0.3 | 1 |
| sgs1Δ | 64 ± 6 | 39 |
| sgs1-hd | 31 ± 3 | 19 |
| sgs1-NΔ | 142 ± 13 | 86 |
| sgs1-N,Δ hd | 70 ± 9 | 42 |
Sgs1 is thought to prevent aberrant crossing-over events during meiosis by suppressing formation of joint molecules [72]. Diploids homozygous for an sgs1 deletion exhibit a meiotic defect that results in decreased sporulation efficiency and spore viability at least partially as a result of missegregation leading to aneuploidy [12, 16, 17, 65, 71]. To investigate the helicase function on meiosis, we examined the effect of sgs1-hd alleles on sporulation and spore viability. As shown in Table 4, the sgs1-hd alleles are recessive, since heterozygous sgs1-hd/SGS1 diploids undergo normal meiosis. However, in homozygous sgs1-hd diploids, like the null, only ∼20% of the cells sporulate after 3 days and show ∼70% spore viability (Table 4), demonstrating that the meiotic defect is caused solely by loss of the helicase activity.
Table 4.
Meiotic defects as measured by sporulation and spore viability
| Strain | Sporulation (%) | Spore Viability (%) |
|---|---|---|
| SGS1/SGS1 | 60 | 94 |
| SGS1/sgs1-hd | 58 | 91 |
| sgs1Δ/sgs1Δ | 22 | 72 |
| sgs1-hd/sgs1-hd | 21 | 68 |
| sgs1-NΔ/SGS1 | 46 | 85 |
| sgs1-NΔ/sgs1-NΔ | <3 | <3 |
| top3Δ/top3Δ | <3 | <3 |
| top3Δ/TOP3 | 58 | 91 |
3.4 Deletion of the Top3-interaction domain of Sgs1 causes a top3-like phenotype
We next analyzed the effect of removing the Top3-interaction domain from a helicase-intact SGS1 gene (sgs1-NΔ). As shown in Fig. 2A, deletion of the N-terminal 82 amino acids of Sgs1 slows growth nearly as much as deletion of TOP3. Similarly, sgs1-NΔ cells are enlarged relative to wild-type though not as much as top3Δ mutant cells (Fig. 3A). Indeed, the cell-cycle distribution of sgs1-NΔ cells is similar to top3 mutant cells, where about 60% of cells are present as large, budded cells with a single nucleus (Fig. 3B).
Furthermore, we looked at various genetic interactions in sgs1-NΔ strains and found that the sgs1-NΔ top3Δ double mutant grows more slowly than either single mutant (Fig. 2A). In addition, sgs1-NΔ top1Δ doubles grow as poorly as sgs1Δ top1Δ double mutants (Fig. 2B). Finally, the genetic interaction between sgs1-NΔ and srs2 resembles that seen for sgs1Δ and srs2Δ and is also suppressed by blocking recombination (Fig. 4A, D and F).
Next, we examined the interaction of the sgs1-NΔ allele with genes involved in the DNA damage response, which are required for reversible DNA damage-dependent cell cycle arrest (for review see [76]). Crosses to the S-phase checkpoint mutant, mec1Δ, and a downstream kinase, dun1Δ, demonstrate that loss of top3, but not sgs1Δ or sgs1-hd, causes a synthetic growth defect or inviability (Table 5, [14]). However, when sgs1-NΔ is introduced into mec1Δ and dun1Δ mutant strains, a synthetic defect identical to that of top3Δ is seen.
Table 5.
Viability with mec1/dun1 mutations
| Inviable/sick | Viable |
|---|---|
| mec1Δ | mec1Δ sml1Δ |
| mec1Δ sml1Δ top3Δ | mec1Δ sml1Δ top3Δ sgs1Δ |
| mec1Δ sml1Δ sgs1-NΔ | mec1Δ sml1Δ sgs1-NΔ,HD |
| dun1Δ top3Δ | dun1Δ top3Δ sgs1Δ |
| dun1Δ sgs1-NΔ | dun1Δ sgs1-NΔ,HD |
Similarly, the recombination frequency at the SUP4-o locus in sgs1-NΔ strains also approximates that seen in top3 strains (Table 3 and [11, 39]). These results are consistent with previous observations of elevated recombination in strains bearing alleles of sgs1 lacking the N terminus [45, 75]. Furthermore, as shown in Table 4, diploids homozygous for sgs1-NΔ have a meiotic defect identical to that of top3Δ mutants and fail to sporulate at significant levels [11, 12, 16, 17]. However, unlike heterozygous top3Δ/TOP3, sgs1Δ/SGS1 or sgs1-hd/SGS1 diploids, sgs1-NΔ has a dominant negative effect on meiosis resulting in decreased sporulation (p < 0.02) and exhibits slightly lowered spore viability.
3.5 The top3-like phenotype of sgs1-NΔ depends on its helicase activity
As described above, loss of sgs1 helicase activity suppresses most top3 defects. Since sgs1-NΔ resembles top3Δ, we mutated the helicase motif of this allele (sgs1-NΔ,hd) to measure its effect in all of the assays described above. Similar to the suppression of top3 by sgs1-hd, eliminating helicase activity from the sgs1-NΔ allele results in the equivalent of an sgs1 null phenotype (see Table 6 for summary). Specifically, the growth defect (Fig. 2), altered cell morphology (Fig. 3A), cell cycle defects (Fig. 3B), hyper-recombination (Table 3) and synthetic defect with mec1 and dun1 mutants (Table 5) are suppressed. There is one case, however, where mutation of the helicase in sgs1-NΔ does not change the phenotype sgs1-NΔ. In combination with a top1 mutation, sgs1-NΔ and sgs1-NΔ,hd exhibit the same growth rate as an sgs1Δ top1Δ double mutant (see Fig. 2B).
Table 6.
Summary of mutant phenotypes
| Strain | Growth | with top3 | with top1 | with srs2 | G2 Delay | Meiosis | Hyperrec | with mec1/dun1 | HUr | MMSr |
|---|---|---|---|---|---|---|---|---|---|---|
| WT | ++++ | + | ++++ | +++ | ++++ | ++++ | - | ++++ | ++++ | ++++ |
| sgs1Δ | +++ | +++ | + | - | +++ | ++ | +++ | ++++ | ++ | ++ |
| sgs1-hd | +++ | +++ | ++ | - | +++ | ++ | ++ | ++++ | + | + |
| sgs1-NΔ | + | - | + | - | + | -- | ++++ | - | ++ | + |
| sgs1-NΔ,hd | +++ | +++ | + | - | +++ | ND | +++ | ++++ | ++ | + |
| top3Δ | + | - (NQ) | - | + | - | ++++ | - | - | - |
3.5 The sgs1 alleles have differential effects on sensitivity to HU and MMS
Lastly, we examined mutant sgs1 strains for their sensitivity to hydroxyurea (HU), which stalls DNA synthesis [77] and methylmethane sulfonate (MMS), a DNA alkylating agent likely to interefere with replication fork progression [78, 79]. Previous studies have shown that plasmid-based helicase-defective sgs1 alleles fail to complement sgs1Δ for MMS and HU [20, 45, 65]. Several reports, however, showed that helicase-defective genomic sgs1 alleles are more sensitive than a null allele to both chemicals [13, 47], suggesting that presence of the non-functional protein is interfering with DNA damage repair. We confirmed that sgs1-hd integrated at its normal locus exhibits increased HU and MMS sensitivity (Fig. 5). Since sgs1-NΔ, sgs1-NΔ,hd and sgs1 null strains are all equally sensitive to HU (Fig. 5), removal of the N terminus, regardless of helicase function, causes HU sensitivity equivalent to deleting the entire protein. Surprisingly, sgs1-NΔ and sgs1-NΔ,hd strains, like the sgs1-hd mutant, are both more sensitive to MMS than the null (Fig. 5). This is the only case we found where an sgs1-NΔ,hd strain does not exhibit a null phenotype.
Fig. 5.

HU and MMS sensitivity of sgs1 and top3 mutants. Mid-log phase YPD-grown cells of mutant strains were spotted in 10-fold serial dilutions on YPD plates and YPD plates containing 50mM HU or 0.02% MMS and grown for 3 days at 30°C.
4. Discussion
4.1 Model for the function of Sgs1 helicase activity and Top3 interaction
Here, using a variety of assays summarized in Table 6, we compare the phenotypic effects of mutations that remove the enzymatic helicase and Top3-interaction functions of Sgs1. All constructs used in this study are under control of the wild-type SGS1 promoter and expressed from the native genomic SGS1 locus. We expand on previous studies to show that point mutation of the SGS1 helicase resembles an sgs1 null allele for suppression of top3 slow growth, for meiotic defects and for synthetic lethality with srs2 mutations. Similarly, synthetic lethal interactions between sgs1 and slx1, mms4, slx3, slx4, slx5, and slx8 are also dependent on Sgs1 helicase activity [34]. However, some of the defects caused by loss of helicase activity are less severe than sgs1 null defects as measured by both growth in a top1 mutant background and recombination at the SUP4-o locus (Fig. 2B, Table 3). For these two assays, the null phenotype is seen only when the Top3 interaction is eliminated from a helicase-deficient protein (sgs1-NΔ,hd). These data suggest that the presence of the Sgs1-Top3 interaction serves some function even in the absence of Sgs1 helicase activity. Moreover, removing the interaction domain from the wild-type helicase active SGS1 allele (sgs1-NΔ) causes a phenotype that is even more severe than an sgs1 null and is similar to a top3 mutant. Thus, a top3-like phenotype likely results from preventing Top3 from interacting with a catalytically active Sgs1.
We note that in several complementation studies, plasmid-borne sgs1-hd alleles were unable to suppress the HR defect of sgs1Δ [65, 80] but behaved like wild-type SGS1 in growth assays of top3Δ sgs1Δ and top1Δ sgs1Δ strains and in its ability to rescue the meiotic defects of homozygous diploid sgs1 mutants [40, 45, 46, 65]. Also, recent studies of the meiotic function of Sgs1 utilized N-terminal fragments of the protein (aa 1-795) entirely lacking the helicase domain since this allele improved sporulation efficiency relative to a null allele [72, 73]. It has been suggested that conflicting observations of the sgs1 mutant phenotype could result from differential expression from plasmid vectors [47]. Such dosage effects, and/or strain differences, may account for these discrepancies. For example, the MR strain used in several of these studies differs from the W303 genetic background used here and in many other studies. In MR, sgs1Δ has a significantly different meiotic defect and shows incomplete epistasis to top3Δ. In addition top3Δ is “semi-lethal” in MR strains. [65, 67].
The data presented here are consistent with the original proposition that the Top3 phenotype results largely from unresolved recombinogenic substrates created by Sgs1 activity [10, 17]. A simplified model that both incorporates these data and includes an alternative pathway that utilizes Top1 is presented in Fig. 6A. In this schematic, a DNA substrate, such as a collapsed or stalled replication fork, is processed to an intermediate, (1), that is converted by the Sgs1 helicase to an intermediate, (2), that is toxic if unresolved into a final product, (3), by Top3. A primary candidate for one of the substrates of the Sgs1-Top3-Rmi1 complex is the Holliday junction (HJ), a key intermediate in recombinational repair and recovery from replication fork damage (see [81, 82] for review.) RecQ, BLM, WRN, RECQL1, RECQ5β, and Sgs1 are all able to bind and efficiently unwind HJ structures in vitro [83-88]. RECQL1, BLM and WRN efficiently promote ATP-dependent HJ branch migration, and, in the case of human and Drosophila BLM/TopIII complexes, ATP-dependent HJ dissolution [28, 85, 86, 89-91]. Alternatively, this intermediate may be a regressed replication fork [25, 47, 76].
Fig. 6.

Model of relationships between Sgs1, Top3 and Top1. Genotype is indicated above each schematic. In wild-type strains (A) a DNA structure (1), e.g., a collapsed or stalled replication fork, can be converted by a series of steps not shown to structures that are acted on by Sgs1 (e.g., a double Holliday junction) and converted into an intermediate (2), e.g., a hemicatenane, which is finally resolved into the final product (3) by Top3. An alternative pathway utilizing Top1 is also shown, which “acts on” an upstream intermediate to generate a viable cell, albeit one with increased recombination and sensitivity to DNA damaging agents (indicated by the smaller size of the number “3”). According to this model, the phenotypes of sgs1-NΔ and top3Δ in (B) and (C), respectively, result from persistence of a toxic intermediate (2*). Elimination of the helicase activity of full-length SGS1 (D) or sgs1-NΔ (E) prevents formation of intermediate (2) and the alternative Top1 pathway is active. Blocking this alternative pathway (F) results in a synergistic growth defect caused by the accumulation of a different toxic intermediate (1*). In the presence of a helicase inactive Sgs1 protein, the synergistic growth defect is less severe (G). However, this improvement in growth depends on the Top3 interaction domain of Sgs1 (H) .
Our model is consistent with the notion that resolution of HJs by a RecQ-Top3 mechanism is a two-step process: ATP-dependent HJ branch migration by a RecQ helicase to form a hemicatenane (Fig. 6A, intermediate 2), immediately followed by decatenation by a type IA topoisomerase via its strand passage activity (Fig. 6A, final product 3). As suggested by others [25, 92], the observation that MMS induced X-molecules persist in sgs1, sgs1 top3, and top3 cells supports the notion that, in sgs1 cells, these X-molecules represent dHJs (substrate 1), whereas those in top3 cells represent hemicatenanes (intermediate 2). Furthermore, novel ternary and quaternary joint molecules and inter-sister dHJs persist in sgs1 mutant cells during meiosis [72]. However, recent observations of Nickoloff and colleagues that crossover and gene conversion tract length may be independent of helicase function, suggest that helicase activity is not involved in HJ branch migration [47]. Thus, intermediate 2 in our model may be a different type of X-shaped intermediate, such as a regressed replication fork.
4.2 Function of the The N terminus of Sgs1
We suggest that, in the absence of Top3, the Sgs1 helicase creates DNA lesions that are the primary cause of the top3 phenotype (intermediate 2* in Fig. 6B). Furthermore, we propose that these lesions also persist when the Sgs1-Top3 interaction is eliminated by deletion of the N terminus of Sgs1, even in the presence of a functional Top3 (2* in Fig. 6C). The similarities between the sgs1-NΔ and top3Δ mutant phenotypes are due to persistence of the same toxic lesions (Fig 6B and C), and in both cases loss of sgs1 helicase activity prevents their formation (Fig. 6D and E). We previously showed that this toxicity is at least partly a result of the activity of HR, since top3Δ defects can be suppressed by mutation of RAD51, 54, 55 and 57 and members of the Shu complex [37, 39, 69]. Our model predicts that mutation of the same genes would also improve the growth of sgs1-NΔ strains and that is indeed the case (data not shown).
Further support for the equivalence of top3 and sgs1-NΔ defects comes from genetic interactions with Pif1, a 5′ to 3′ helicase involved in nuclear and mitochondrial DNA metabolism. We previously demonstrated that overexpression of the Pif1 helicase suppresses slow growth, MMS and HU sensitivity of top3Δ, and that loss of pif1 is synthetically lethal with loss of top3 but not sgs1 [93]. Similarly, overexpression of Pif1 suppresses sgs1-NΔ slow growth, HU and MMS sensitivity. Moreover, sgs1-NΔ is synthetically lethal with pif1 and this lethality is dependent on the sgs1-NΔ helicase activity [93]. Thus, Pif1 helicase activity is essential to counteract Sgs1 helicase activity and promote survival in the absence of Top3, or the absence of the Sgs1-Top3 interaction.
If the only function of the N terminus of Sgs1 were to interact with Top3, then removal of this domain should not alter the phenotype of a top3Δ strain. However, the growth defect of sgs1-NΔ top3Δ strains is greater than top3Δ single mutant strains (Fig. 2A). One hypothesis for this difference is that removal of the interaction frees the Sgs1 helicase to act ectopically at inappropriate sites, thereby causing a more severe phenotype. Another possibility, first suggested by Mullen and colleagues, is that the N-terminal region of Sgs1 is involved in regulating its helicase activity and that removal of this domain creates a hyperactive allele [45]. Thus, in addition to eliminating the Top3 interaction, deletion of the N terminus could alter the enzymatic activity of the helicase (e.g., processivity) leading to an increase of the toxic intermediate.
Although there is no biochemical evidence for the N-terminal deletion directly influencing Sgs1 helicase activity, some genetic data support the notion that Sgs1-NΔ contains a hyperactive helicase. The DNA damage sensitivity of the sgs1-NΔ alleles can be suppressed by overexpressing Top3, and overexpression of Top3 improves the growth of sgs1-NΔ strains ([64] and data not shown) suggesting that the increase in topoisomerase activity counteracts the toxic effects of excess helicase activity. Additionally, functional interactions are known to regulate other RecQ helicases, such as the stimulation or attenuation of WRN and BLM helicase activities by interaction with TRF2 [94] or p53 [95], respectively.
4.3 Genetic interactions between sgs1 and top1
We propose that a subset of structures processed by the Sgs1-Top3 pathway can also be processed via an alternative Top1-dependent pathway (Fig. 6A). Unlike the epistatic relationship between sgs1Δ and top3Δ indicating that the two genes function in a single pathway (Fig. 2A), sgs1Δ and top1Δ exhibit a synergistic defect suggesting that these two genes function in separate but overlapping pathways (Fig. 2B and Fig. 6F). Notably, mutant sgs1 alleles cause different effects in a top1Δ background than in a top3Δ background. Although growth of both top3Δ and top1Δ strains is slowed by the sgs1-NΔ allele, mutation of the sgs1-NΔ helicase eliminates this effect in top3Δ strains but not in a top1Δ background (Fig. 2A and 2B and Fig. 6D and 6H). One interpretation of these data is that removal of the N terminus is responsible for the sgs1Δ top1Δ synergistic growth defect and loss of helicase activity is unimportant. However, Sgs1 helicase activity clearly plays a role in the absence of top1 as seen by comparing the growth of SGS1 top1Δ strains with sgs1-hd top1Δ strains (Fig. 2B and 6G). In addition, the N-terminal Top3 interaction of Sgs1 is important for growth in a top1Δ background regardless of helicase activity, as indicated by the growth difference between sgs1-hd top1Δ and sgs1-NΔ,hd top1Δ strains (Fig. 2B and 6G and 6H). These results are in agreement with previous studies, including observations that Sgs1 N-terminal fragments fully lacking the helicase domain improve the growth of sgs1Δ top1Δ strains ([41, 45] and our unpublished data).
4.4 Differences in allele sensitivities to HU and MMS
The effects of the sgs1 mutant alleles in this study exhibited slight differences for HU and MMS sensitivities. The increased sensitivity to HU seen in sgs1-hd is relieved by removal of the Top3 interaction (Fig. 5). Perhaps removing the interaction frees Top3 from binding catalytically inactive Sgs1, and improves the ability of Top3 to act in repairing HU damage as well as improving growth in the absence of top1. The observation that the sgs1-NΔ,hd allele has a greater MMS sensitivity than complete deletion of SGS1 suggests that the helicase-inactive allele is interfering with the cellular response to MMS damage in a Top3-independent fashion. The inactive sgs1-NΔ,hd could, for example, interact with other proteins to essentially sequester them in an inactive complex. Thus, a protein fragment that is unable to interact with Top3 and is incapable of catalyzing DNA unwinding likely preserves another aspect(s) of Sgs1 function, e.g., interaction with Top2 or Rmi1 [16, 18, 19]. In the null allele, this interaction is missing accounting for its less severe phenotype.
The response of sgs1 mutant alleles to HU and MMS are of interest, particularly in light of the discovery of a new class of Sgs1/Top3 suppressors known as the “Shu” genes (suppressor of sgs1 HU sensitivity) that includes Shu1, Shu2, Csm2, and Psy3 proteins [69, 96]. Mutation of any one of these genes suppresses the HU sensitivity of Top3 and Sgs1 but not their MMS sensitivity. These proteins interact in pair-wise combinations, and likely form a complex since the quadruple mutant has the same phenotype as any single or various double mutant combinations. [69, 96].
In conclusion, this study elucidates the central roles of two major elements of the Sgs1 protein: its catalytic helicase activity and its Top3 interaction. Our data highlight the significance of the physical interaction of Sgs1 and Top3 in suppressing deleterious Sgs1 helicase activity. Bloom, Werner, and Rothmund-Thompson syndromes are all distinct human diseases with an increased incidence of cancers caused by mutations in the human homologs of SGS1 and RECQ, and in many ways sgs1 mutants phenocopy several of these diseases. Thus, the evolutionary conservation of the RecQ family interactions with topoisomerase III isozymes raises the possibility that this protein-protein interaction may be important for tumor suppression in humans as well.
Acknowledgments
We thank Uffe Mortensen, Frederic Foucault, Greg Freyer, Morten Dunø, Michael Lisby, Erika Shor, Naz Erdeniz, Robert Reid, Marisa Wagner, and Lorraine Symington for helpful discussions concerning this work. We especially thank Kara Bernstein for careful reading of the manuscript, Qi Feng for her assistance in preparation of this manuscript and Ivana Sunjaveric for technical support. J.W. is particularly indebted to Shan Sockanathan, Floria Lupu, and Jonathan Eggenschweiler for providing additional materials and support throughout the course of this work. This work was supported by GM07088, a pre-doctoral training grant (J.W.) and by NIH grant GM50237 (R.R.).
Abbreviations
- MMS
methyl methanesulfonate
- HU
hydroxyurea
- HR
homologous recombination
- HJ
Holliday junction
- dHJ
double Holliday junction
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Umezu K, Nakayama K, Nakayama H. Escherichia coli RecQ protein is a DNA helicase. Proc Natl Acad Sci U S A. 1990;87:5363–5367. doi: 10.1073/pnas.87.14.5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Cobb JA, Bjergbaek L. RecQ helicases: lessons from model organisms. Nucleic Acids Res. 2006;34:4106–4114. doi: 10.1093/nar/gkl557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Sharma S, Doherty KM, Brosh RM., Jr. Mechanisms of RecQ helicases in pathways of DNA metabolism and maintenance of genomic stability. Biochem J. 2006;398:319–337. doi: 10.1042/BJ20060450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ellis NA, Groden J, Ye T-Z, Straughen J, Lennon DJ, Ciocci S, Proytcheva M, German J. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell. 1995;83:655–666. doi: 10.1016/0092-8674(95)90105-1. [DOI] [PubMed] [Google Scholar]
- [5].Yu CE, Oshima J, Fu YH, Wijsman EM, Hisama F, Alisch R, Matthews S, Nakura J, Miki T, Ouais S, Martin GM, Mulligan J, Schellenberg GD. Positional cloning of the Werner’s syndrome gene. Science. 1996;272:258–262. doi: 10.1126/science.272.5259.258. [DOI] [PubMed] [Google Scholar]
- [6].Kitao S, Shimamoto A, Goto M, Miller RW, Smithson WA, N.M L, Furuichi Y. Mutations in RECQL4 cause a subset of cases of Rothmund-Thomson syndrome. Nat. Genet. 1999;1:82–84. doi: 10.1038/8788. [DOI] [PubMed] [Google Scholar]
- [7].Siitonen HA, Kopra O, Kaariainen H, Haravuori H, Winter RM, Saamanen AM, Peltonen L, Kestila M. Molecular defect of RAPADILINO syndrome expands the phenotype spectrum of RECQL diseases. Hum Mol Genet. 2003;12:2837–2844. doi: 10.1093/hmg/ddg306. [DOI] [PubMed] [Google Scholar]
- [8].Van Maldergem L, Siitonen HA, Jalkh N, Chouery E, De Roy M, Delague V, Muenke M, Jabs EW, Cai J, Wang LL, Plon SE, Fourneau C, Kestila M, Gillerot Y, Megarbane A, Verloes A. Revisiting the craniosynostosis-radial ray hypoplasia association: Baller-Gerold syndrome caused by mutations in the RECQL4 gene. J Med Genet. 2006;43:148–152. doi: 10.1136/jmg.2005.031781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hanada K, Hickson ID. Molecular genetics of RecQ helicase disorders. Cell Mol Life Sci. 2007;64:2306–2322. doi: 10.1007/s00018-007-7121-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gangloff S, McDonald JP, Bendixen C, Arthur L, Rothstein R. The yeast type I topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog: a potential eukaryotic reverse gyrase. Mol Cell Biol. 1994;14:8391–8398. doi: 10.1128/mcb.14.12.8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wallis JW, Chrebet G, Brodsky G, Rolfe M, Rothstein R. A hyper-recombination mutation in S. cerevisiae identifies a novel eukaryotic topoisomerase. Cell. 1989;58:409–419. doi: 10.1016/0092-8674(89)90855-6. [DOI] [PubMed] [Google Scholar]
- [12].Arthur L. Ph.D. dissertation. Columbia University College of Physicians & Surgeons; New York, NY: 1991. Characterization of a novel eukaryotic topoisomerase (TOP3) in Saccharomyces cerevisiae that affects recombination and gene expression. [Google Scholar]
- [13].Saffi J, Pereira VR, Henriques JA. Importance of the Sgs1 helicase activity in DNA repair of Saccharomyces cerevisiae. Curr Genet. 2000;37:75–78. doi: 10.1007/s002940050012. [DOI] [PubMed] [Google Scholar]
- [14].Chakraverty RK, Kearsey JM, Oakley TJ, Grenon M, de La Torre Ruiz MA, Lowndes NF, Hickson ID. Topoisomerase III acts upstream of Rad53p in the S-phase DNA damage checkpoint. Mol Cell Biol. 2001;21:7150–7162. doi: 10.1128/MCB.21.21.7150-7162.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Tsai HJ, Huang WH, Li TK, Tsai YL, Wu KJ, Tseng SF, Teng SC. Involvement of topoisomerase III in telomere-telomere recombination. J Biol Chem. 2006;281:13717–13723. doi: 10.1074/jbc.M600649200. [DOI] [PubMed] [Google Scholar]
- [16].Watt PM, Louis EJ, Borts RH, Hickson ID. Sgs1: a eukaryotic homolog of E. coli RecQ that interacts with topoisomerase II in vivo and is required for faithful chromosome segregation. Cell. 1995;81:253–260. doi: 10.1016/0092-8674(95)90335-6. [DOI] [PubMed] [Google Scholar]
- [17].Gangloff S, de Massy B, Arthur L, Rothstein R, Fabre F. The essential role of yeast topoisomerase III in meiosis depends on recombination. EMBO J. 1999;18:1701–1711. doi: 10.1093/emboj/18.6.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Chang M, Bellaoui M, Zhang C, Desai R, Morozov P, Delgado-Cruzata L, Rothstein R, Freyer GA, Boone C, Brown GW. RMI1/NCE4, a suppressor of genome instability, encodes a member of the RecQ helicase/Topo III complex. EMBO J. 2005;24:2024–2033. doi: 10.1038/sj.emboj.7600684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mullen JR, Nallaseth FS, Lan YQ, Slagle CE, Brill SJ. Yeast Rmi1/Nce4 controls genome stability as a subunit of the Sgs1-Top3 complex. Mol Cell Biol. 2005;25:4476–4487. doi: 10.1128/MCB.25.11.4476-4487.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Frei C, Gasser SM. The yeast Sgs1p helicase acts upstream of Rad53p in the DNA replication checkpoint and colocalizes with Rad53p in S-phase-specific foci. Genes Dev. 2000;14:81–96. [PMC free article] [PubMed] [Google Scholar]
- [21].Fabre F, Chan A, Heyer WD, Gangloff S. Alternate pathways involving Sgs1/Top3, Mus81/ Mms4, and Srs2 prevent formation of toxic recombination intermediates from single-stranded gaps created by DNA replication. Proc Natl Acad Sci U S A. 2002;99:16887–16892. doi: 10.1073/pnas.252652399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ira G, Malkova A, Liberi G, Foiani M, Haber JE. Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell. 2003;115:401–411. doi: 10.1016/s0092-8674(03)00886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cobb JA, Bjergbaek L, Shimada K, Frei C, Gasser SM. DNA polymerase stabilization at stalled replication forks requires Mec1 and the RecQ helicase Sgs1. EMBO J. 2003;22:4325–4336. doi: 10.1093/emboj/cdg391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rockmill B, Fung JC, Branda SS, Roeder GS. The Sgs1 helicase regulates chromosome synapsis and meiotic crossing over. Curr Biol. 2003;13:1954–1962. doi: 10.1016/j.cub.2003.10.059. [DOI] [PubMed] [Google Scholar]
- [25].Liberi G, Maffioletti G, Lucca C, Chiolo I, Baryshnikova A, Cotta-Ramusino C, Lopes M, Pellicioli A, Haber JE, Foiani M. Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev. 2005;19:339–350. doi: 10.1101/gad.322605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chan KL, North PS, Hickson ID. BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J. 2007;26:3397–3409. doi: 10.1038/sj.emboj.7601777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cheok CF, Bachrati CZ, Chan KL, Ralf C, Wu L, Hickson ID. Roles of the Bloom’s syndrome helicase in the maintenance of genome stability. Biochem Soc Trans. 2005;33:1456–1459. doi: 10.1042/BST0331456. [DOI] [PubMed] [Google Scholar]
- [28].Raynard S, Bussen W, Sung P. A double Holliday junction dissolvasome comprising BLM, topoisomerase IIIalpha, and BLAP75. J Biol Chem. 2006;281:13861–13864. doi: 10.1074/jbc.C600051200. [DOI] [PubMed] [Google Scholar]
- [29].Wu L, Bachrati CZ, Ou J, Xu C, Yin J, Chang M, Wang W, Li L, Brown GW, Hickson ID. BLAP75/RMI1 promotes the BLM-dependent dissolution of homologous recombination intermediates. Proc Natl Acad Sci U S A. 2006;103:4068–4073. doi: 10.1073/pnas.0508295103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yin J, Sobeck A, Xu C, Meetei AR, Hoatlin M, Li L, Wang W. BLAP75, an essential component of Bloom’s syndrome protein complexes that maintain genome integrity. EMBO J. 2005;24:1465–1476. doi: 10.1038/sj.emboj.7600622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lee SK, Johnson RE, Yu SL, Prakash L, Prakash S. Requirement of yeast SGS1 and SRS2 genes for replication and transcription. Science. 1999;286:2339–2342. doi: 10.1126/science.286.5448.2339. [DOI] [PubMed] [Google Scholar]
- [32].Schmidt KH, Kolodner RD. Requirement of Rrm3 helicase for repair of spontaneous DNA lesions in cells lacking Srs2 or Sgs1 helicase. Mol Cell Biol. 2004;24:3213–3226. doi: 10.1128/MCB.24.8.3213-3226.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Torres JZ, Schnakenberg SL, Zakian VA. Saccharomyces cerevisiae Rrm3p DNA helicase promotes genome integrity by preventing replication fork stalling: viability of rrm3 cells requires the intra-S-phase checkpoint and fork restart activities. Mol Cell Biol. 2004;24:3198–3212. doi: 10.1128/MCB.24.8.3198-3212.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Mullen JR, Kaliraman V, Ibrahim SS, Brill SJ. Requirement for three novel protein complexes in the absence of the Sgs1 DNA helicase in Saccharomyces cerevisiae. Genetics. 2001;157:103–118. doi: 10.1093/genetics/157.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Fricke WM, Brill SJ. Slx1-Slx4 is a second structure-specific endonuclease functionally redundant with Sgs1-Top3. Genes Dev. 2003;17:1768–1778. doi: 10.1101/gad.1105203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kaliraman V, Mullen JR, Fricke WM, Bastin-Shanower SA, Brill SJ. Functional overlap between Sgs1-Top3 and the Mms4-Mus81 endonuclease. Genes Dev. 2001;15:2730–2740. doi: 10.1101/gad.932201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gangloff S, Soustelle C, Fabre F. Homologous recombination is responsible for cell death in the absence of the Sgs1 and Srs2 helicases. Nat Genet. 2000;25:192–194. doi: 10.1038/76055. [DOI] [PubMed] [Google Scholar]
- [38].Oakley TJ, Goodwin A, Chakraverty RK, Hickson ID. Inactivation of homologous recombination suppresses defects in topoisomerase III-deficient mutants. DNA Repair. 2002;1:463–482. doi: 10.1016/s1568-7864(02)00032-0. [DOI] [PubMed] [Google Scholar]
- [39].Shor E, Gangloff S, Wagner M, Weinstein J, Price G, Rothstein R. Mutations in Homologous Recombination Genes Rescue top3 Slow Growth in Saccharomyces cerevisiae. Genetics. 2002;162:647–662. doi: 10.1093/genetics/162.2.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lu J, Mullen JR, Brill SJ, Kleff S, Romeo AM, Sternglanz R. Human homologues of yeast helicase. Nature. 1996;383:678–679. doi: 10.1038/383678a0. [DOI] [PubMed] [Google Scholar]
- [41].Bennett RJ, Noirot-Gros MF, Wang JC. Interaction between yeast sgs1 helicase and DNA topoisomerase III. J Biol Chem. 2000;275:26898–268905. doi: 10.1074/jbc.M003137200. [DOI] [PubMed] [Google Scholar]
- [42].Gray MD, Shen JC, Kamath-Loeb AS, Blank A, Sopher BL, Martin GM, Oshima J, Loeb LA. The Werner syndrome protein is a DNA helicase. Nat Genet. 1997;17:100–103. doi: 10.1038/ng0997-100. [DOI] [PubMed] [Google Scholar]
- [43].Karow JK, Chakraverty RK, Hickson ID. The Bloom’s syndrome gene product is a 3′-5′ DNA helicase. J Biol Chem. 1997;272:30611–30614. doi: 10.1074/jbc.272.49.30611. [DOI] [PubMed] [Google Scholar]
- [44].Bennett RJ, Sharp JA, Wang JC. Purification and characterization of the Sgs1 DNA helicase activity of Saccharomyces cerevisiae. J Biol Chem. 1998;273:9644–9650. doi: 10.1074/jbc.273.16.9644. [DOI] [PubMed] [Google Scholar]
- [45].Mullen JR, Kaliraman V, Brill SJ. Bipartite structure of the SGS1 DNA helicase in Saccharomyces cerevisiae. Genetics. 2000;154:1101–1114. doi: 10.1093/genetics/154.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ui A, Satoh Y, Onoda F, Miyajima A, Seki M, Enomoto T. The N-terminal region of Sgs1, which interacts with Top3, is required for complementation of MMS sensitivity and suppression of hyper-recombination in sgs1 disruptants. Mol Genet Genomics. 2001;265:837–850. doi: 10.1007/s004380100479. [DOI] [PubMed] [Google Scholar]
- [47].Lo YC, Paffett KS, Amit O, Clikeman JA, Sterk R, Brenneman MA, Nickoloff JA. Sgs1 regulates gene conversion tract lengths and crossovers independently of its helicase activity. Mol Cell Biol. 2006;26:4086–4094. doi: 10.1128/MCB.00136-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Tong AH, Evangelista M, Parsons AB, Xu H, Bader GD, Page N, Robinson M, Raghibizadeh S, Hogue CW, Bussey H, Andrews B, Tyers M, Boone C. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001;294:2364–2368. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- [49].Wu L, Davies SL, North PS, Goulaouic H, Riou JF, Turley H, Gatter KC, Hickson ID. The Bloom’s syndrome gene product interacts with topoisomerase III. J Biol Chem. 2000;275:9636–9644. doi: 10.1074/jbc.275.13.9636. [DOI] [PubMed] [Google Scholar]
- [50].Wu L, Hickson ID. The Bloom’s syndrome helicase stimulates the activity of human topoisomerase III-alpha. Nucleic Acids Res. 2002;30:4823–4829. doi: 10.1093/nar/gkf611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Bennett RJ, Wang JC. Association of yeast DNA topoisomerase III and Sgs1 DNA helicase: Studies of fusion proteins. Proc Natl Acad Sci U S A. 2001;98:11108–11113. doi: 10.1073/pnas.201387098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Duno M, Thomsen B, Westergaard O, Krejci L, Bendixen C. Genetic analysis of the Saccharomyces cerevisiae Sgs1 helicase defines an essential function for the Sgs1-Top3 complex in the absence of SRS2 or TOP1. Mol Gen Genet. 2000;264:89–97. doi: 10.1007/s004380000286. [DOI] [PubMed] [Google Scholar]
- [53].Erdeniz N, Mortensen UH, Rothstein R. Cloning-free PCR-based allele replacement methods. Genome Res. 1997;7:1174–1183. doi: 10.1101/gr.7.12.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Reid R, Lisby M, Rothstein R. Cloning-free genome alterations in Saccharomyce cerevisiae using adaptamer-mediated PCR. Methods in Enzymology. 2002;350:258–277. doi: 10.1016/s0076-6879(02)50968-x. [DOI] [PubMed] [Google Scholar]
- [55].Reid RJD, Sunjevaric I, Keddache M, Rothstein R. Efficient PCR-based gene disruption in Saccharomyces strains using intergenic primers. Yeast. 2002;19:319–328. doi: 10.1002/yea.817. [DOI] [PubMed] [Google Scholar]
- [56].Thomas BJ, Rothstein R. Elevated recombination rates in transcriptionally active DNA. Cell. 1989;56:619–630. doi: 10.1016/0092-8674(89)90584-9. [DOI] [PubMed] [Google Scholar]
- [57].Zhao X, Muller EG, Rothstein R. A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol Cell. 1998;2:329–340. doi: 10.1016/s1097-2765(00)80277-4. [DOI] [PubMed] [Google Scholar]
- [58].Guthrie C, Fink GR. Guide to yeast genetics and molecular biology. In: Abelson JM, Simon MI, editors. Methods in Enzymology. Vol. 194. Academic Press, Inc.; San Diego: 1991. [PubMed] [Google Scholar]
- [59].Sherman F, Fink GR, Hicks JB. Methods in Yeast Genetics. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1986. [Google Scholar]
- [60].Klapholz S, Esposito RE. A new mapping method employing a meiotic rec-mutant of yeast. Genetics. 1982;100:387–412. doi: 10.1093/genetics/100.3.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Rothstein R, Helms C, Rosenberg N. Concerted deletions and inversions are caused by mitotic recombination between delta sequences in Saccharomyces cerevisiae. Molecular & Cellular Biology. 1987;7:1198–1207. doi: 10.1128/mcb.7.3.1198. published erratum appears in Mol Cell Biol 9 (1989) 3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].James P, Halladay J, Craig EA. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics. 1996;144:1425–1436. doi: 10.1093/genetics/144.4.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kurtz S, Lindquist S. Subcellular differentiation in sporulation yeast cells. Cell. 1986;45:771–779. doi: 10.1016/0092-8674(86)90791-9. [DOI] [PubMed] [Google Scholar]
- [64].Fricke WM, Kaliraman V, Brill SJ. Mapping the DNA topoisomerase III binding domain of the Sgs1 DNA helicase. J Biol Chem. 2001;276:8848–8855. doi: 10.1074/jbc.M009719200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Miyajima A, Seki M, Onoda F, Ui A, Satoh Y, Ohno Y, Enomoto T. Different domains of Sgs1 are required for mitotic and meiotic functions. Genes Genet Syst. 2000;75:319–326. doi: 10.1266/ggs.75.319. [DOI] [PubMed] [Google Scholar]
- [66].Mankouri HW, Morgan A. The DNA helicase activity of yeast Sgs1p is essential for normal lifespan but not for resistance to topoisomerase inhibitors. Mech Ageing Dev. 2001;122:1107–1120. doi: 10.1016/s0047-6374(01)00253-6. [DOI] [PubMed] [Google Scholar]
- [67].Onodera R, Seki M, Ui A, Satoh Y, Miyajima A, Onoda F, Enomoto T. Functional and physical interaction between Sgs1 and Top3 and Sgs1- independent function of Top3 in DNA recombination repair. Genes Genet Syst. 2002;77:11–21. doi: 10.1266/ggs.77.11. [DOI] [PubMed] [Google Scholar]
- [68].Ui A, Seki M, Ogiwara H, Onodera R, Fukushige S, Onoda F, Enomoto T. The ability of Sgs1 to interact with DNA topoisomerase III is essential for damage-induced recombination. DNA Repair (Amst) 2005;4:191–201. doi: 10.1016/j.dnarep.2004.09.002. [DOI] [PubMed] [Google Scholar]
- [69].Shor E, Weinstein J, Rothstein R. A genetic screen for top3 suppressors in Saccharomyces cerevisiae identifies SHU1, SHU2, PSY3 and CSM2: four genes involved in error-free DNA repair. Genetics. 2005;169:1275–89. doi: 10.1534/genetics.104.036764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Klein HL. Mutations in recombinational repair and in checkpoint control genes suppress the lethal combination of srs2Delta with other DNA repair genes in Saccharomyces cerevisiae. Genetics. 2001;157:557–565. doi: 10.1093/genetics/157.2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Rockmill B, Voelkel-Meiman K, Roeder GS. Centromere-proximal crossovers are associated with precocious separation of sister chromatids during meiosis in Saccharomyces cerevisiae. Genetics. 2006;174:1745–1754. doi: 10.1534/genetics.106.058933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Oh SD, Lao JP, Hwang PY, Taylor AF, Smith GR, Hunter N. BLM Ortholog, Sgs1, prevents aberrant crossing-over by suppressing formation of multichromatid joint molecules. Cell. 2007;130:259–272. doi: 10.1016/j.cell.2007.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Jessop L, Rockmill B, Roeder GS, Lichten M. Meiotic chromosome synapsis-promoting proteins antagonize the anti-crossover activity of sgs1. PLoS Genet. 2006;2:1402–1412. doi: 10.1371/journal.pgen.0020155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Watt PM, Hickson ID, Borts RH, Louis EJ. SGS1, a homologue of the Bloom’s and Werner’s syndrome genes, is required for maintenance of genome stability in Saccharomyces cerevisiae. Genetics. 1996;144:935–945. doi: 10.1093/genetics/144.3.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Spell RM, Jinks-Robertson S. Examination of the roles of Sgs1 and Srs2 helicases in the enforcement of recombination fidelity in Saccharomyces cerevisiae. Genetics. 2004;168:1855–1865. doi: 10.1534/genetics.104.032771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Branzei D, Foiani M. The DNA damage response during DNA replication. Curr Opin Cell Biol. 2005;17:568–575. doi: 10.1016/j.ceb.2005.09.003. [DOI] [PubMed] [Google Scholar]
- [77].Slater ML. Effect of reversible inhibition of deoxyribonucleic acid synthesis on the yeast cell cycle. Journal of Bacteriology. 1973;113:263–270. doi: 10.1128/jb.113.1.263-270.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Lundin C, North M, Erixon K, Walters K, Jenssen D, Goldman AS, Helleday T. Methyl methanesulfonate (MMS) produces heat-labile DNA damage but no detectable in vivo DNA double-strand breaks. Nucleic Acids Res. 2005;33:3799–3811. doi: 10.1093/nar/gki681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Wyatt MD, Pittman DL. Methylating agents and DNA repair responses: Methylated bases and sources of strand breaks. Chem Res Toxicol. 2006;19:1580–1594. doi: 10.1021/tx060164e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Onoda F, Seki M, Miyajima A, Enomoto T. Elevation of sister chromatid exchange in Saccharomyces cerevisiae sgs1 disruptants and the relevance of the disruptants as a system to evaluate mutations in Bloom’s syndrome gene. Mutat Res. 2000;459:203–209. doi: 10.1016/s0921-8777(99)00071-3. [DOI] [PubMed] [Google Scholar]
- [81].Heyer WD. A new deal for Holliday junctions. Nat Struct Mol Biol. 2004;11:117–119. doi: 10.1038/nsmb0204-117. [DOI] [PubMed] [Google Scholar]
- [82].Liu Y, West SC. Happy Hollidays: 40th anniversary of the Holliday junction. Nat Rev Mol Cell Biol. 2004;5:937–944. doi: 10.1038/nrm1502. [DOI] [PubMed] [Google Scholar]
- [83].Harmon FG, Kowalczykowski SC. RecQ helicase, in concert with RecA and SSB proteins, initiates and. Genes Dev. 1998;12:1134–1344. doi: 10.1101/gad.12.8.1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Garcia PL, Liu Y, Jiricny J, West SC, Janscak P. Human RECQ5beta, a protein with DNA helicase and strand-annealing activities in a single polypeptide. EMBO J. 2004;23:2882–2891. doi: 10.1038/sj.emboj.7600301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Karow JK, Constantinou A, Li JL, West SC, Hickson ID. The Bloom’s syndrome gene product promotes branch migration of Holliday junctions. Proc Natl Acad Sci U S A. 2000;97:6504–6508. doi: 10.1073/pnas.100448097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Constantinou A, Tarsounas M, Karow JK, Brosh RM, Bohr VA, Hickson ID, West SC. Werner’s syndrome protein (WRN) migrates Holliday junctions and co- localizes with RPA upon replication arrest. EMBO Rep. 2000;1:80–84. doi: 10.1093/embo-reports/kvd004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Sharma S, Sommers JA, Choudhary S, Faulkner JK, Cui S, Andreoli L, Muzzolini L, Vindigni A, Brosh RM., Jr. Biochemical analysis of the DNA unwinding and strand annealing activities catalyzed by human RECQ1. J Biol Chem. 2005;280:28072–28084. doi: 10.1074/jbc.M500264200. [DOI] [PubMed] [Google Scholar]
- [88].Bennett RJ, Keck JL, Wang JC. Binding specificity determines polarity of DNA unwinding by the Sgs1 protein of S. cerevisiae. J Mol Biol. 1999;289:235–248. doi: 10.1006/jmbi.1999.2739. [DOI] [PubMed] [Google Scholar]
- [89].LeRoy G, Carroll R, Kyin S, Seki M, Cole MD. Identification of RecQL1 as a Holliday junction processing enzyme in human cell lines. Nucleic Acids Res. 2005;33:6251–6257. doi: 10.1093/nar/gki929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Wu L, Hickson ID. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature. 2003;426:870–874. doi: 10.1038/nature02253. [DOI] [PubMed] [Google Scholar]
- [91].Plank JL, Wu J, Hsieh TS. Topoisomerase III-alpha and Bloom’s helicase can resolve a mobile double Holliday junction substrate through convergent branch migration. Proc Natl Acad Sci U S A. 2006;103:11118–11123. doi: 10.1073/pnas.0604873103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Mankouri HW, Hickson ID. Top3 processes recombination intermediates and modulates checkpoint activity after DNA damage. Mol Biol Cell. 2006;17:4473–4483. doi: 10.1091/mbc.E06-06-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Wagner M, Price G, Rothstein R. The absence of Top3 reveals an interaction between the Sgs1 and Pif1 DNA helicases in Saccharomyces cerevisiae. Genetics. 2006;174:555–573. doi: 10.1534/genetics.104.036905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Opresko PL, Laine JP, Brosh RM, Jr., Seidman MM, Bohr VA. Coordinate action of the helicase and 3′ to 5′ exonuclease of Werner syndrome protein. J Biol Chem. 2001;276:44677–44687. doi: 10.1074/jbc.M107548200. [DOI] [PubMed] [Google Scholar]
- [95].Yang Q, Zhang R, Wang XW, Spillare EA, Linke SP, Subramanian D, Griffith JD, Li JL, Hickson ID, Shen JC, Loeb LA, Mazur SJ, Appella E, Brosh RM, Jr., Karmakar P, Bohr VA, Harris CC. The processing of Holliday junctions by BLM and WRN helicases is regulated by p53. J Biol Chem. 2002;277:31980–31987. doi: 10.1074/jbc.M204111200. [DOI] [PubMed] [Google Scholar]
- [96].Mankouri HW, Ngo HP, Hickson ID. Shu Proteins Promote the Formation of Homologous Recombination Intermediates that Are Processed by Sgs1-Rmi1-Top3. Mol Biol Cell. 2007;18:4062–4073. doi: 10.1091/mbc.E07-05-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]