

Abstract
Certain peptides derived from the α1 domain of the major histocompatibility class I antigen complex (MHC-I) inhibit receptor internalization, increasing the steady-state number of active receptors on the cell surface and thereby enhancing the sensitivity to hormones and other agonists. These peptides self-assemble, and they also bind to MHC-I at the same site from which they are derived, suggesting that they could bind to receptor sites with significant sequence similarity. Receptors affected by MHC-I peptides do, indeed, have such sequence similarity, as illustrated here by insulin receptor (IR) and insulin-like growth factor-1 receptor. A synthetic peptide with sequence identical to a certain extracellular receptor domain binds to that receptor in a ligand-dependent manner and inhibits receptor internalization. Moreover, each such peptide is selective for its cognate receptor. An antibody to the IR peptide not only binds to IR and competes with the peptide but also inhibits insulin-dependent internalization of IR. These observations, and binding studies with deletion mutants of IR, indicate that the sequence QILKELEESSF encoded by exon 10 plays a key role in IR internalization. Our results illustrate a principle for identifying receptor-specific sites of importance for receptor internalization, and for enhancing sensitivity to hormones and other agonists.
Keywords: major histocompatibility complex, self-assembly, endocytosis, peptide, hormone
Our previous work showed that peptides derived from the α1 domain of major histocompatibility class I antigen complex (MHC-I) inhibit the internalization of certain receptors, including insulin receptor (IR) and insulin-like growth factor-1 receptor (IGF-1R) (1–6). A biological consequence is to increase the steady-state number of active receptors on the cell surface, and thus to make cognate agonists more potent. Bioactivity of these peptides is determined by their self-assembly and formation of aggregates with highly ordered conformation and β-sheet structure (7–9). Molecular self-assembly often is required for protein function (10) and specific molecular recognition (11). The same phenomenon has been demonstrated with some oligopeptides, in which a β-sheet motif has been exploited to achieve self-assembly into large aggregates (12, 13).
A biologically active peptide binds to its corresponding sequence in the α1 helix of MHC-I (4), suggesting that this self-interaction interferes with a putative role of MHC-I in the receptor internalization pathway (14, 15). [In this context, the term “MHC-I” may include both classical and nonclassical forms of the molecule (16, 17).] Alternatively, such a peptide might bind directly to a similar sequence on the receptor and thereby block interaction with MHC-I. The self-assembly property of a bioactive MHC-I peptide, its binding to identical or similar sequences, and its ability to inhibit internalization of certain receptors suggested the following hypothesis: A receptor that is affected, as described, by an MHC-I peptide contains a homologous sequence in its extracellular domain, which acts as a binding site for the α1 domain of MHC-I. If such a site on a receptor could be identified, a peptide of identical sequence would bind (as though by self-assembly), and thus inhibit internalization. The α1 sequence of MHC-I affects several receptors (1, 2, 4). We suppose that a synthetic peptide identical to the postulated receptor sequence might be more selective for its cognate receptor. As most of our previous work (3, 18) focused on the effects of MHC-I peptides on IR, we now have tested our hypothesis with IR and the closely related IGF-1R.
MATERIALS AND METHODS
Peptide Binding to Immunocaptured IR.
IR-derived peptide (IRp) and IR truncated peptide [IRp(trunc)] with the sequence YSQILKELEESSFR [to allow iodination, N-terminal aspartic acid in IRp(trunc) was replaced by tyrosine] were iodinated using a standard procedure (4). Assays of IRp binding to IR [overexpressed in Chinese hamster ovary (CHO) or NIH 3T3 cells] were performed using a modification of an immunocapture assay (19). Plates with immunocaptured receptors were incubated 90 min at 37°C with [125I]IRp (1 × 106 cpm/50 μl; sp.act. 200 Ci/mmol) and unlabeled insulin (as indicated) in Krebs–Ringer Hepes with 1% BSA binding buffer (80 mM NaCl/6 mM KCl/2 mM CaCl2/1 mM MgSO4/50 mM Hepes, pH 7.4/1% BSA), washed three times with Krebs–Ringer Hepes buffer, cut, and counted. Nonspecific binding was determined by competition with 500 nM unlabeled peptides. Binding of IGF-1 receptor-derived peptide (IGF-1Rp) to IR was performed under the same conditions. IGF-1R was immunocaptured using mAb Ab-1 (Oncogene) or 2C8, (Santa Cruz Biotechnology) and conditions as described for IR. IRp binding to IGF-1R was carried out in the presence or absence of 5 nM insulin or IGF-1.
Molecular Cloning and Mutant Expression.
Deletion mutants were constructed using the PCR and standard cloning techniques (20), and subcloned into a mammalian expression vector pCR3.1 (Invitrogen). A 2.0-kb fragment of the C-terminal portion of the gene (starting at L696) was created by using the PCR primers: IRC, 5′-CCCGTCTAGATAGGCACTGTTAGGAAGG-3′ and IRMP2, 5′-ATGGAGGAGTACTCGTTTAAAAAGACGTTTGGGCCCTACCTGCACAACGTTAACTTCGTCCCCAGGCCATCTCGG-3′. Underlined bases represent the exchanged nucleotides used to introduce two new restriction sites: HpaI and XbaI. Upstream sequences, (N348-L693 for Δ22 and N348-T704 for Δ11), were obtained by two different set of primers: IRNP and IRMP1 for Δ11 construct and IRNP and IRMPH for Δ22. IRNP, 5′-AACAATCTGGCAGCTGAGCTAGAAGCC-3′; IRMP1, 5′-CGTCTTTTTAAACGAGTACTCCTCCATATGCTTATCGATCTGAGAGTCCGTCTGTCGACAGGAGCAGCATTCGCCG-3′; IRMPH, 5′-CAGGATGTTAACGTCAGTCTTTGGACAGGAGCA-3′. New restriction sites used for the further cloning (DraI and HpaI) were introduced by IRMP1 and IRMPH primers, respectively. The entire ORF of the wild-type gene and the mutants was sequenced. CHO cells were stably transfected using Lipofectin reagent (GIBCO/BRL). Pools of transfected cells grown in F12 containing 10% calf serum, 2 mM glutamine, and 600 mg/liter G418 were expanded in 6-well plates, and stable transfectants were identified by their ability to synthesize IR.
Insulin Binding and Internalization.
Procedures to measure insulin binding and ligand-induced receptor internalization have been described (18). The stably transfected CHO cells were grown to confluence, dissociated from the flask, washed once with PBS and twice with KRHB binding buffer. Aliquots (50 μl) were preincubated at 37°C for 45 min, followed by the addition of 50 μl buffer containing about 100 pM [125I]insulin or [125I]IGF-1 (sp.act. 2,000 Ci/mmol, Amersham) and a peptide, and were incubated 35 min at 37°C. Nonspecific binding was defined as cell-associated radioactivity when 1 μM unlabeled insulin or 0.1 μM unlabeled IGF-1 was added together with the corresponding tracer. Internalized tracer was defined as cell associated radioactivity after acid wash (pH = 4.0) for 5 min on ice. Cell surface binding was calculated as total cell-associated minus internalized radioactivity. Cells were spun through an oil mixture (dibutyl phthalate/phthalic acid bis(2-ethylhexyl ester) = 7:3; Sigma) to remove free ligand. In the experiments where αIRp antibody was used, cells were preincubated with 1:1,000 dilution of antibody or the preimmune sera for 1 hr at 37°C.
Glucose Uptake.
Procedures to measure insulin binding and insulin-stimulated glucose uptake in rat adipocytes are described elsewhere (3, 18). Single-cell suspensions of adipocytes were incubated at 37°C for 30 min in the absence or presence of IRp and harvested on silicone oil, and the amount of cell-associated radiolabel was determined. [14C]glucose uptake assays were as described elsewhere (1, 3). Cells were incubated with [14C]glucose 30 min before harvest on silicone oil, and the amount of incorporated glucose was determined as described.
Immunoprecipitations and Western Blot Analysis.
Cells in late growth phase were solubilized in lysis buffer (19) and immunoprecipitated overnight at 4°C with 10 μg each of the anti-IR mAb 29B4 (Santa Cruz Biotechnology.) bound to protein G Sepharose (Pharmacia). Samples were electrophoresed on 8% SDS-polyacrylamide gels and transferred to Immobilon-P membrane (Millipore). The blots were incubated with polyclonal antibody C-19 against the IR β chain (Santa Cruz Biotechnology), anti-phosphotyrosine, 4G10 (Upstate Biotechnology), or rabbit polyclonal anti-IRp antibody in 1:1,000 dilution. This antibody was produced by Zymed laboratories using the full-length IRp as an antigen. For the immunoprecipitation of IGF-1R, 2C8 antibody was used (Santa Cruz Biotechnology), whereas N-20 antibody (Santa Cruz Biotechnology) was applied for Western blot analysis.
RESULTS
IR and IGF-1 Receptor Sequences with Homology to Bioactive MHC-I Peptide Sequence.
The entire IR sequence was searched for a match to the bioactive MHC-I peptide Dk-(61–85) (3). A single match was found, with 57% similarity and 35% identity (Fig. 1); it was in the C-terminal extracellular portion of the α subunit of IR at residues 687–710, encoded by exon 10 (21, 22), in close proximity to the membrane and different from the ligand binding site. The result of a similar search on IGF-1R also is shown in Fig. 1. Again, only a single match was found, in a domain corresponding to that on IR. It is noteworthy that the MHC-I peptide matching domains on IR and IGF-1R, respectively, are considerably less alike (47% identity) than the IR and IGF-1R sequences as a whole (67% identity) (23). Thus, although these receptor sequences are similar to the same MHC-I sequence, they nevertheless differ considerably from each other. Corresponding peptides (designated IRp and IGF-1Rp) were synthesized, and their bioactivity was analyzed. A shorter, 14-residue version of IRp, [IRp(trunc),DSQILKELEESSFR], was synthesized, corresponding to residues 689–702 in IR; this segment retains most of the sequence that is identical to Dk-(61–85) (sequence identity 50%).
Figure 1.

Localization and amino acid sequences for receptor sites with homology to MHC-I peptide. Amino acid sequences, identities, similarities: MHC-I peptide, IR, IGF-1R. Sequence alignment of MHC-I-derived peptide Dk-(61–85) (3) to IR (21, 22) and IGF-1R (23). Comparison was performed using the default parameters of bestfit program of the GCG Software package, University of Wisconsin, which identified the portion of each compared sequence that shared highest similarity. Positions of the matching sequences in MHC-I, IR, and IGF-1R are indicated in parentheses. Black boxes indicate identity, gray boxes indicate similarity. Data for similarity include identity.
Binding Sites for IRp and IGF-1Rp.
Given that the newly synthesized peptides include all the structure required for self-assembly, we would expect each peptide to bind to its identical sequence on its cognate receptor. Radiolabeled IRp (radioiodinated at Y708) was used to study binding of IRp to IR, partially purified from CHO-T cells by immunocapture (19); receptor thus immobilized is fully functional in ligand binding and kinase activity.
Fig. 2 shows that IRp and IRp(trunc) bind to IR but only when insulin is added. Both peptides reached maximum binding at insulin concentration about 3 nM and showed little or no binding if the hormone was omitted. Fig. 2 (Inset) further demonstrates the insulin-dependency of the peptide binding. We used NIH 3T3 cell line that overexpresses IR, with point mutation at S323L, which interferes with the capacity to bind insulin (24). As compared with wild type, this mutant also has lost the ability to bind IRp. The binding selectivities of IRp and IGF-1Rp are shown in Table 1; each peptide binds preferentially to its cognate receptor. This is seen also in competition experiments, where 73% ± 4% (mean ± SEM, n = 5) of radiolabeled IRp binding could be prevented by competition with 500 nM unlabeled IRp, but only 14% ± 6% with unlabeled IGF-1Rp. The insulin dependence of IRp and IRp(trunc) binding implies that the insulin-induced conformational change of IR exposes the IRp binding site.
Figure 2.
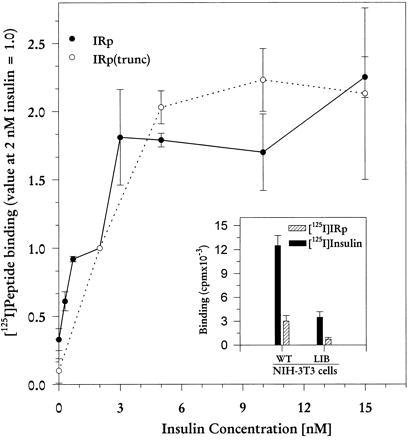
Peptide binding to partially purified, immunocaptured receptors. 125I-labeled IRp and IRp(trunc) (106 cpm/50 μl) were used (4) for binding to IR overexpressed in CHO cells. To compare numerous experiments in which different batches of labeled peptides were used, data were normalized to binding at 2 nM insulin. Data are means ± SEM for three to five experiments, with each point done in triplicate. (Inset) Binding to IR variants overexpressed in NIH 3T3 cells. WT, wild-type IR. LIB, low insulin binding IR with point mutation at S323L (24). The immunocaptured receptors were stimulated with 2 nM insulin, and the radiolabeled peptide was bound as described. The same conditions were used for insulin binding, except that radioactive insulin (30,000 cpm/50 μl) was added. For competition, 100 nM insulin was used. Data are means ± SEM for n = 3, with each data point done in triplicate.
IR Deletion Mutants and IRp Binding.
To further test if IRp binds to the identical sequence on IR, two deletion mutants of IR, Δ11 and Δ22, were constructed (Fig. 3A) and stably overexpressed in CHO cells. These internal in-frame deletions did not impair the posttranslational processing or cell surface expression of IR; Western blot analysis of immunoprecipitated IR showed equal levels of wild-type and mutant overexpression (Fig. 3A densitometry data). The deletions did not affect insulin-mediated activation of IR tyrosine kinase, as seen from Western blots for phosphorylation of the IR β chain. Moreover, the deletions did not significantly compromise the receptor’s ability to bind insulin with apparent Kd about 2 nM, as in the wild type, as determined from binding isotherms. The corresponding Bmax values were only slightly decreased in the two mutants, more in Δ22 than in Δ11.
Figure 3.
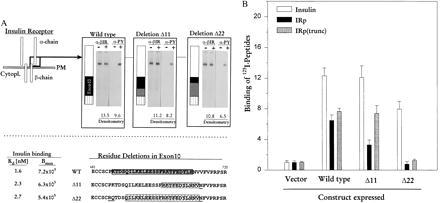
Deletions introduced in the IR gene and their effects on peptide binding. (A) (Upper) The deletions are depicted schematically and below as primary sequence data. Open black square at C-terminal end of α chain depicts the position of the deletions, with enlargement of that region shown at right. Black box represents exon 10, gray box indicates extent of deletions (11 or 22 residues), and striped box shows position of exon 11 (22). Western blot analysis was performed as described. α-βIR, anti-IR β chain antibody. α-PY, anti-phosphotyrosine antibody. Insulin stimulation of cells is indicated by (−) or (+). Densitometry data represent multiples of IR expression over endogenous levels of IR in cells transfected with pCR3.1 vector alone. (Lower) Primary sequence of IR, residues 681–720 and internal deletions. Hatched boxes indicate deletions for Δ11 (R702-V712) and Δ22 (Q 691-V712). Underlined amino acids represent point mutations introduced to create the necessary restriction sites. Shaded box in wild type is sequence of IRp. Kd and Bmax are calculated values derived from binding isotherms in four different experiments. (B) Binding of [125I]insulin, [125I]IRp, and [125I]IRp(trunc) to IR mutants expressed in CHO cells. A number of clones were generated for wild type, the deletion variants, and the vector alone. Binding was tested on three clones each for wild type, Δ11 and Δ22, one clone for vector alone. Histogram shows the binding to one clonal population each, in which vector, wild type, Δ11 or Δ22 variants were expressed; other clones differed by less than 10%. Peptide binding data represent multiples of binding over the endogenous levels; vector is given the value 1.0. All cells expressing IR variants bound 49–65% of added [125I]insulin, which corresponds to approximately 3–5 × 105 receptors per cell. Cells transfected with vector alone bound 5–8% of added [125I]insulin, corresponding to about 1 × 103 receptors per cell. Immunocaptured receptors were stimulated with 5 nM insulin for binding assays. Data are mean ± SEM (n = 9) for insulin and IRp binding, and n = 4 for IRp(trunc) binding with each data point done in triplicate.
Specific binding of insulin, IRp, and IRp(trunc) was measured in several arbitrarily selected cell pools, using the immunocapture procedure as described in Materials and Methods (Fig. 3B). Receptors carrying the 11-residue in-frame deletion bound 35% less IRp, but bound IRp(trunc) almost as well as did wild-type receptors. Binding of IRp and IRp(trunc) was completely abolished in the 22-residue deletion; the small amount bound represents no more than would be expected from endogenous levels of IR. These results indicate that IRp does, indeed, bind to its identical sequence on IR. They also strongly suggest that the site of peptide-receptor interaction corresponds to the 14 residues contained in IRp(trunc), most likely the sequence QILKELEESSF [a shorter sequence than IRp(trunc) itself], which is contained in the Δ11 mutant and in the truncated peptide, and which is 56% identical to the MHC-I peptide.
Bioactivity and Selectivity of IRp and IGF-1Rp.
To examine the biological selectivity of IRp for IR and IGF-1Rp for IGF-1R, we compared their effects on ligand-induced internalization in CHO cells overexpressing each receptor. IRp inhibited internalization of IR in a dose-related manner, whereas IGF-1Rp was much less effective (Fig. 4A). IGF-1Rp strongly inhibited internalization of IGF-1R, whereas IRp had no effect (Fig. 4B). The bioactive MHC-I peptide affected internalization of both IR and IGF-1R, but without selectivity; at 30 μM, Dk-(61–85) reduced internalization 37% ± 5% for IR, 43% + 9% for IGF-1R (mean ± SEM, n = 3), as previously published (3, 4).
Figure 4.
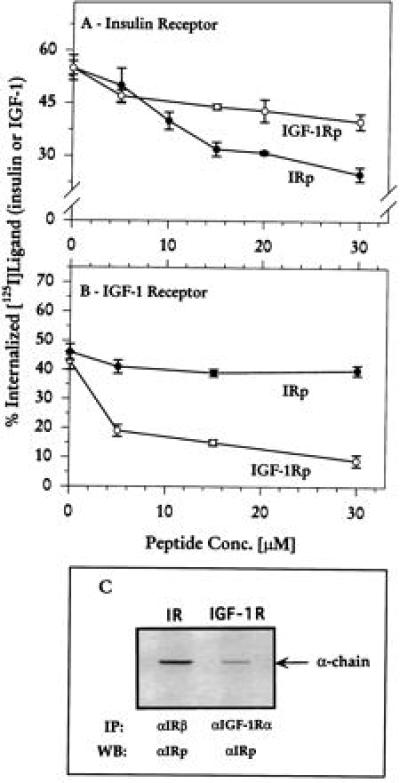
Selectivity of IRp and IGF-1Rp. (A and B) Internalization of IR and IGF-1R, respectively, was measured in CHO cells overexpressing each receptor (26, 27), as described. Data are mean ± SEM of triplicates from four to six experiments. (C) Analysis of cell lysates overexpressing IR or IGF-1R. Receptors were immunoprecipitated using the corresponding antibody and the blot was probed with αIRp antibody. IP, immunoprecipitation; WB, Western blot; αIRβ, antibody against IR β chain; αIRp, antibody against IRp; αIGF-1Rα antibody against IGF-1R α chain.
To further characterize the receptor site on IR, a polyclonal rabbit antibody was raised against IRp (αIRp, produced by Zymed Laboratories). On Western blots, this antibody recognizes the α chain of IR but not of IGF-1R (Fig. 4C). The band seen in the IGF-1R lane probably represents endogenous level of IR α chain. Moreover, the antibody selectively inhibits the ligand-dependent internalization of IR; 31% ± 2% for IR and 49% ± 1.5% for IGF-1R (mean + SEM, n = 4). Preimmune serum showed no effect on IR (63% ± 2%) or IGF-1R (53% ± 3%) internalization.
IRp enhanced the effect of insulin on glucose uptake in fresh rat adipocytes, in a dose-dependent manner. Maximal effect was obtained at about 3 μM peptide concentration, with EC50 300–400 nM (Fig. 5A), and without effect on insulin binding (Fig. 5B). Maximal enhancement was obtained after 120-min incubation (data not shown).
Figure 5.
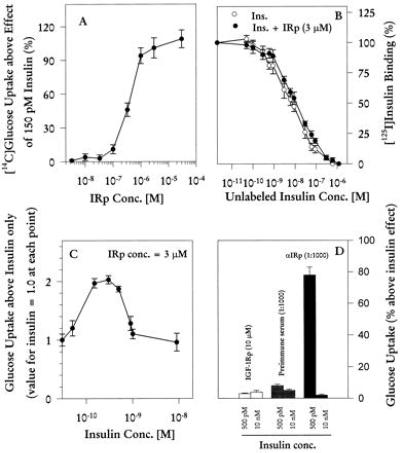
Biological activity of IRp in rat adipocytes. (A) Dose response for effect of IRp at 150 pM insulin after 90-min incubation, as percent increase over insulin alone. Incorporation of [14C]glucose into cells was measured. Data are mean ± SEM of four experiments, each point done in triplicate. (B) Competition curves for binding of [125I]insulin in the absence (○) or presence (•) of 3 μM IRp. Each point is mean ± SEM of four to six experiments. In all experiments, measurements at each point were done in triplicate. (C) Relative effect of 3 μM IRp on insulin-stimulated glucose uptake at various insulin concentrations, measured after 90-min incubation. Uptake without peptide at each insulin concentration is given the value 1.0, and each point is mean ± SEM of seven experiments. (D) Effect of IGF-1Rp and αIRp antibody in glucose uptake. Experiments were performed as described in C, except only two insulin concentrations were tested. Data are mean ± SEM of four experiments.
The IRp-induced enhancement of insulin action was seen only at submaximal insulin concentrations (e.g., 150 pM; Fig. 5C), in contrast to MHC-I peptides, which increase glucose uptake both at submaximal and maximal insulin concentrations. At 10 nM insulin, for example, IRp-induced glucose uptake was no different from that with insulin alone, whereas MHC-I peptide enhances glucose uptake about 50% further (2). This difference reflects the selectivity of IRp in inhibiting internalization of IR, whereas MHC-I peptides affect both IR and the glucose transporter GLUT4 (1, 6).
Further evidence of IRp selectivity was obtained in glucose uptake experiments with IGF-1Rp and the αIRp antibody. IGF-1Rp had very little effect at several insulin and peptide concentrations (Fig. 5D and data not shown), whereas the antibody enhanced insulin-stimulated glucose uptake as well as did IRp.
The concentration of IRp required for the effect on receptor internalization in cultured cells was about 7-fold higher than for enhancing glucose uptake in the adipocyte studies. This apparent potency difference probably is due to peptide degradation in the two kinds of cells. Using 125I-labeled IRp, we found that only about 30% was undegraded in the experiments with CHO cells, whereas more than 95% remained intact after 120-min incubation with adipocytes.
Identification of IR Sequence Important for Receptor Internalization.
The ligand-dependent internalization of the Δ22 deletion mutant is substantially reduced and the effect of insulin on glucose uptake is enhanced as shown on Table 2. The Δ11 deletion mutant shows an intermediate internalization but no difference in enhancement of glucose uptake compared with the wild type.
Recognition of the specific sequence on IR required for receptor internalization also was confirmed with αIRp antibody, which recognizes the α chain of wild-type IR and of the Δ11 deletion, but not of Δ22 (Fig. 6). Western blot analysis confirms that antibody binds to IR in an insulin-dependent manner, and to wild-type and Δ11 mutant, but not to Δ22. We conclude that the binding site for the antibody is identical to (or very close to) that for IRp—presumably the sequence maintained on Δ11 but deleted on Δ22, namely, QILKELEESSF. We suggest that this extracellular sequence involved in IR internalization affects endocytosis through a different mechanism than the motif GPLYXXXNPEY, required for ligand-dependent internalization and present in the cytoplasmic domain of IR (25).
Figure 6.
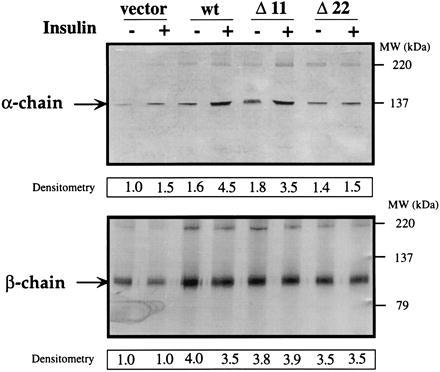
Recognition of IR α chain by anti-peptide antibody. Analysis by immunoprecipitation and Western blot of CHO cells overexpressing different IR variants were performed as described. Blots were probed with αIRp (Upper) and C-19, an anti-IR β-chain antibody (Lower). Insulin stimulation of cells is indicated by (−) or (+). Migration positions of IR α or β chain are indicated by arrows, molecular weight markers are shown on the right. Densitometry data represent multiples of IR expression over endogenous levels of IR in cells transfected with vector alone and not stimulated with insulin. The amount of α or β chain was determined by scanning the blots, which then were analyzed using NIH Image 1.55 software. Data representative of four experiments are shown.
DISCUSSION
Our previous work showed that Dk-(61–85) and related peptides inhibit receptor internalization, increasing the steady-state number of active receptors on the cell surface, and resulting in increased sensitivity to a hormone (1, 4). As a result of the tendency of these bioactive peptides to self-assemble, they bind to an identical (or nearly identical) sequence, both when interacting with the MHC-I protein and when forming peptide self-aggregates (8, 9). We hypothesized that just as peptides with the same sequence as α1 bind to α1, receptors affected by the peptides might have extracellular domains that are homologous to MHC-I α1 domain.
Accordingly, we searched IR and IGF-1R for sequence similarity to Dk-(61–85). We identified such sites in the extracellular domains; despite their similarity to the MHC-I peptide, the domains on IR and IGF-1R do not share a common motif. Further, recent work (unpublished data) has shown that growth hormone receptor, leptin receptor, and some other membrane receptors also contain extracellular domains homologous to the α1 domain of MHC-I, but again without a common motif among them. Moreover, as with IR and IGF-1R, their internalization is inhibited selectively by cognate peptides.
In summary, we show here: (i) A synthetic peptide, IRp, derived from an IR sequence homologous to Dk-(61–85) binds to IR in an insulin-dependent manner. IR bearing a point mutation, which no longer binds insulin, also has lost the ability to bind IRp. IRp inhibits internalization of IR without affecting insulin binding, thus increasing the steady-state number of active IR on the cell surface and enhancing insulin-stimulated glucose uptake in rat adipocytes. (ii) Deletions in this same extracellular domain of IR reduce the binding of IRp partially or wholly (according to the extent of the deletion), suggesting that the binding site is contained in the deleted domain. (iii) IRp and IGF-1Rp (the peptide similarly derived from IGF-1R) are selective for their cognate receptors, both in binding and in inhibiting internalization. (iv) An antibody to IRp binds to IR (but not to a deletion mutant of IR or to IGF-1R), inhibits insulin-induced internalization of IR, enhances insulin-stimulated glucose uptake, and competes with IRp for binding. Taken together, these findings are decisive in showing that a peptide of identical sequence to a certain extracellular domain of either IR or IGF-1R can bind to the same sequence on the cognate receptor, consequently inhibiting receptor internalization. For IR, this important sequence is likely to be QILKELEESSF. Furthermore, our observations on the ligand dependency of binding imply that this binding site for IRp is accessible only in the ligand-induced conformation of the receptor.
One way to interpret our findings is to suppose that MHC-I itself plays some role in receptor endocytosis by interacting with a receptor and that this interaction is competitively blocked by the bioactive peptides. The underlying principle seems to be self-assembly, i.e., interaction of identical or similar sequences. The affected receptors do not share a single common motif for interaction with the α1 sequence of MHC-I, but the latter—like a master key—can recognize distinctively different sequences among different receptors. We suggest that the principle and method described here may be generally applicable to identifying receptor domains that play a role in receptor endocytosis. Novel compounds that interact with such domains may have therapeutic potential for selectively altering the steady-state number of chosen cell surface receptors, and thus enhancing sensitivity to hormones or other agonists.
Table 1.
Selectivity of IRp and IGF-1Rp binding
| Peptide | Specific peptide binding, cpm
|
Selectivity
|
|
|---|---|---|---|
| IR | IGF-1R | IR/IGF-1R | |
| IRp | 4,590 ± 424 | 780 ± 140 | 5.9 |
| IGF-1Rp | 674 ± 171 | 3,380 ± 484 | 0.2 |
Binding of radiolabeled IRp and IGF-1Rp was performed in the presence of 5 nM insulin. Data are mean ± SEM of six experiments, each point done in triplicate.
Table 2.
Insulin-induced IR internalization and glucose uptake in wild-type and deletion mutants
| Cell type | Internalization, % at 40 min* | Glucose uptake at 10 nM insulin, basal = 1.0† |
|---|---|---|
| Wild type | 36 ± 3.5 | 1.43 ± 0.05 |
| Δ11 mutant | 27 ± 3.3 | 1.48 ± 0.05 |
| Δ22 mutant | 14 ± 1.7 | 2.28 ± 0.17 |
Insulin-induced internalization was measured as described in Materials and Methods. Data are mean ± SEM of four experiments, each point done in triplicate.
Insulin-stimulated glucose uptake was measured upon incubation of the cells for 1 hr with 10 nM insulin followed by additional 30-min incubation with [14C] glucose. Data are mean ± SEM of five to seven experiments, each point done in triplicate.
Acknowledgments
We thank Drs. Carol Haft-Renfrew, Simeon I. Taylor, and Richard A. Roth for providing us with cDNA clones and cell lines; Dr. Grant Hendrick for performing HPLC analyses; and Amy E. Sawyer and Alan F. Paredes for excellent technical assistance.
ABBREVIATIONS
- MHC-I
major histocompatibility complex class I protein
- IR
insulin receptor
- IRp
insulin receptor-derived peptide
- IRp(trunc)
IR truncated peptide
- IGF
insulin-like growth factor
- IGF-1R
IGF-1 receptor
- IGF-1Rp
IGF-1 receptor-derived peptide
- CHO
Chinese hamster ovary
References
- 1.Stagsted J, Olsson L, Holman G D, Cushman S W, Satoh S. J Biol Chem. 1993;268:22809–22813. [PubMed] [Google Scholar]
- 2.Stagsted J, Ziebe S, Satoh S, Holman G D, Cushman S W, Olsson L. J Biol Chem. 1993;268:1770–1774. [PubMed] [Google Scholar]
- 3.Stagsted J, Reaven G M, Hansen T, Goldstein A, Olsson L. Cell. 1990;62:297–307. doi: 10.1016/0092-8674(90)90367-n. [DOI] [PubMed] [Google Scholar]
- 4.Olsson L, Goldstein A, Stagsted J. Proc Natl Acad Sci USA. 1994;91:9086–9090. doi: 10.1073/pnas.91.19.9086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hsu D, Olefsky J M. Endocrinology. 1993;133:1247–1251. doi: 10.1210/endo.133.3.8365366. [DOI] [PubMed] [Google Scholar]
- 6.Shibata H, Suzuki Y, Omaka W, Tanaka S, Kojima I. J Biol Chem. 1995;270:11489–11495. doi: 10.1074/jbc.270.19.11489. [DOI] [PubMed] [Google Scholar]
- 7.Stagsted J, Mapelli C, Meyers C, Matthews B W, Anfinsen C B, Goldstein A, Olsson L. Proc Natl Acad Sci USA. 1993;90:7686–7690. doi: 10.1073/pnas.90.16.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stagsted J, Baase W A, Goldstein A, Olsson L. J Biol Chem. 1991;266:12844–12847. [PubMed] [Google Scholar]
- 9.Weaver L, Stagsted J, Behnke O, Matthews B W, Olsson L. J Struct Biol. 1996;117:165–172. doi: 10.1006/jsbi.1996.0080. [DOI] [PubMed] [Google Scholar]
- 10.Warnock D E, Hinshaw J E, Schmid S L. J Biol Chem. 1996;271:22310–22314. doi: 10.1074/jbc.271.37.22310. [DOI] [PubMed] [Google Scholar]
- 11.Lees J F, Tasab M, Bulleid N J. EMBO J. 1997;16:908–916. doi: 10.1093/emboj/16.5.908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krejchi M T, Atkins E D T, Waddon A J, Fournier M J, Mason T L, Tirrell D A. Science. 1994;265:1427–1432. doi: 10.1126/science.8073284. [DOI] [PubMed] [Google Scholar]
- 13.Aggeli A, Bell M, Boden N, Keen J N, Knowles P F, McLeish T C B, Pitkeathly M, Radford S E. Nature (London) 1997;386:259–262. doi: 10.1038/386259a0. [DOI] [PubMed] [Google Scholar]
- 14.Simonsen M, Olsson L. Ann Immunol (Paris) 1983;134D:85–92. doi: 10.1016/s0769-2625(83)80059-2. [DOI] [PubMed] [Google Scholar]
- 15.Due C, Simonsen M, Olsson L. Proc Natl Acad Sci USA. 1986;83:6007–6011. doi: 10.1073/pnas.83.16.6007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raulet D H. Adv Immunol. 1994;55:381–421. doi: 10.1016/s0065-2776(08)60514-3. [DOI] [PubMed] [Google Scholar]
- 17.Wang C R, Fischer Lindahl K. Proc Natl Acad Sci USA. 1993;90:2784–2788. doi: 10.1073/pnas.90.7.2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stagsted J, Hansen T, Roth R A, Goldstein A, Olsson L. J Pharmacol Exp Ther. 1993;267:997–1001. [PubMed] [Google Scholar]
- 19.Steele-Perkins G, Roth R A. J Biol Chem. 1990;265:9458–9463. [PubMed] [Google Scholar]
- 20.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, New York: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 21.Ullrich A, Bell J R, Chen E Y, Herrera R, Petruzelli L M, Dull T J, Gray A, Coussens L, Liao Y C, Tsubokawa M, Mason A, Seeburg P H, Grunfeld C, Rosen O M, Ramachandran J. Nature (London) 1985;313:756–761. doi: 10.1038/313756a0. [DOI] [PubMed] [Google Scholar]
- 22.Ebina Y, Ellis L, Jarnagin K, Edery M, Graf L, Clausner E, Ou J, Masiarz F, Kan Y W, Goldfine I D, Roth R A, Rutter W. Cell. 1985;40:747–758. doi: 10.1016/0092-8674(85)90334-4. [DOI] [PubMed] [Google Scholar]
- 23.Ullrich A, Gray A, Tam A W, Yang-Feng T, Tsubokawa M, Collins C, Henzel W, Le Bon T, Kathuria S, Chen E, Jacobs S, Francke U, Ramachandran J, Fujita-Yamaguchi Y. EMBO J. 1986;5:2503–2512. doi: 10.1002/j.1460-2075.1986.tb04528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roach P, Zick Y, Formisano P, Accili D, Taylor S I, Gorden P. Diabetes. 1994;43:1096–1102. doi: 10.2337/diab.43.9.1096. [DOI] [PubMed] [Google Scholar]
- 25.Rajagopalan M, Neidigh J L, McClain D A. J Biol Chem. 1991;266:23068–23073. [PubMed] [Google Scholar]
- 26.Zhang B, Roth R. Proc Natl Acad Sci USA. 1991;88:9858–9862. doi: 10.1073/pnas.88.21.9858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ellis L, Clausner E, Morgan D O, Edery M, Roth R A, Rutter W J. Cell. 1986;45:721–732. doi: 10.1016/0092-8674(86)90786-5. [DOI] [PubMed] [Google Scholar]