

Abstract
Steroidogenic factor 1 (SF1, NR5A1) is a nuclear receptor that regulates multiple genes involved in adrenal and gonadal development, steroidogenesis, and the reproductive axis. Human mutations in SF1 were initially found in two 46,XY female patients with severe gonadal dysgenesis and primary adrenal failure. However, more recent case reports have suggested that heterozygous mutations in SF1 may also be found in patients with 46,XY partial gonadal dysgenesis and underandrogenization but normal adrenal function. We have analyzed the gene encoding SF1 (NR5A1) in a cohort of 27 patients with 46,XY disorders of sex development (DSD) from the German network of DSD. Heterozygous SF1 mutations were found in 5 out of 27 (18.5%) of cases. Four patients with SF1 mutations presented with the similar phenotype of mild gonadal dysgenesis, severe underandrogenization, and absent Müllerian structures. Of these, two patients harbored missense mutations within the DNA-binding region of SF1 (p.C33S, p.R84H), one patient had a nonsense mutation (p.Y138X) and one patient had a frameshift mutation (c.1277dupT) predicted to disrupt RNA stability or protein function. One additional patient ([c.424_427dupCCCA]+[p.G146A]) displayed a more marked phenotype of severe gonadal dysgenesis, normal female external genitalia, and Müllerian structures. Functional studies of the missense mutants (p.C33S, p.R84H) and of one nonsense mutant (p.Y138X) revealed impaired transcriptional activation of SF1-responsive target genes. To date, adrenal insufficiency has not occurred in any of the patients. Thus, SF1 mutations are a relatively frequent cause of 46,XY DSD in humans.
Keywords: steroidogenic factor-1, SF1, NR5A1, gonadal dysgenesis, disorders of sex development (DSD), male pseudohermaphroditism, nuclear receptor
INTRODUCTION
Steroidogenic factor–1 (SF1/Ad4BP, NR5A1; MIM# 184757) is a nuclear receptor that was first identified following the search for a common regulator of the cytochrome P450 steroid hydroxylase family of enzymes [Lala et al., 1992]. Subsequent studies have revealed that SF1 regulates a great number of genes involved in gonadal and adrenal development, reproduction and steroidogenesis [Parker et al., 2002].
SF1 is believed to bind to the response elements of target genes as a monomer. Critical regions of SF1 involved in transcriptional regulation include a two zinc finger DNA–binding domain (DBD), an “A” box region found in monomeric nuclear receptors, a hinge region, and a helical ligand–binding domain (LBD) region containing an activation function–2 (AF–2) domain [Parker and Schimmer, 1997]. Crystalization of the LBD of SF1 has revealed that phospholipids may act as ligands for SF1 [Krylova et al., 2005].
SF1 is essential for gonadal and adrenal differentiation in mammals. In mice and humans, SF1 is expressed very early in the urogenital ridge and continues to be highly expressed in the developing adrenal, gonad, ventromedial hypothalamus, and pituitary [Hatano et al., 1994; Ingraham et al., 1994; Hanley et al., 1999]. Targeted disruption of Sf1 (Ftzf1) in mice results in gonadal and adrenal agenesis, persistence of Müllerian structures and abnormalities of the hypothalamus and pituitary gonadotropes [Luo et al., 1994; Majdic et al., 2002]. Heterozygous animals have a milder phenotype including an impaired adrenal stress response and reduced testicular size [Bland et al., 2000].
In humans, SF1 mutations were first described in patients with 46,XY disorders of sex development (DSD), Müllerian structures, and primary adrenal failure (MIM# 184757) [Achermann et al., 1999, 2002]. The first of these mutations (p.G35E) was a de novo heterozygous change affecting a critical amino acid in the “P” box region of the first zinc–finger of the DNA binding domain that severely affects SF1 function [Achermann et al., 1999; Ito et al., 2000]. The second mutation was a recessively–inherited homozygous change (p.R92Q) affecting the “A” box of SF1 and causing partial loss of function [Achermann et al., 2002]. A heterozygous SF1 mutation has also been reported in a 46,XX female with primary adrenal failure and apparently normal ovarian development [Biason-Lauber and Schoenle, 2000].
Recently, heterozygous SF1 mutations have been described in seven 46,XY patients with ambiguous genitalia, gonadal dysgenesis, but no adrenal insufficiency [Correa et al., 2004; Mallet et al., 2004; Hasegawa et al., 2004; Lin et al., 2007]. The potential dosage–dependent actions of SF1 suggest that these heterozygous SF1 changes might lead to a partial phenotype of 46,XY DSD with normal adrenal function. Here, we report five novel SF1 mutations identified in a cohort of 27 patients with severe underandrogenization and variable degrees of gonadal dysgenesis. These results suggest that heterozygous SF1 changes are a relatively frequent finding in patients with 46,XY gonadal dysgenesis/DSD.
MATERIALS AND METHODS
Patients
The cohort studied consisted of 27 46,XY patients from the “German Network of Disorders of Sex Development.” The patients had presented with phenotypes ranging from ambiguous genitalia to normal female genitalia and had been diagnosed with variable degrees of gonadal dysgenesis. Müllerian structures were reported in 17 patients. Adrenal insufficiency was not present in any of them. Dysmorphic or systemic features (e.g., kidney, heart, limbs) were absent. Informed consent for the genetic studies was obtained from all parents or patients according to institutional guidelines.
Molecular Analysis of NR5A1
Genomic DNA was extracted from peripheral blood leukocytes and exons 2–7 of the gene encoding SF1 (NR5A1) were PCR–amplified (primer sequences available on request). PCR products were purified using exonuclease 1 and shrimp alkaline phosphatase (New England Biolabs, Cambridge, UK; USB, Cleveland, OH) and sequenced using a V3 kit and 3130x analyzer (Applied Biosystems, Foster City, CA). DNA mutation numbering is based on GenBank reference DNA sequence NM_004959.3, with the A of the ATG initiation codon designated +1 (www.hgvs.org/mutnomen).
Eletromobility Shift Assays
Wild–type (WT) and mutant SF1 proteins were in-vitro translated from the pCMX expression vector using the TNT reticulocyte system (Promega, Southampton, UK). Synthetic oligonucleotide probes corresponding to the 30 SF1 binding site on the Cyp11a minimal promoter were labeled with [32P]dCTP by Klenow polymerase and electromobility shift assays were performed as described previously [Ito et al., 2000].
Studies of SF1 Expression and Nuclear Localization
Mutant green fluorescent protein (GFP)–SF1 constructs were generated by site–directed mutagenesis in a pAcGFP–C1 vector (Clontech, Oxford, UK) to produce mutant fusion proteins with a GFP tag at the amino–terminus of SF1. Studies of protein expression and cellular localization were performed by transfecting WTand mutant pAcGFP–C1SF1 expression vectors (0.8 µg) into tsa 201 human embryonic kidney cells. After 24 hr, cells were fixed and nuclear counterstaining performed with Vectashield containing DAPI (Vector Laboratories, Peterborough, UK).
Transient Gene Express Assays
Expression vectors containing the p.C33S and p.R84H missense mutants and pY138X nonsense mutant were generated by sitedirected mutagenesis (QuikChange, Stratagene, Amsterdam, The Netherlands) using WT pCMXSF1 as a template. Transient transfection studies were performed in 96–well plates using Lipofectamine 2000 (Invitrogen, Paisley, UK) and a Dual–Luciferase reporter assay system (Promega). Initial studies were performed in tsa 201 cells by transfecting empty, WT, or mutant SF1 expression vectors (2ng/well) (p.G35E, p.C33S, p.R84H, p.Y138X) with SF1–responsive minimal promoters linked to luciferase (100ng/well) (data shown for Cyp11a). In addition, synergistic activation of the LHβ promoter by SF1 and Egr1 (2ng/well) was studied in tsa 201 cells, as described previously [Lin et al., 2007]. Finally, WT or mutant SF1 vectors (10ng/well) were cotransfected with Cyp11a and MIS reporters into TM3 (mouse Leydig) and TM4 (mouse Sertoli) cells, respectively. Cells were lysed for 24 hr following transfection and assayed for luciferase activity (FLUOstar Optima; BMG Labtech, Aylesbury, UK), with standardization for Renilla coexpression.
Studies of Potential Dominant Negative Interactions
Studies of WT/mutant interactions were performed by transfecting increasing amounts of pCMXWT or mutant SF1 expression vector (0, 1, 2, 5, 10 ng) with either 1 ng empty vector or 1 ng WT SF1 and Cyp11a reporter (100 ng) in tsa 201 cells. Luciferase assays were performed as described above.
RESULTS
Cohort Analysis
SF1 mutations were found in 5 out of 27 (18.5%) patients studied. An overview of these changes and clinical phenotypes is provided in Table 1 and Figure 1.
TABLE 1.
Phenotypes, Genotypes, and Hormonal Data of the FIve Patients*
| Patient | Age (years) | External genitalia | Uterus | Epididymis and vas deferens | Gonada histology | Testosterone levels at diagnosis | Gender assignment | Adrenal investigations (at current age) | Mutation | Parents |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 4 | Clitoromegaly urogenital sinus, inguinal gonads | No | Yes | Bilateral testes, Sertoli rich, few spermatogonia and Leydig cells | hCG stimulation at birth: testosterone <0.08→0.07 ng/ml | ♀ | Cortisol 3.6 µg/dl DHEAS 50 ng/ml ACTH 25 pg/ml | p.C33S(c. 98G>C, exon 2) | Mother: normal; father: normal |
| 2 | 17 | Citoromegaly urogenital sinus, inguinal gonads | No | Yes | Bilateral testes | hCG stimulation at 2 years: testosterone <0.01→2.5ng/ml | ♀ | Corsitol 7.4 to 24.9µg/dlaDHEAS 1230 ng/ml | p.R84H (c.251G>A, exon 4) | Mother: normal; father; NA |
| 3 | 10 | Clitoromegaly, urogenital sinus inguinal gonads | No | Yes | Bilateral testes Sertoli cells only | At birth: testosterone 0.25ng/ml | ♀ | Cortisol 7.2µg/dl DHEAS 891ng/ml | pY138X(c414C>A, exon 4) | Mother: normal; father; NA |
| 4 | 8 | Clitoromegaly urogenital sinus, inguinal gonads | No | Yes | Bilateral testes | hCG stimulation at birth;testostrrone 0.86→.086ng/ml | ♀ | Cortisol 6.7 µg/dl DHEAS 548ng/ml | c1277dupT(exon 7) | Mother: c1277 dupT; father: NA |
| 5 | 22 | Normal female, no aplpable gonads | Yes | Not known | Bilateral streak gonads | At 22 years: LH 25.8U/I FSH 111.9U/I estradiol 7pg/ml testosterone 0.2 nd/ml | ♀ | NA | c424_427 dupCCCA+p.G146A polymorphism(c.437G>C)(both exon 4) | Mother: NA; father; NA |
Normal ranges: basal cortisol, 3–15 µg/dl (<0.5–10 years); DHEAS, <50–352 ng/ml (0.5–7 years), 75–1,312 ng/ml (7–10 years), 150–4,400 ng/ml (>10 years); ACTH, 9–50 pg/ml. Conversion to SI units: testosterone ng/ml×3.47 for nmol/l; cortisol µg/dl×27.6 for nmol/l; DHEAS ng/ml×2.56 for nmol/l; ACTH pg/ml×0.22 for pmol/l.DNA mutation numbering is based on the GenBank reference DNA sequence NM_004959.3 using current guidelines (www.hgvs.org/mutnomen) and with the A of the ATG initiation codon designated +1.
Peak cortisol following synacthen stimulation.“At birth” refers to the first week of life. ACTH, adrenocorticotropin; NA, not analyzed.
FIGURE 1.
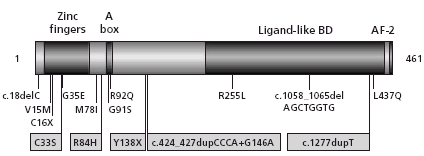
Structure of SF1 including the position of SF1 mutations in 46,XYpatients with disorders of sex development. p.G35E and p.R92Q are the first mutations described in SF1 in 46,XY individuals with gonadal dysgenesis and adrenal failure. p.R255L was found in a normal 46,XX female with adrenal failure.The othermutations have been identified in individuals with 46,XY DSD without adrenal insufficiency.The five novel mutations reported here are boxed. DNA RefSeq NM_004959.3.
Case Histories
Patients 1–4 were diagnosed with ambiguous genitalia at birth. All these individuals presented with a similar genital phenotype consisting of phallic hypoplasia/clitoromegaly, fusion of the labioscrotal folds, a single perineal opening representing a urogenital sinus, and palpable inguinal gonads. The karyotype was found to be 46,XY in each case, but Müllerian structures (uterus, upper vagina) were absent. Gonadectomy–performed before the age of 2 years–revealed bilateral testes with Wolffian structures (epididymides, vasa deferentia). Endocrine evaluation at diagnosis showed low testosterone levels before and after human chorionic–gonadotropin (hCG) stimulation in Patients 1, 2 and 4, and low basal testosterone in Patient 3 (Table 1). In all patients, female gender assignment was made because of the significant undervirilization and clitoroplasty were undertaken following discussion and with parental consent.
Patient 5 presented as a young adult with primary amenorrhea. She had normal female external genitalia. Gonadotropins (folliclestimulating hormone [FSH] 111.9 U/l; luteinizing hormone [LH] 25.8 U/l) were elevated, karyotype was found to be 46,XY, and Müllerian structures and bilateral streak gonads were identified on ultrasound (Table 1).
Basal cortisol and dehydroepiandrosterone sulfate (DHEAS) were within normal ranges in Patients 1–4 and no symptoms or signs of adrenal insufficiency have emerged during follow–up (Table 1). A synacthen stimulation test performed at 17 years of age in Patient 2 showed a very good cortisol response.
SF1 (NR5A1)Mutations
An overview of the SF1 (NR5A1) mutations detected is shown in Table 1 and Figure 1. Patients 1 and 2 harbor heterozygous missense mutations in SF1 (Patient 1: c.98G>C, p.C33S; Patient 2: c.251G>A, p.R84H), whereas Patient 3 has a heterozygous nonsense mutation (c.414C4A, p.Y138X). Patient 4 is heterozygous for a single nucleotide duplication (c.1277dupT), which is predicted to result in a frameshift and disruption of the ligand binding domain of SF1. Her mother also carries this heterozygous change. Patient 5 has a 4 base pair duplication (c.424_427dupCCCA) in exon 4 in one allele, which is predicted to result in a frameshift and premature stop codon after seven amino acids. A c.437G>C polymorphism (p.G146A) was present on the second allele, which was confirmed by subcloning the patient's DNA. Analysis of DNA from 100 healthy control subjects did not detect any of these key novel changes by restriction analysis (data not shown). The p.G146A change is a recognized nonsynonymous polymorphism in SF1 (rs1110061).
Electromobility Shift Assays
Studies of the 3′ Cyp11a promoter showed absent binding for the p.C33S mutant consistent with its position within the “P” box of the DBD. The p.R84H mutant and the p.Y138X truncated protein both showed reduced binding (Fig. 2A).
FIGURE 2.
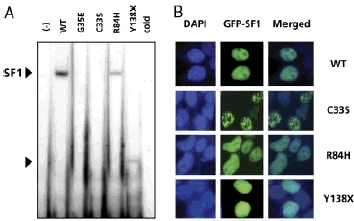
DNA binding, expression, and cellular localization of SF1. A: Electromobility shift assay showing wild–type (WT) and mutant SF1 binding to a labeled probe corresponding to the 3′ SF1 binding site of the Cyp11a promoter. in-vitro translated empty vector (−) (lane 1) and an excess of unlabeled cold probe (with WT SF1 protein) (lane 7) were used as controls. B:Cellular localization of GFP–SF1 fusion proteins (green), generated and expressed in tsa 201 cells using a pAcGFP–C1 vector. Nuclear counterstaining was performed with DAPI (blue), and images merged to conform nuclear localization.WT SF1 showed strong nuclear localization, with relative nucleolar exclusion and very occasional nuclear subfoci. A similar expression and localization pattern to WT was seen for the p.R84H mutant. Clustering in larger subnuclear foci was seen in the cells transfected with the p.C33S mutant.The p.Y138 Xmutant showed di¡use nuclear localization.
SF1 Expression and Cellular Localization
GFP–tagged WT SF1 showed strong nuclear localization with nucleolar exclusion. A similar pattern was seen with the p.R84H mutant vector. The p.C33S construct showed marked subnuclear focal aggregation in many cells, whereas the p.Y138X nonsense mutant appeared to be located in a diffuse pattern throughout the nucleus (Fig. 2B).
Functional Studies of SF1 Activity
All mutants studied (p.C33S, p.R84H, p.Y138X) showed markedly impaired transcriptional activity (Fig. 3A–D). Cotransfection of mutant with WT SF1 did not show a dominant negative effect even when 10:1 ratios of mutant:WT vector were transfected (Fig. 3E). The p.G146A polymorphism in SF1 has been shown previously to be associated with approximately 80% WT activity [WuQiang et al., 2003].
FIGURE 3.
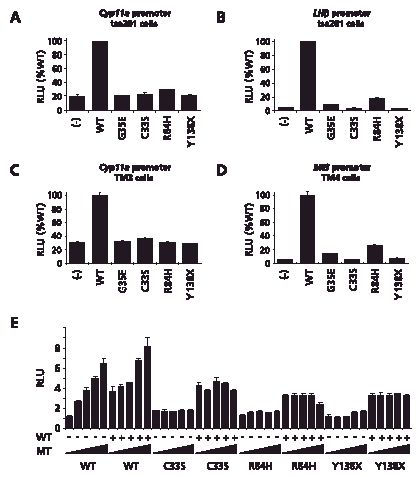
A: Effect of the SF1 mutants on transcriptional activity of the minimal promoters of Cyp11a in tsa 201 cells as described in themethods section. B: Effect of the SF1 mutants on synergistic activation of the LH beta promoter bySF1 and Egr1.C,D: Effect of the SF1 mutants on transcriptional activity of the Cyp11a and MIS promoter in TM3 (mouse Leydig) and TM4 (mouse Sertoli) cells, respectively. E: Studies of a potential dominant negative e¡ect ofmutant SF1 were performed by transfecting increasing amounts of wild–type (WT) ormutant SF1 (p.C33S, p.R84H, p.Y138X) expression vector (0,1,2,5,10 ng) with1–ng empty vector (−) or1–ngWT (1) and a Cyp11a promoter reporter (100 ng) in tsa 201cells. Additional empty vector was transfected to keep DNA quantities consistent.Data represent the mean 7± standard error of themean (SEM) of three independent experiments, each performed in triplicate.
DISCUSSION
Here, we present five novel heterozygous SF1 mutations in a cohort of 27 46,XY patients (18.5%) with severe underandrogenization but without adrenal insufficiency. These findings substantially increase the number of SF1 (NR5A1) mutations reported in humans, and show that mutations in SF1 can be found in patients with a range of phenotypic features (Table 2).
TABLE 2.
Molecular and Clinical Features of 46, XY Individuals With in SF1(NR5A1)
| Mutatiion | Occurence | Inheritance | Adrenal failure | External genitalia | Testicular dysgenesis | Müllerian structures | Reference |
|---|---|---|---|---|---|---|---|
| p.G35E | Heterozygous | De novo | + | Female | Severe | + | Achermann etal[1999] |
| p.R92Q | Homozygous | Recessive | + | Female | Not known | + | Achermann etal [2002] |
| c.424_427 dupCCCA+p.G146A | Heterozygous+ polymorphismxs | Not known | − | Female | Severe | + | This report |
| p.M781 | Heterozygous | SLD | − | Female | Mild | (+) | Liin et al [2007] |
| p.V15M | Heterozygous | De novo | − | Female | Mild | − | Liin et al [2007] |
| p.C16X | Heterozygous | De novo | − | Ambiguous | Mild | + | Mallet et al [2004] |
| p.C91S | Heterozygous | SLD | − | Ambiguous | Mild | (+) | Lin et al [2007] |
| c.18delC | Heterozygous | De novo | − | Amdiguous | Mild | − | Has egawa et al [2004] |
| c.1058_1065del AGCTGGTG | Heterozygous | De novo | − | Ambiguous | Regresseda | − | Correa et al.[2004] |
| p.C33S | Heterozygous | De novo | − | Ambiguous | Mild | − | This report |
| p.R84H | Heterozygous | De novo | − | Ambiguous | Normal | − | This report |
| p.Y138X | Heterozygous | De novo | − | Ambiguous | Mild | − | This report |
| c.1277dupT | Heterozygous | SLD | − | Ambiguous | Normal | − | This report |
| p.L437Q | Heterozygous | De novo | − | Hypospadias | Mild | − | Lin et al.[2007] |
Gonadal tissue not found at laparoscopy at age 31 years
SLD, sex-limited dominant
+, present
−, absent
(+), remnant present
Within our series, four patients (heterozygous p.C33S, p.R84H, p.Y138X, and c.1277dupT) presented with a similar phenotype consisting of severe underandrogenization of the external genitalia, bilateral testes, and Wolffian structures, but absence of Muüllerian structures (uterus, fallopian tubes). These findings suggest that androgen biosynthesis and action were compromised during early fetal development (9–16 weeks), but that sufficient secretion of anti–Müllerian hormone (AMH) occurred from Sertoli cells to allow regression of Müllerian structures during this critical developmental period. The presence of well–formed Wolffian structures in Patients 1–4 is unusual given the degree of underandrogenization observed. Whether the development of Wolffian structures with relatively impaired androgenization of the external genitalia will be a feature that distinguishes SF1 mutations from other diagnoses, such as partial gonadal dysgenesis or partial androgen insensitivity, remains to be seen.
The identification of naturally–occurring missense mutations in SF1 is also helping to reveal important functional domains for nuclear receptor action. The amino acids of the missense mutations (p.C33S, p.R84H) are highly conserved in different species. The mutated cysteine (p.C33S) plays a central role in formation of the first zinc finger of the DNA binding domain of SF1 (Fig. 1). The C33S change results in impaired DNA binding to SF1 response elements (Fig. 2A), a complete loss of transcriptional activation without dominant negative activity (Fig. 3A–E) and abnormal clustering in sub7–nuclear aggregations (Fig. 2B). These foci likely represent promyelocytic leukemia (PML) or nuclear domain 10 (ND10) bodies, and have been reported previously with several nuclear receptor mutations associated with human disease [Baumann et al., 2001; Vottero et al., 2002; Black et al., 2004]. The arginine at position 84 lies between the zinc–fingers and “A”–box of SF1 in the carboxylterminal DNA–binding region (Fig. 1). This change also disrupts DNA binding and transcriptional activation, with little effect on cellular localization. Finally, the nonsense mutation (p.Y138X) results in a truncated protein with loss of the LBD, and the frameshift mutation resulting from the duplication c.1277dupT is predicted to lead to abnormal protein sequence. However, it is feasible that RNA containing the Y138X change is subject to nonsense–mediated decay.
Compared to the first four individuals, Patient 5 has a more severe phenotype, as she has normal female external genitalia and presented only in adolescence with absent breast development and primary amenorrhea. A uterus and streak gonads were found. The mutation c.424_427dupCCCA results in a frameshift and premature stop codon after seven amino acids. In addition to this heterozygous frameshift change, Patient 5 also harbors a heterozygous p.G146A polymorphism on the other allele. This p.G146A is reported to have approximately 80% of WT function and to be associated with micropenis or cryptorchidism [WuQiang et al., 2003; Wada et al., 2005, 2006]. Thus, we hypothesize that combination of the hypomorphic polymorphism, together with the heterozygous frameshift mutation, reduces gene dosage of SF1 below haploinsufficiency, resulting in the more severe phenotype observed in this patient.
While SF1 has a key role in testis development and function, the effects of reduced SF1 activity in the ovary are less clear [Hanley et al., 1999; Biason-Lauber and Schoenle, 2000]. The identification of a heterozygous frameshift mutation in SF1 in the mother of Patient 4 provides evidence that heterozygous SF1 mutations may be inherited in a sex–limited dominant fashion. Mothers may carry such changes, but have preserved ovarian function, whereas 46,XY offspring are affected. This finding has important implications for counseling, as heterozygous females may resemble “carriers” in an X–linked disorder.
Taken together, our findings show that heterozygous SF1 mutations are a relatively frequent finding in individuals with 46,XY DSD, and that the testis may be more sensitive to partial loss of SF1 function than the adrenal gland in humans [Jameson,2004; Lin et al., 2006]. Whether these patients will develop adrenal insufficiency with time remains to be seen. Thus, determining the genetic cause in 46,XY DSD can have implications for investigation, counseling of families, and longterm follow–up.
Acknowledgments
We are grateful to the patients and families, members of the network, Daniel Kelberman, Ron Evans, Meera Ramayya, J. Larry Jameson, and Masafumi Ito for technical support and vectors. B.K. holds a grant from the Bundesministerium für Bildung und Forschung (BMBF) Network of Disorders of Sex Development (01GM0311) and J.C.A. holds a Wellcome Trust Senior Research Fellowship in Clinical Science (079666). Research at the Institute of Child Health and Great Ormond Street Hospital for Children National Health Service (NHS) Trust benefits from R&D funding received from the NHS Executive.
REFERENCES
- Achermann JC, Ito M, Ito M, Hindmarsh PC, Jameson JL. A mutation in the gene encoding steroidogenic factor-1 causes XY sex reversal and adrenal failure in humans. Nat Genet. 1999;22:125–126. doi: 10.1038/9629. [DOI] [PubMed] [Google Scholar]
- Achermann JC, Ozisik G, Ito M, Orun UA, Harmanci K, Gurakan B, Jameson JL. Gonadal determination and adrenal development are regulated by the orphan nuclear receptor steroidogenic factor-1, in a dose-dependent manner. J Clin Endocrinol Metab. 2002;87:1829–1833. doi: 10.1210/jcem.87.4.8376. [DOI] [PubMed] [Google Scholar]
- Baumann CT, Ma H, Wolford R, Reyes JC, Maruvada P, Lim C, Yen PM, Stallcup MR, Hager GL. The glucocorticoid receptor interacting protein 1 (GRIP1) localizes in discrete nuclear foci that associate with ND10 bodies and are enriched in components of the 26S proteasome. Mol Endocrinol. 2001;15:485–500. doi: 10.1210/mend.15.4.0618. [DOI] [PubMed] [Google Scholar]
- Biason-Lauber A, Schoenle EJ. Apparently normal ovarian differentiation in a prepubertal girl with transcriptionally inactive steroidogenic factor 1 (NR5A1/SF-1) and adrenocortical insufficiency. Am J Hum Genet. 2000;67:1563–1568. doi: 10.1086/316893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black BE, Vitto MJ, Gioeli D, Spencer A, Afshar N, Conaway MR, Weber MJ, Paschal BM. Transient, ligand-dependent arrest of the androgen receptor in subnuclear foci alters phosphorylation and coactivator interactions. Mol Endocrinol. 2004;18:834–850. doi: 10.1210/me.2003-0145. [DOI] [PubMed] [Google Scholar]
- Bland ML, Jamieson CA, Akana SF, Bornstein SR, Eisenhofer G, Dallman MF, Ingraham HA. Haploinsufficiency of steroidogenic factor-1 in mice disrupts adrenal development leading to an impaired stress response. Proc Natl Acad Sci USA. 2000;97:14488–14493. doi: 10.1073/pnas.97.26.14488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa RV, Domenice S, Bingham NC, Billerbeck AE, Rainey WE, Parker KL, Mendonca BB. A microdeletion in the ligand binding domain of human steroidogenic factor 1 causes XY sex reversal without adrenal insufficiency. J Clin Endocrinol Metab. 2004;89:1767–1772. doi: 10.1210/jc.2003-031240. [DOI] [PubMed] [Google Scholar]
- Hanley NA, Ball SG, Clement-Jones M, Hagan DM, Strachan T, Lindsay S, Robson S, Ostrer H, Parker KL, Wilson DI. Expression of steroidogenic factor 1 and Wilms' tumour 1 during early human gonadal development and sex determination. Mech Dev. 1999;87:175–180. doi: 10.1016/s0925-4773(99)00123-9. [DOI] [PubMed] [Google Scholar]
- Hasegawa T, Fukami M, Sato N, Katsumata N, Sasaki G, Fukutani K, Morohashi K, Ogata T. Testicular dysgenesis without adrenal insufficiency in a 46,XY patient with a heterozygous inactive mutation of steroidogenic factor-1. J Clin Endocrinol Metab. 2004;89:5930–5931. doi: 10.1210/jc.2004-0935. [DOI] [PubMed] [Google Scholar]
- Hatano O, Takayama K, Imai T, Waterman MR, Takakusu A, Omura T, Morohashi KR. Sex-dependent expression of a transcription factor, Ad4BP, regulating steroidogenic P-450 genes in the gonads during prenatal and postnatal rat development. Development. 1994;120:2787–2797. doi: 10.1242/dev.120.10.2787. [DOI] [PubMed] [Google Scholar]
- Ingraham HA, Lala DS, Ikeda Y, Luo X, Shen WH, Nachtigal MW, Abbud R, Nilson JH, Parker KL. The nuclear receptor steroidogenic factor 1 acts at multiple levels of the reproductive axis. Genes Dev. 1994;8:2302–2312. doi: 10.1101/gad.8.19.2302. [DOI] [PubMed] [Google Scholar]
- Ito M, Achermann JC, Jameson JL. A naturally occurring steroidogenic factor-1 mutation exhibits differential binding and activation of target genes. J Biol Chem. 2000;275:31708–31714. doi: 10.1074/jbc.M002892200. [DOI] [PubMed] [Google Scholar]
- Jameson JL. Of mice and men: the tale of steroidogenic factor-1. J Clin Endocrinol Metab. 2004;89:5927–5929. doi: 10.1210/jc.2004-2047. [DOI] [PubMed] [Google Scholar]
- Krylova IN, Sablin EP, Moore J, Xu RX, Waitt GM, MacKay JA, Juzumiene D, Bynum JM, Madauss K, Montana V, Bebedeva L, Suzawz M, Williams JD, Williams SP, Guy RK, Thornton JW, Fletterick RJ, Willson TM, Ingraham HA. Structural analyses reveal phosphatidyl inositols as ligands for the NR5 orphan receptors SF-1 and LRH-1. Cell. 2005;120:343–355. doi: 10.1016/j.cell.2005.01.024. [DOI] [PubMed] [Google Scholar]
- Lala DS, Rice DA, Parker KL. Steroidogenic factor I, a key regulator of steroidogenic enzyme expression, is the mouse homolog of fushi tarazu-factor I. Mol Endocrinol. 1992;6:1249–2458. doi: 10.1210/mend.6.8.1406703. [DOI] [PubMed] [Google Scholar]
- Lin L, Gu WX, Ozisik G, To WS, Owen CJ, Jameson JL, Achermann JC. Analysis of DAX1 (NR0B1) and steroidogenic factor-1 (SF1/Ad4BP, NR5A1) in children and adults with primary adrenal failure: ten years' experience. J Clin Endocrinol Metab. 2006;91:3048–3054. doi: 10.1210/jc.2006-0603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Philibert P, Ferraz-de-Souza B, Kelberman D, Homfray T, Albanese A, Molini V, Sebire NJ, Einaudi S, Conway GS, Hughes IA, Jameson JL, Sultan C, Dattani MT, Achermann JC. Heterozygous missense mutations in steroidogenic factor 1 (SF1/Ad4BP, NR5A1) are associated with 46,XY disorders of sex development with normal adrenal function. J Clin Endocrinol Metab. 2007;92:991–999. doi: 10.1210/jc.2006-1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Ikeda Y, Parker KL. A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell. 1994;77:481–490. doi: 10.1016/0092-8674(94)90211-9. [DOI] [PubMed] [Google Scholar]
- Majdic G, Young M, Gomez-Sanchez E, Anderson P, Szczepaniak LS, Dobbins RL, McGarry JD, Parker KL. Knockout mice lacking steroidogenic factor 1 are a novel genetic model of hypothalamic obesity. Endocrinology. 2002;143:607–614. doi: 10.1210/endo.143.2.8652. [DOI] [PubMed] [Google Scholar]
- Mallet D, Bretones P, Michel-Calemard L, Dijoud F, David M, Morel Y. Gonadal dysgenesis without adrenal insufficiency in a 46, XY patient heterozygous for the nonsense C16X mutation: a case of SF1 haploinsufficiency. J Clin Endocrinol Metab. 2004;89:4829–4832. doi: 10.1210/jc.2004-0670. [DOI] [PubMed] [Google Scholar]
- Parker KL, Schimmer BP. Steroidogenic factor 1: a key determinant of endocrine development and function. Endocr Rev. 1997;18:361–377. doi: 10.1210/edrv.18.3.0301. [DOI] [PubMed] [Google Scholar]
- Parker KL, Rice DA, Lala DS, Ikeda Y, Luo X, Wong M, Bakke M, Zhao L, Frigeri C, Hanley NA, Stallings N, Schimmer BP. Steroidogenic factor 1: an essential mediator of endocrine development. Recent Prog Horm Res. 2002;57:19–36. doi: 10.1210/rp.57.1.19. [DOI] [PubMed] [Google Scholar]
- Vottero A, Kino T, Combe H, Lecomte P, Chrousos GP. A novel, C-terminal dominant negative mutation of the GR causes familial glucocorticoid resistance through abnormal interactions with p160 steroid receptor coactivators. J Clin Endocrinol Metab. 2002;87:2658–2667. doi: 10.1210/jcem.87.6.8520. [DOI] [PubMed] [Google Scholar]
- Wada Y, Okada M, Hasegawa T, Ogata T. Association of severe micropenis with Gly146Ala polymorphism in the gene for steroidogenic factor-1. Endocr J. 2005;52:445–448. doi: 10.1507/endocrj.52.445. [DOI] [PubMed] [Google Scholar]
- Wada Y, Okada M, Fukami M, Sasagawa I, Ogata T. Association of cryptorchidism with Gly146Ala polymorphism in the gene for steroidogenic factor-1. Fertil Steril. 2006;85:787–790. doi: 10.1016/j.fertnstert.2005.09.016. [DOI] [PubMed] [Google Scholar]
- WuQiang F, Yanase T, Wei L, Oba K, Nomura M, Okabe T, Goto K, Nawata H. Functional characterization of a new human Ad4BP/SF-1 variation, G146A. Biochem Biophys Res Commun. 2003;311:987–994. doi: 10.1016/j.bbrc.2003.10.096. [DOI] [PubMed] [Google Scholar]