

Abstract
The key enzyme of the fermentation of glutamate by Acidaminococcus fermentans, 2-hydroxyglutarylcoenzyme A dehydratase, catalyzes the reversible syn-elimination of water from (R)-2-hydroxyglutaryl-coenzyme A, resulting in (E)-glutaconylcoenzyme A. The dehydratase system consists of two oxygen-sensitive protein components, the activator (HgdC) and the actual dehydratase (HgdAB). Previous biochemical and spectroscopic studies revealed that the reduced [4Fe–4S]+ cluster containing activator transfers one electron to the dehydratase driven by ATP hydrolysis, which activates the enzyme. With a tenfold excess of titanium(III) citrate at pH 8.0 the activator can be further reduced, yielding about 50% of a superreduced [4Fe–4S]0 cluster in the all-ferrous state. This is inferred from the appearance of a new Mössbauer spectrum with parameters δ = 0.65 mm/s and ΔEQ = 1.51–2.19 mm/s at 140 K, which are typical of Fe(II)S4 sites. Parallel-mode electron paramagnetic resonance (EPR) spectroscopy performed at temperatures between 3 and 20 K showed two sharp signals at g = 16 and 12, indicating an integer-spin system. The X-band EPR spectra and magnetic Mössbauer spectra could be consistently simulated by adopting a total spin St = 4 for the all-ferrous cluster with weak zero-field splitting parameters D = −0.66 cm−1 and E/D = 0.17. The superreduced cluster has apparent spectroscopic similarities with the corresponding [4Fe–4S]0 cluster described for the nitrogenase Fe-protein, but in detail their properties differ. While the all-ferrous Fe-protein is capable of transferring electrons to the MoFe-protein for dinitrogen reduction, a similar physiological role is elusive for the superreduced activator. This finding supports our model that only one-electron transfer steps are involved in dehydratase catalysis. Nevertheless we discuss a common basic mechanism of the two diverse systems, which are so far the only described examples of the all-ferrous [4Fe–4S]0 cluster found in biology.
Electronic supplementary material
The online version of this article (doi:10.1007/s00775-008-0345-z) contains supplementary material, which is available to authorized users.
Keywords: 2-Hydroxyglutaryl-coenzyme A dehydratase, Activator, Iron–sulfur protein, Superreduced, All-ferrous
Introduction
The anaerobic bacterium Acidaminococcus fermentans ferments glutamate via the 2-hydroxyglutarate pathway to ammonium, carbon dioxide, acetate, butyrate, and molecular hydrogen [1]. A key enzyme in this fermentation is 2-hydroxyglutaryl-CoA dehydratase (CoA is coenzyme A), which catalyzes the reversible syn-elimination of water from (R)-2-hydroxyglutaryl-CoA, resulting in (E)-glutaconyl-CoA. This reaction is of considerable interest since a nonactivated β-C–H bond has to be cleaved (pK ≈ 40); for reviews see [2–4]. The enzyme system from A. fermentans is composed of two protein components, the homodimeric activator or archerase (HgdC) and the heterodimeric dehydratase (HgdAB), whose genes have been cloned and sequenced. After overexpression of hgdC in Escherichia coli, the activator could be purified to homogeneity aided by a C-terminal Strep-tag. The protein [γ2, 2 × (27 ± 1) kDa] contains 4.0 ± 0.1 iron and 3.8 ± 0.1 acid-labile sulfur, which form one [4Fe–4S]+/2+ cluster between the two subunits [5]. The activator can be readily reduced with dithionite or titanium(III) citrate. Most likely in vivo a reduced clostridial [4Fe–4S]-type ferredoxin [6] or the hydroquinone form of a flavodoxin [7], both isolated from A. fermentans, can act as reducing agents for the activator. The activator is extremely oxygen-sensitive; its half life under air is only about 10 s. The oxygen sensitivity results from destruction of the [4Fe–4S] cluster, which is exposed to the solvent [8]. Furthermore, the activator also precipitates in the presence of the dyes that are commonly used for the determination of redox potentials; herefore, the redox potential of the [4Fe–4S]+/2+ cluster (E0′ ≈ −320 mV) can only be roughly estimated from its almost complete reduction by ferredoxin or flavodoxin (E0′ = −420 mV) [7]. The native 2-hydroxyglutaryl-CoA dehydratase HgdAB (αβ, 54 + 42 kDa) contains 4.5 ± 0.3 iron, 3.5 ± 0.4 acid-labile sulfur and 1.0 FMNH2 [5]. In order to obtain catalysis, incubation of the dehydratase with reduced activator in the [4Fe–4S]+ state, ATP, and MgCl2 is necessary. Spectroscopic studies revealed that thereby one electron is transferred from the activator to the [4Fe–4S] cluster of the dehydratase [5, 7, 9], which cannot be reduced by any other means, except by deazaflavin and light in the presence of EDTA as shown for the related 2-hydroxyisocarproyl-CoA dehydratase (J. Kim and W. Buckel, unpublished results).
A mechanism has been proposed in which the dehydration is initiated by injection of this electron into the thiol ester carbonyl. The resulting substrate-derived ketyl radical eliminates the adjacent hydroxyl group. The enoxy radical formed can now be deprotonated to the product-related allylic ketyl radical, because radical formation has lowered the pK of the β-hydrogen by about 26 U [10]. In the last step the electron is returned from the allylic ketyl radical to the [4Fe–4S] cluster of the dehydratase and a new cycle can start. With the related 2-hydroxyisocaproyl-CoA dehydratase from Clostridium difficile this radical has been identified by electron paramagnetic resonance (EPR) spectroscopy [11] and it has been shown that such a cycle can endure for about 10,000 turnovers until a new ‘shot’ by the activator is required [12].
Homologous activators with around 60% sequence identity have been characterized from 2-hydroxyglutaryl-CoA dehydratase in Fusobacterium nucleatum [13], lactyl-CoA dehydratase in C. propionicum (T. Selmer and W. Buckel, unpublished results), phenyllactyl-CoA dehydratase in C. sporogens [14, 15], and 2-hydroxyisocaproyl-CoA dehydratase in C. difficile [12]. Furthermore, each genome of almost all anaerobically thriving bacteria and archaea, even that of E. coli (yjiL gene), encodes up to four different homologues of the A. fermentans activator with 30–40% sequence identity. Only the function of the two homologues in Thauera aromatica and other anaerobic benzoate-degrading bacteria could be assessed as the α- and δ-subunits of the four subunits comprising benzoyl-CoA reductase. The catalysis of this enzyme, which in addition requires ATP and reduced ferredoxin as substrates, has been proposed to proceed also via a ketyl radical [16].
Hence such activators, which probably all contain a single [4Fe–4S] cluster between two subunits with a helix–cluster–helix architecture [8], are widespread in the anaerobic world. Proteins with almost the same architecture but with unrelated sequences are the nitrogenase reductases, which appear to have the same function as the activators [17]. The proteins of both families increase the reductive power of an electron by ATP hydrolysis. Nitrogenase reductase from Azotobacter vinelandii, also called Fe-protein or Av2, has been shown to become superreduced to the all-ferrous state by titanium(III) citrate [18–26]. The energy efficiency of nitrogenase would be twofold enhanced if the catalysis proceeded via superreduced Av2, owing to the transfer of two electrons from the corresponding [4Fe–4S]0 cluster per hydrolysis of two ATP molecules [27, 28]. However, the low redox potential Em = −790 mV reported for the (+1/0) couple of the iron–sulfur cluster virtually excludes any physiological relevance [measured with Cr(II)EDTA as a reductant] [29]. Interestingly, a much higher value, Em = −460 mV, was initially obtained by using methyl viologen [26]. Moreover, a recent report indicates that nitrogenase catalysis actually can be supported by superreduced Av2 and may proceed via the (+2/0) couple of the iron–sulfur cluster, if the in vivo reductant flavodoxin hydroquinone (FldHQ) is used [30]. In this new state of the Fe protein, the all-ferrous [4Fe–4S]0 cluster is difficult to discriminate from the +1 and +2 redox states using the absorption spectra, and it appears to be diamagnetic. In contrast, the Av2 [4Fe–4S]0 cluster made with titanium(III) citrate has a distinct absorption spectrum [18], and an unusually high spin state S = 4, as was inferred from elaborate integer-spin EPR and magnetic Mössbauer studies [18, 19, 31, 32] and theoretically explored by density functional theory studies [33, 34]. Here we report an all-ferrous state for the [4Fe–4S] cluster in the activator from A. fermentans after treatment with a tenfold excess of titanium(III) citrate. To our knowledge this is the second biological system which stabilizes a [4Fe–4S]0 cluster. Remarkable spectroscopic and biochemical similarities between activator and the A. vinelandii Fe-protein give reason to favor a common functional principle of these exciting proteins.
Materials and methods
Metallic 57Fe was purchased from Chemotrade (Düsseldorf, Germany). The metal (20 mg) was dissolved in 0.5 mL 8 M HCl at 80 °C for 12 h. This stock solution was added directly to the medium. Standard I nutrient broth was from Merck (Darmstadt, Germany). E. coli strain XL1-blue was purchased from Stratagene (Heidelberg, Germany). Streptavidin–Sepharose and the plasmid pASK-IBA3 were obtained from IBA (Göttingen, Germany). All other chemicals and substances were from Sigma or Fluka (Deisenhofen, Germany).
Anaerobic manipulations were performed in an anaerobic chamber (Coy Laboratories, Ann Arbor, MI, USA) with an atmosphere of 5% H2 in N2. X-band EPR measurements were performed using a Bruker E500 ELEXSYS continuous-wave spectrometer with the Bruker dual-mode cavity (ER4116DM) or a standard cavity (ER4102ST) and an Oxford Instruments helium-flow cryostat (ESR 910). Microwave frequencies were measured with a Hewlett–Packard frequency counter (HP5352B), and the field control was calibrated with a Bruker NMR field probe (ER035M). The spectra were simulated on the basis of a spin-Hamiltonian description of the electronic ground state:
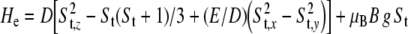 |
1 |
where St is the spin of the coupled system and D and E/D are the axial and rhombic zero-field parameters. The simulations were performed with a program which was developed from the S = 5/2 routines of Gaffney and Silverstone [35] and which specifically makes use of the Newton–Raphson iterative search method for transition fields as described there. Frequency-dependent Gaussian line shapes were used with full-width-at-half-maximum σν given in millitesla at g = 1. Alternatively also the Bruker XSOPHE suite [36] was used for the calculation of energy-level schemes and transition roadmaps. Mössbauer spectra were recorded using two spectrometers with alternating constant acceleration. The minimum experimental line width was Γfwhm = 0.24 mm/s (full width at half maximum). The sample temperature was maintained constant either in an Oxford Variox or an Oxford Mössbauer-Spectromag cryostat. The latter is a split-pair superconducting magnet system for fields up to 8 T in which the sample temperature can be kept constant in the range 1.5–250 K. The field is perpendicular to the γ-beam. The 57Co/Rh source was positioned at room temperature inside the gap of the magnet in a reentrant bore tube at a distance of about 85 mm from the sample. The field is zero at this position. All isomer shifts are quoted relative to iron metal at 300 K. Magnetic Mössbauer spectra were simulated with a computer program based on the same spin-Hamiltonian for St = 4 as for the EPR simulations (Eq. 1). The hyperfine interactions for 57Fe were calculated by using the usual nuclear Hamiltonian [37].
Purification of recombinant activator from A. fermentans
Standard I medium (8–10 L, Merck, Germany) was inoculated with 25 mL overnight culture of recombinant E. coli (XL1-blue/hgdC in pASK-IBA3) and grown in tightly closed 2-L bottles at 30 °C. When an optical density at 600 nm of about 0.3 was reached, cells were induced with anhydrotetracycline (30 μg/L). After anaerobic growth for a further 3 h, cells were harvested by centrifugation and suspended in 20 mM 3-(N-morpholino)propanesulfonic acid, pH 7.2, 100 mM NaCl, 10 mM MgCl2, 1 mM ATP, 2 mM dithiothreitol or 4 mM dithionite (buffer A). The bacteria were lysed anaerobically using a French press system. Cell debris and membranes were removed by ultracentrifugation for 60 min at 100,000g. The filtered supernatant was loaded with gravity flow onto an 8-mL streptavidin–Sepharose column, which was equilibrated with buffer A. After complete binding, the column was washed with at least ten column volumes of buffer A. The activator was eluted with 2.5 mM d-desthiobiotin in buffer A, then concentrated with an Amicon stirred cell, and immediately used for further experiments.
Results
Preparation and characterization of superreduced activator
The activator as isolated was concentrated to 60–80 mg protein/mL (1.2–1.6 mM) in 50 mM tris(hydroxymethyl)aminomethane hydrochloride pH 8.0. A tenfold excess of titanium(III) citrate was added, which yielded a color change from brown to red. Correspondingly, a new shoulder appeared in the UV–vis spectrum at about 500 nm (Fig. 1). The orange-red color apparently is a typical feature of superreduced [4Fe–4S]0 clusters, and does not originate from titanium(III) citrate directly, because this was removed by gel filtration on a Sephadex G-25 desalting column. It is noteworthy that the same color change was also observed upon full reduction of the nitrogenase Fe-protein from A. vinelandii [18].
Fig. 1.

UV–vis absorption spectrum of activator reduced with dithionite, and superreduced with titanium(III) citrate
As described previously, the activator had to be used right after purification for biochemical studies because of its tendency to precipitate within 4 days [5]. To our surprise, the superreduced activator was nearly as stable as the singly reduced or oxidized enzyme; the activity decayed only with a half life of 4–5 days or more. Remarkably, dehydratase activity could not be observed with 2-hydroxyglutaryl-CoA when titanium(III) citrate reduced activator was used in the activity assay [38].
EPR spectroscopy
In a previous EPR and Mössbauer study we demonstrated that dithionite-reduced activator1 possesses a [4Fe–4S]+ cluster with spin S = 3/2 [5]. The unusual magnetic ground state was inferred from X-band EPR spectra as shown in the inset in Fig. 2. The EPR resonances extend over a wide field range of at least 800 mT with a dominating strong absorption-like peak at g ≈ 4.5 and a broad trough at g ≈ 1.3–2. The large anisotropy and particularly the presence of high effective g values indicate the high spin multiplicity with large zero-field splitting (D > gμBB). Although the broad derivative spectrum does not have the classical appearance known for a quartet species from a transition metal complex [39], the pattern could be nicely simulated by using the usual spin-Hamiltonian for S = 3/2 with reasonable zero-field splitting parameters D = 1.3 cm−1 and E/D = 0.213. Just a simple Gaussian distribution of the rhombicity parameter with half width σ(E/D) = 0.11 was sufficient to account for the observed line broadening caused by g strain [5]. Moreover, a minor fraction of the [4Fe–4S]+ clusters (approximately 3%) was found to have the more common spin state S = 1/2, which gives rise to the narrower resonances around g = 2. The corresponding simulations for both spin states of [4Fe–4S]+, S = 1/2 and 3/2, are shown as gray and blue lines in the inset in Fig. 2.
Fig. 2.

Normal-mode (B1⊥B0) X-band electron paramagnetic resonance (EPR) spectrum of titanium(III) citrate reduced activator (low-field part, microwave frequency 9.6337 GHz; power 1.6 mW; modulation 2 mT at 100 kHz). The simulation for St = 4 (green line) is obtained with D = −0.66 cm−1, E/D = 0.165, g = (2.1, 1.99, 2.03) (Footnote 2) and a frequency-dependent Gaussian line width of σν = 70 mT. The inset shows the full range X-band spectrum of dithionite-reduced activator at 10 K with simulations for the [4Fe–4S]+ clusters as published previously [5]. The arrows in the main panel and in the inset indicate the field position of the strongest integer-spin signal from [4Fe–4S]0 clusters with St = 4
The strong absorption-like band at g ≈ 4.5 from S = 3/2 persists also in the EPR spectrum of titanium(III) citrate treated activator (Fig. 2, bold trace). We assign it to [4Fe–4S]+ clusters remaining from incomplete conversion of the reduced activator. The relative abundance of this contribution is estimated by corresponding the Mössbauer spectra to account for about 50% of the total iron (see below). Formation of superreduced clusters, however, is also discernible in the EPR spectrum from the appearance of two new lines in the low-field range at g ≈ 11 and g ≈ 16. Narrow absorption-like signals at such high g values without related strong derivative lines at higher field are completely unexpected for half-integer-spin systems, but rather suggest integer-spin transitions. The interpretation is corroborated by parallel-mode measurements (B1||B0) for which the new low-field resonances are the strongest signals, whereas the spectrum from the reduced activator, particularly the absorption-like band at g ≈ 4.5, is completely attenuated, as shown in Fig. 3. The striking difference of the two detection modes results from the different selection rules Δm = ±1 for B1⊥B0 and Δm = 0 for B1||B0, which essentially discriminate half-integer-spin and integer-spin transitions [40, 41]. Therefore, the low-field transitions at g ≈ 11 and g ≈ 16 must be assigned to an all-ferrous [4Fe–4S]0, cluster for which coupling of the four Fe(II) sites with local spin SFe = 2 can yield only an integer total spin. This clearly indicates superreduction of the activator by titanium(III) citrate. The ground state for such a tetrameric all-ferrous cluster can have, in principle, any total spin ranging from St = 0 to St = 8, depending on the six spin coupling constants between the four ferrous ions. But sophisticated quantum chemical studies for the titanium(III) citrate reduced [4Fe–4S]0 cluster from the Fe protein suggest only St = 0, 4, or 8 as possible solutions for that particular system [33, 34]. Below we show that the all-ferrous [4Fe–4S]0 cluster in superreduced activator actually has total spin St = 4, with properties that closely resemble those of the superreduced state of the Fe protein from A. vinelandii [18, 31].
Fig. 3.

Parallel-mode (B1||B0) X-band EPR spectra of titanium(III) citrate reduced activator recorded at 3, 10, and 20 K (microwave frequency 9.3404 GHz; power 1.6 mW; modulation 0.57 mT at 100 kHz). The green lines represent simulations for St = 4 with D = −0.66 cm−1, E/D = 0.165, g = (2.1, 2.0, 2.1) (Footnote 2), and a frequency-dependent Gaussian line width of σν = 30 mT
The integer-spin signals are remarkably sharp in the parallel-mode EPR spectra with full width at half maximum of only Γfwhm ≈ 4–5 mT for the main derivative peaks, but with very broad and asymmetric underlying shoulders. Such a superposition of broad and narrow powder signals may be caused by complex distributions of spin-Hamiltonian parameters, which owe their origin to microheterogeneity in the surrounding of the [4Fe–4S] clusters. In normal mode, however, the corresponding lines appear to be almost Gaussians of moderate 9–12 mT line width (Fig. 2). The difference may be related to the different orientational selections that contribute to the respective powder spectra [41]. Other ms components of the wave functions determine the transition probabilities between the zero-field split sublevels of the integer-spin system for parallel-mode detection, and thus may respond differently on the structural distortions. In the simulations, however, we could not reproduce the complex experimental line shape of the parallel-mode spectra of the superreduced activator by a simple distribution model for any of the spin-Hamiltonian parameters. Since we wanted to refrain from further sophistications other than Gaussian distributions, we just focused on the main features of the EPR spectra, i.e., the well-resolved sharp lines in the parallel-mode spectra and the Gaussian lines of the normal-mode spectra recorded at base temperature.
The EPR spectra of superreduced activator recorded in normal and parallel mode can be consistently simulated with total spin St = 4, as shown in Figs. 2 and 3. The zero-field parameters obtained from global optimization are D = −0.66 (±0.1) cm−1 and E/D = 0.165 (±0.01), with g = (2.2, 1.99, 2.03).2 Particularly the unusual temperature dependence of the two narrow parallel-mode signals in Fig. 3 is basically well reproduced with those values, i.e., at 3 K the simulated line at g ≈ 11 is stronger than the line at g ≈ 16, whereas the intensity ratio is inverted at 20 K. According to the energy splitting scheme shown on the left in Fig. 4, the derivative signals arise from transitions within the low-lying “ms = ±4” and the excited “ms = ±3” non-Kramers doublets, respectively. At the actual conditions of D and E/D, both of these ms doublets are virtually unsplit at zero magnetic field. For fields applied in the molecular z-direction, their X-band resonances are expected at apparent g values of 16 and 12, respectively. However, the transition probability is zero for the “ms = ±4” transition at exact B0||z and is very low for the “ms = ±3” transition, but the Zeeman effect is very anisotropic and causes mixing of the ms wave functions for both doublets, so EPR transitions become allowed for B0 oriented between the principal axes. The major intensity in a parallel-mode powder spectrum actually arises from orientations close to the z-axis. The variation of the resonance field as a function of the field orientation is visualized by the transition roadmaps shown on the right in Fig. 4. Powder derivative signals are expected from these diagrams only at the turning points of the respective traces. The corresponding g values for the “ms = ±3” and “ms = ±4” derivative spectra are indicated by the vertical dotted lines and g labels in diagram B in Fig. 4. Interestingly, there is a third resonance traced in the diagrams, coming from the highest pair of ms levels for the field close to the y-axis (marked with an asterisk). That also has a turning point at g ≈ 16, so the contribution in the EPR spectrum entirely overlaps the “ms = ±4” transitions.
Fig. 4.

A Energy level scheme for St = 4 with D = −0.66 cm−1, E/D = 0.165, g = 2.03, and B0 applied in the z-direction. The ms values given in quotation marks are labels for the magnetic sublevels but do not reflect the true composition of the wave functions. The vertical bold bars indicate parallel-mode transitions for ν = 9.6 GHz. B–D Transition roadmap diagrams for the same system, which are plots of the resonance fields for hypothetical single-crystal rotation experiments. The resonances of the “ms = ±4” and “ms = ±3” non-Kramers doublets are shown in red and blue, whereas transitions within the highest two ms levels are marked with an asterisk: B rotation of B0 in the z → x-plane; C rotation in the y → x-plane; and D rotation in the y → z-plane
The roadmap diagram also explains why the g ≈ 11 and the g ≈ 16 derivative signals have different intensities in the parallel-mode spectra at 20 K, although the Boltzmann law arrogates almost equal population of the corresponding non-Kramers doublets at this condition of temperature and field strength. Apparently, the ground-state “ms = ±4” doublet has a higher magnetic anisotropy, so the resonances become spread over a wider field range than for the “ms = ±3” transitions and the signal amplitudes are attenuated. Additionally, also lower intrinsic transition probabilities can be found at g ≈ 16, because higher ms functions become less mixed as a result of the competition between Zeeman- and zero-field interaction. At 3 K, in contrast, the g ≈ 11 signal fades out because the excited states become mostly depopulated. The effect yields substantial information about the magnitude of D and E/D, although the constraint on the parameters obtained from the intensity variations in the parallel-mode spectra in Fig. 3 may be somewhat weakened by the odd overall experimental line shapes of the two signals. However, the actual D and E/D values are nicely supported by the successful simulation of the resonance lines in the normal-mode spectrum in Fig. 2.
The parallel-mode spectra of the superreduced activator (Fig. 3) resemble remarkably well the EPR features of the all-ferrous cluster in the Fe-protein [21, 24] with a ground-state spin St = 4 [18, 31]. This cluster also shows a very sharp line in the parallel-mode X-band spectra around g ≈ 16 (actually at g = 16.4), but at g ≈ 11 only a weak, broad feature is observed at elevated temperature, whereas for the activator the line at g ≈ 11 is sharp and appears already at 3 K as a strong sharp signal. The inherent similarity of the spectra illustrates the same spin state for the all-ferrous clusters of the activator and the Fe protein and reveals similar physical properties. The differences in the EPR spectra arise from only small differences in the zero-field interaction, which slightly modulate the properties of the resonating non-Kramers doublets as shown in Fig. 4 (D = −0.75 cm−1, E/D = 0.30 for the Fe protein [31] compared with −0.66 cm−1 and 0.165 for the activator). In view of the fact that the zero-field splitting of the cluster spin manifold is inherited from the single-ion properties of four ferrous sites [42], and that there are 15 different ways to obtain St = 4 from such a spin tetrade [31], the close spectral similarity suggests very similar electronic properties of the all-ferrous clusters from activator and the Fe protein.
Mössbauer spectroscopy
We used 57Fe Mössbauer spectroscopy to quantify superreduction of the activator samples and to further substantiate the comparison with the Fe protein. Figure 5 shows the zero-field Mössbauer spectrum of titanium(III) citrate reduced activator recorded at 140 K. The asymmetric pattern could be fitted with three quadrupole doublets (I, IIa, and IIb), of which the prominent subspectrum I can be clearly assigned to [4Fe–4S]+ clusters from remaining starting material, because its isomer shift, δ = 0.51 mm/s, and quadrupole splitting, ΔEQ = 0.94 mm/s, are virtually identical to those observed for reduced activator at 80 K (δ = 0.53 mm/s, ΔEQ = 0.95 mm/s) [5]. The relative intensity of subspectrum I accounts for 54% of the total iron.
Fig. 5.

Zero-field Mössbauer spectrum of titanium(III) citrate reduced activator at 140 K. Subspectrum I and the superposition of subspectra IIa and IIb represent the contributions from reduced [4Fe–4S]+ and superreduced [4Fe–4S]0 clusters, respectively. δ(I) = 0.51 mm/s, ΔEQ(I) = 0.94 mm/s, 54% relative intensity; δ(IIa, IIb) = 0.65 mm/s, ΔEQ(IIa) = 1.51 mm/s, ΔEQ(IIb) = 2.59 mm/s; intensity ratio of IIa and IIb fixed at 3:1
Superreduction of the activator and formation of all-ferrous [4Fe–4S]0 clusters is revealed by the presence of the wide split component with higher isomer shift shown as the shaded area in Fig. 5. The corresponding all-ferrous [4Fe–4S]0 clusters from Av2 showed a similar pattern, but one that could be fitted with four subspectra according to four different iron sites. However, three subspectra were strongly overlapping and had similar quadrupole splittings in the range ΔEQ = 1.2–1.7 mm/s, but one deviated, showing ΔEQ = 3.08 mm/s [18, 31]. Such distinct differences cannot be resolved from the spectra of the all-ferrous cluster of the activator because of broad lines and limited experimental resolution, but the basic distribution pattern of three similar sites and one different site seems to persist also for the activator. We could fit our data with two Lorentzian doublets, IIa and IIb, having identical isomer shifts and a fixed intensity ratio of 3:1, so subspectrum IIa accounts for three similar iron sites of the all-ferrous cluster with unresolved signals, and subspectrum IIb represents a fourth, discernibly different site. Details on the significance of the fit model are given in the electronic supplementary material.3 The Mössbauer parameters obtained are δ = 0.65 mm/s for IIa and IIb and ΔEQ = 1.51 mm/s for IIa and ΔEQ = 2.59 mm/s for IIb, respectively. The high isomer shift and large quadrupole splittings are typical for tetrahedral Fe(II)S4 sites with localized valences. Particularly the isomer shift, which is the key parameter for the assignment of valence states, compares very well with that of the monomeric Fe(II) centers in reduced rubredoxin (δ = 0.70 mm/s at 4.2 K) [32, 43] or with that of the diferrous [2Fe–2S] cluster in a plant-type ferredoxin (δ = 0.71 mm/s at 4.2 K) [44]. Finally it matches exactly the value found for the all-ferrous cubane clusters in the superreduced Fe-protein (δ = 0.65 mm/s at 130 K). We assume that the rather strong line broadening found for the activator, Γfwhm(IIa) = 0.7 mm/s, Γfwhm(IIb) = 0.47 mm/s, is caused by further heterogeneity of the cluster sites in the frozen protein sample. We can exclude broadening by paramagnetic relaxation at 140 K since basically the same pattern was observed at 80 K.
It was not possible to achieve higher yields of superreduced [4Fe–4S]0 clusters than the approximately 50% mixture with remaining [4Fe–4S]+ clusters observed in the Mössbauer spectra shown here. Prolonged incubation with titanium(III) citrate or a higher excess of reducing agent did not improve the result. All attempts failed to fully convert a sample by the use of any chemical means, whereas photolytic reduction of the activator with deazaflavin led to cluster deterioration and precipitation of the protein. Because of this restriction, the midpoint of the [4Fe–4S]+ → [4Fe–4S]0 transition could not be exactly determined, but we estimate that the redox potential is lower than that of titanium(III) citrate, for which the value E0′1/2 = −720 mV against the standard hydrogen electrode is reported [45] (E0′ = −420 mV; or E0 = 0 mV). This estimation is based on the fact that most likely two protons per electron are released during the oxidation of TiIII to TiIV, which forms a titanyl ion:
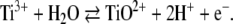 |
Hence at pH 8.0, at which the all-ferrous cluster was prepared, the redox potential of titanium(III) citrate should be—according to the Nernst equation—2 × 60 mV more negative, i.e., E0′ = −840 mV.
Magnetic Mössbauer measurements of titanium(III)-treated activator were performed at 4.2 K with applied fields of 0.2, 4, and 7 T. They yielded a complex magnetic hyperfine pattern with a large overall splitting of about 10 mm/s as shown in Fig. 6. On the basis of the model applied for the fit of the zero-field data, the spectra could be decomposed into three components. The inner part in the range from −2.5 to 3 mm/s is mostly determined by the contribution from the reduced activator having [4Fe–4S]+ clusters with spin S = 3/2 [5]. The component could be reasonably well simulated (subspectrum I, blue lines) by using isomer shift, quadrupole splitting, zero-field splitting, and magnetic hyperfine parameters as published previously for the S = 3/2 species4 [5]. The relative intensity of subspectrum I was fixed to 54% as taken from the zero-field spectrum.
Fig. 6.

Applied-field Mössbauer spectra of titanium(III) citrate reduced activator recorded at 4.2 K with B = 0.2, 4 and 7 T. The blue lines are spin-Hamiltonian simulations for subspectrum I from reduced [4Fe–4S]+ clusters with spin S = 3/2 [5] (Footnote 3). The thick green lines and the shaded areas mark the superposition of the two subspectra IIa and IIb from superreduced [4Fe–4S]0 clusters with the parameters as given in Table 1. The individual contribution of subspectrum IIb from the unique Fe(II) site with antiparallel aligned local spin is additionally shown as a thin green line above the other spectra
The outer hyperfine lines in the magnetic Mössbauer spectra found at about −4 and +5.5 mm/s are the best indicators for the paramagnetic contribution from the all-ferrous [4Fe–4S]0 clusters, which must have large internal fields at the iron nuclei because of the large spin. The magnetic splitting of the corresponding spectra is clearly outside the typical range of hyperfine splitting known for [4Fe–4S]+ clusters with spin S = 3/2 or 1/2. We could readily simulate that contribution from the superreduced activator (Fig. 6, green lines with shaded areas) by using the spin-Hamiltonian for spin St = 4 (Eq. 1), amended with the 57Fe hyperfine interaction with four sites grouped in the two subspectra IIa and IIb introduced above. Particularly the field dependence of the Mössbauer spectra, which depends critically on the zero-field splitting, is well reproduced with the corresponding zero-field splitting parameters D = −0.66 cm−1 and E/D = 0.165 obtained from the EPR analysis. The complete set of Mössbauer parameters used in the simulation of the magnetic spectra are summarized in Table 1. We have to mention, however, that particularly the results obtained for subspectrum IIb are not unique, because of the relatively poor signal-to-noise ratio of the broad spectra and the strong overlap with the remaining starting material, which perturb particularly the analysis of the otherwise informative weak-field spectra. Therefore, the solution given for the anisotropy of the A tensors and the components of the electric field gradient tensors (η and the Euler angles α, β, and γ) have to be regarded as generic. The main objective of presenting the analysis of the magnetic Mössbauer spectra is to show that the data are nicely consistent with the EPR analysis.
Table 1.
Mössbauer parameters for the all-ferrous [4Fe–4S]0 cluster of superreduced activator with spin St = 4 at 4.2 K
| Subspectrum IIa (three sites, 75% intensity) | Subspectrum IIb (one site, 25% intensity) | |
|---|---|---|
| δ (mm/s) | 0.71 (3) | 0.71 (6) |
| ΔEQ (mm/s) | +1.80 (5) | +2.6 (1) |
| η | 0.8 (4) | 0.7 (4) |
| α (°) | 90a | 90a |
| β (°) | 54 (10) | 125 (40) |
| Ax/gNβN (T) | −10.2 (1) | +5.0 (5) |
| Ay/gNβN (T) | −6.4 (1) | +8.0 (5) |
| Az/gNβN (T) | −7.23 (2) | +7.8 (1) |
| A0/gNβN (T)b | −7.95 (7) | +6.6 (4) |
The zero-field parameters used for the simulation were D = −0.66 cm−1 and E/D = 0.165. The Euler angles α, β, and γ rotate the electric field gradient tensor with respect to the principal axes system of the zero-field splitting. The numbers given in parentheses are rough estimates of the error margins obtained by single-parameter variation and visual inspection
aThe angles α and γ were fixed at 90° and 0° to reduce the number of parameters
bIsotropic value is 1/3 tr(A)
The three iron sites of subspectrum IIa that are indistinguishable at zero field also could not be further discriminated with applied fields, neither by sign or size of the quadrupole splitting nor by the magnetic hyperfine interaction. The isomer shift and quadrupole splitting appear to be uniform, with values close to those at 140 K with appropriate corrections for the lower temperature, and the hyperfine splitting can be described by only a single A tensor. Any possible heterogeneity in subspectrum IIa is masked by the anisotropy of A and the rather poor experimental resolution. The isotropic part A0 of the hyperfine tensor is negative for subspectrum IIa, which is best seen from the behavior of the resolved outer magnetic lines. They both have a strong component that is moving “inward” with increasing strength of the applied field from B = 0.2–7 T, owing to opposing internal and applied fields. Since the corresponding negative sign of A0 is like the sign intrinsically expected for iron(II) with respect to the local spin SFe = 2, the three iron sites forming subspectrum IIa must be basically coupled “parallel” to the total spin St = 4 of the [4Fe–4S]0 cluster.
The isotropic value A0 for subspectrum IIb, in contrast, is taken to be positive. This causes the increasing magnetic splitting of a part of the outer hyperfine lines moving outward with increasing applied field. For demonstration of this effect the corresponding subspectrum IIb is also shown separately as a thin green line above the superposition of subspectra IIa and IIb and the experimental traces in Fig. 6. Thus, the local spin of the corresponding unique Fe(II) site is predominantly opposite to the total spin of the [4Fe–4S]0 cluster. The specific scheme of negative and positive A0 values was adopted from the study of the [4Fe–4S]0 cluster in superreduced Fe-protein [31]. There it was also demonstrated that the occurrence of one positive and three negative A values is a necessary common property for any ground multiplet with total spin St = 4 that one can obtain from a tetrade of four spins SFe,i = 2 with isotropic exchange interaction [31].
The effective A-tensor components found for the [4Fe–4S]0 cluster of the superreduced activator are relatively weak compared with the intrinsic a0 value for high-spin Fe(II) sites like that of rubredoxins (isotropic part A0/gNβN = −7.94 and 6.6 T for the activator cluster compared with a0/gNβN = −16.2 T for rubredoxin) [32, 43], but they are in the range of those of the [4Fe–4S]0 centers from the Fe-protein [A0 = −11.2 (±0.2) and 5 MHz [31], which corresponds to A0/gNβN = −8.1 (±0.14) and +3.6 T, respectively]. The apparent reduction of the effective hyperfine coupling in the cubane clusters is caused by competing antiferromagnetic spin coupling. In the corresponding coupling scheme invoked for the all-ferrous cluster of the Fe-protein [31], the spin-projection factors Ki that describe the effective hyperfine interaction with respect to the total spin of the individual sites, A0(i) = Kiao(i), have been obtained as K1 = K2 = K3 = 7/15 (subsumed in subspectrum IIa here) and K4 = −2/5 (subspectrum IIb). With the a0 value for rubredoxin, a0/gNβN = −16.2 T, one obtains A0(IIa)/gNβN = −7.6 T and A0(IIb)/gNβN = 6.5 T. Both values are in reasonable agreement with the experimental result found for the superreduced activator, similar to what was found for the Fe-protein. In summary the spectroscopic properties found for the all-ferrous [4Fe–4S]0 cluster from the superreduced activator resemble quite closely those of the superreduced Fe-protein from A. vinelandii nitrogenase.
Discussion
In the past, cubane [4Fe–4S]n+ clusters had only been known to undergo one-electron redox reactions using either the [4Fe–4S]+/2+ couple or the [4Fe–4S]2+/3+ couple, but not both [46]. According to which couple the clusters are supporting, the iron–sulfur proteins were graded as ferredoxin-type proteins or high-potential iron proteins (HIPIPs) [47]. The latter are most abundant in phototrophic organisms. Except for the case of the [4Fe–4S]n+ cluster in the HIPIP from Chromatium vinosum, which can be artificially cycled from the +3 to the +1 state [48], the classification appeared to be unique and generally valid, until Watt and Reddy [26] reported that the [4Fe–4S]+ cluster of the nitrogenase Fe-protein from A. vinelandii could be reversibly reduced by methyl viologen to an all-ferrous [4Fe–4S]0 state where all irons have oxidation state +2. The midpoint potential for this reaction was initially given as −0.46 V versus the standard hydrogen electrode (SHE), but later studies with other reductants reported −0.79 V versus SHE [29], in contrast to −0.31 V for the first reduction (+2 ↔ +1). Titanium(III) citrate reduced nitrogenase Fe-protein was the subject of elaborate EPR and Mössbauer spectroscopic investigations, which yielded detailed insight in the electronic structure of the all-ferrous cluster and revealed spin S = 4 for the ground state [18, 31]. More recently, the formation of another superreduced state of Av2 with a diamagnetic [4Fe–4S]0 cluster and a different electronic absorption spectrum was observed, if the in vivo reductant FldHQ was used [30]. Notably, that system has a sufficiently high redox potential to support nitrogenase catalysis with two-electron transfer by using the (+2/0) couple of the iron–sulfur cluster.
Hitherto, nitrogenase Fe-protein was the only known enzyme which supported the all-ferrous [4Fe–4S]0 cluster. Also the first example of a synthetic all-ferrous [4Fe–4S]0 complex with ligands other than phosphane was reported only recently [49, 50]. It represents a very valuable analogue of the biological all-ferrous cluster, since it has been structurally and spectroscopically characterized. The Mössbauer spectra of the compound closely match those of titanium(III) citrate reduced Av2. In this paper we have demonstrated that superreduction in biological systems is not a unique phenomenon of the Fe-protein, but also the activator from 2-hydroxyglutaryl-CoA-dehydratase is able to support the [4Fe–4S]2+/+ as well as the [4Fe–4S]+/0 redox cycles of a single cubane cluster.
The electronic structures of the superreduced [4Fe–4S]0 cluster from the Fe-protein and from the activator appear to have a series of remarkable spectroscopic similarities. First, the UV–vis spectra of both systems exhibit a shoulder-type signal around 500–530 nm which causes distinct color changes from brown to orange-red upon reduction. We note that reduced iron(II) rubredoxin does not exhibit a similar absorption in the visible region [51]. Moreover, both [4Fe–4S]0 clusters exhibit the same total spin St = 4 in their ground state, and even the zero-field-splitting parameters compare remarkably well (activator, D = −0.66 cm−1, E/D = 0.165; Fe-protein, D = −0.75 cm−1, E/D = 0.33 [31]). The moderate difference of the rhombicity parameters for the superreduced activator, deviating from the almost fully rhombic limit encountered with superreduced Fe-protein, gives rise to the two distinct lines in the parallel-mode EPR spectra of the activator instead of the strong single line found for the Fe protein.
The Mössbauer isomer shifts for both centers are very similar and uniform for the respective cluster sites, owing to the same ferrous character of the metal ions. Our data are consistent with the specific distribution pattern of three iron(II) sites with similar quadrupole splitting and similar magnetic hyperfine coupling, and one different iron(II) site with larger quadrupole splitting and positive A tensor found for the Fe protein. In both enzymes the clusters are suspended at the interface of a protein dimer, but the spectra lack any indication of twofold symmetry, as one might expect from the respective protein structures [8, 52]. Since the electric field gradient, which gives rise to the quadrupole splitting, senses only local charge asymmetries, the more uniform appearance of the activator might reflect a smaller asymmetry of the actual cluster coordination in the protein and solvent moiety. Apart from that, the Mössbauer lines of the superreduced activator are unusually broad. Therefore we suppose that the molecular structure is probably not very well defined in the frozen solutions and exhibits microheterogeneity at the cluster site which leads to a distribution of quadrupole splittings for each of the iron(II) sites.
Microheterogeneity seems also to be the reason for the unusual shape of the EPR signals measured from the superreduced activator. The resonances show a superposition of sharp lines and very broad shoulders centered at similar resonance fields. The corresponding distribution of spin-Hamiltonian parameters, like the distribution of electric field gradients, may be related to the location of the cluster at a sensitive position in the protein structure between two protein monomers [8], where variations in the conformational substates of the protein may have a particularly strong impact on the geometry and electronic properties of the cluster. Such a distribution of spin-Hamiltonian parameters has been observed previously also for the dithionite-reduced activator with [4Fe–4S]+ clusters having the unusual spin state S = 3/2 [5] (the unusually broad EPR spectrum is redepicted in the inset in Fig. 2). Here, it is interesting to mention that the [4Fe–4S]+ cluster of the Fe-protein has spin S = 1/2 under physiologically reduction conditions, but it also changes to S = 3/2 if it is reduced in the presence of urea [53].
In contrast to the situation for the Fe protein from nitrogenase, it was never possible to obtain a complete superreduction of the activator; at best a ratio close to 1:1 could be achieved for [4Fe–4S]+ and [4Fe–4S]0 clusters. One may speculate whether this is related to the conformational distributions invoked above for interpretation of the spectroscopic heterogeneities. Probably, some fraction within the ensemble of variations may impose such unfavorable conditions on the molecular structure of the cluster and the resulting electronic properties such that further reduction is practically impossible. This interpretation seems to be supported by the observation that hard photoreduction by using deazaflavin as a potent electron source leads to inactivation and precipitation of the protein. However, for the time being also the trivial explanation cannot be excluded, namely, the midpoint redox potential for the [4Fe–4S]+/0 couple could be lower than that of titanium(III) citrate at pH 8.0 (see above), and thus even with a tenfold excess of this agent the cluster only becomes half reduced. We also cannot readily explain why titanium(III) citrate reduced activator is found to be enzymatically inactive, whereas the EPR and Mössbauer spectra reveal the presence of about 50% [4Fe–4S]+ clusters remaining, which should be catalytically competent. We presume that in the enzymatic assay the activator is fully reduced to the [4Fe–4S]0 state because of the very low protein concentration in micromolar range, in contrast to the situation in the highly concentrated spectroscopic samples where the protein is in millimolar abundance.
Since the ability of a [4Fe–4S] cluster to undergo two one-electron redox reactions using both the [4Fe–4S]+/2+ and the [4Fe–4S]+/0 couples should be basically related to its geometry, i.e., the metric details of molecular structure, as well as the (dielectric) properties of the protein site harboring the metal center. In this sense it may not be accidental that both proteins, the nitrogenase Fe-protein and the activator of 2-hydroxyglutaryl-CoA dehydratase, have very similar quaternary structures: homodimeric proteins in which a [4Fe–4S] cluster bridges the subunits. In both proteins an α-helix from each subunit points with its N-terminus towards the cluster and thus may stabilize with its dipole a negative charge, which should increase the redox potential. In the Fe-protein of nitrogenase this helix–cluster–helix architecture forms an angle of 150°, which upon binding of ATP and interaction with the iron–molybdenum protein opens to 180° [17]. Whether this conformational change further influences the redox potential of the cluster remains to be established. The helix–cluster–helix architecture of the activator of 2-hydroxyglutaryl-CoA dehydratase forms an angle of 105°, which is 45° closer than that of the Fe-protein of nitrogenase [8]. Upon ATP binding, the activator angle is thought to open in a similar manner [54], but a crystal structure of the complex between the activator and dehydratase stabilized by AlF4−-ADP (J. Kim and W. Buckel, unpublished results) is not available yet. Most likely the common electronic properties of the [4Fe–4S] clusters as well as the common structural features favor a related mechanism of both enzyme systems. Redox potentials are lowered by hydrolysis of ATP in order to achieve chemically difficult reductions of the substrates molecular nitrogen and 2-hydroxyglutaryl-CoA, respectively. However, owing to the observed inability of the superreduced activator to catalyze the enzyme reaction, the [4Fe–4S]2+ → [4Fe–4S]+ cluster transition and the subsequent electron transfer to the dehydratase component (HgdAB) appears to be the only biologically relevant charge transfer. The situation seems to be more complex for the nitrogenase mechanism. It was postulated that the all-ferrous nitrogenase Fe-protein indeed is an active redox state for nitrogen reduction. First, Watt and Reddy [26] reported that the ATP consumption for nitrogen fixation using all-ferrous Fe-protein is only half of that reported for the [4Fe–4S]+ cluster containing Fe-protein (one ATP per two transferred electrons compared with one ATP per one electron) [26]. In the meantime, the principal physiological relevance of the [4Fe–4S]0 cluster appears to have been corroborated for the nitrogenase system [30]. Maybe the enzyme can switch the redox pathway to choose between different electron donors for nitrogen reduction available in the cell. A similar role for the all-ferrous cluster of the 2-hydroxyglutaryl-CoA dehydratase system is still elusive.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Comparison of the fit model for the zero-field Mössbauer spectra of Ti(III) citrate-treated activator with one and two Lorentzian doublets for the all-ferrous cluster. (PDF 374 kb)
Acknowledgments
The authors thank Iris Schall (Philipps-Universität, Marburg) for growth of A. fermentans in large amounts, and Bernd Mienert and Frank Reikowski (MPI for Bioinorganic Chemistry) for the EPR and Mössbauer measurements. The work was supported by the Deutsche Forschungsgemeinschaft and the Fonds der Chemischen Industrie.
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
In the following the term “dithionite-reduced” will refer to the [4Fe–4S]+ state of the cluster and “superreduced” will refer to the [4Fe–4S]0 state of the cluster. “Ti(III)-reduced” refers to the treatment of the enzyme with titanium(III) citrate which for the activator generates a mixture of clusters in the [4Fe–4S]0 and the [4Fe–4S]+ state.
The value of gx can be hardly determined because the x-direction does not contribute significantly to the spectra; the errors of gy and gz are about 0.03 and 0.1. Actually, for the simulation of the normal-mode spectrum a slightly higher gz value of 2.1 was applied than the value of 2.03 for the parallel-mode spectrum. We assume that the difference is due to the broad distribution of zero-field parameters and the potential consequence that in the integer-spin system different parts of that contribution may contribute differently in normal-mode and parallel-mode spectra.
The presence of a distinct subspectrum IIb as separate contribution to the broad signals coming from the all-ferrous clusters of titanium(III) citrate treated activator is consistent with the spectrum from Av2, but it is not so significant in our data. We mention that a basic fit with only one subspectrum for the whole superreduced fraction is also possible. However, the least-squares error sum is clearly lower for two doublets IIa and IIb (0.791 compared with 0.939), and some systematic fit deviations are seen at the shoulders of the main lines if only one doublet II is invoked. Details are given in the electronic supplementary material.
Subspectrum I is a superposition of two components according to two types of iron sites for the [4Fe–4S]+ clusters with S = 3/2, as found previously. The parameters are D = 1.3 cm−1, E/D = 0.22, g = 1.873, δ = 0.55 mm/s, ΔEQ = 0.98 mm/s, and η = 0.63, A/gNβN = −11.93 T for the first component (50%), and η = 1, A/gNβN = −9.23 T for the second component. The minor 3% contribution from clusters with S = 1/2, previously found from EPR, was neglected.
In memory of Prof. Helmut Beinert (1913–2007), who was a pioneer in spectroscopy and biochemistry of iron–sulfur clusters.
Electronic supplementary material
The online version of this article (doi:10.1007/s00775-008-0345-z) contains supplementary material, which is available to authorized users.
References
- 1.Buckel W, Barker HA (1974) J Bacteriol 117:1248–1260 [DOI] [PMC free article] [PubMed]
- 2.Buckel W, Hetzel M, Kim J (2004) Curr Opin Chem Biol 8:462–467 [DOI] [PubMed]
- 3.Buckel W, Martins BM, Messerschmidt A, Golding BT (2005) Biol Chem 386:951–959 [DOI] [PubMed]
- 4.Kim J, Hetzel M, Boiangiu CD, Buckel W (2004) FEMS Microbiol Rev 28:455–468 [DOI] [PubMed]
- 5.Hans M, Buckel W, Bill E (2000) Eur J Biochem 267:7082–7093 [DOI] [PubMed]
- 6.Thamer W, Cirpus I, Hans M, Pierik AJ, Selmer T, Bill E, Linder D, Buckel W (2003) Arch Microbiol 179:197–204 [DOI] [PubMed]
- 7.Hans M, Bill E, Cirpus I, Pierik AJ, Hetzel M, Alber D, Buckel W (2002) Biochemistry 41:5873–5882 [DOI] [PubMed]
- 8.Locher KP, Hans M, Yeh AP, Schmid B, Buckel W, Rees DC (2001) J Mol Biol 307:297–308 [DOI] [PubMed]
- 9.Hans M, Sievers J, Müller U, Bill E, Vorholt JA, Linder D, Buckel W (1999) Eur J Biochem 265:404–414 [DOI] [PubMed]
- 10.Smith DM, Buckel W, Zipse H (2003) Angew Chem Int Ed Engl 42:1867–1870 [DOI] [PubMed]
- 11.Kim J, Darley D, Buckel W, Pierik AJ (2008) Nature (in press) [DOI] [PubMed]
- 12.Kim J, Darley D, Buckel W (2005) FEBS J 272:550–561 [DOI] [PubMed]
- 13.Tamannaei A (2003) Diploma thesis, Philipps-Universität, Marburg
- 14.Dickert S, Pierik AJ, Buckel W (2002) Mol Microbiol 44:49–60 [DOI] [PubMed]
- 15.Dickert S, Pierik AJ, Linder D, Buckel W (2000) Eur J Biochem 267:3874–3884 [DOI] [PubMed]
- 16.Boll M, Fuchs G (2005) Biol Chem 386:989–997 [DOI] [PubMed]
- 17.Schindelin H, Kisker C, Schlessman JL, Howard JB, Rees DC (1997) Nature 387:370–376 [DOI] [PubMed]
- 18.Angove HC, Yoo SJ, Burgess BK, Münck E (1997) J Am Chem Soc 119:8730–8731 [DOI]
- 19.Angove HC, Yoo SJ, Münck E, Burgess BK (1998) J Biol Chem 273:26330–26337 [DOI] [PubMed]
- 20.Erickson JA, Nyborg AC, Johnson JL, Truscott SM, Gunn A, Nordmeyer FR, Watt GD (1999) Biochemistry 38:14279–14285 [DOI] [PubMed]
- 21.Musgrave KB, Angove HC, Burgess BK, Hedman B, Hodgson KO (1998) J Am Chem Soc 120:5325–5326 [DOI]
- 22.Nyborg AC, Erickson JA, Johnson JL, Gunn A, Truscott SM, Watt GD (2000) J Inorg Biochem 78:371–381 [DOI] [PubMed]
- 23.Nyborg AC, Johnson JL, Gunn A, Watt GD (2000) J Biol Chem 275:39307–39312 [DOI] [PubMed]
- 24.Strop P, Takahara PM, Chiu HJ, Angove HC, Burgess BK, Rees DC (2001) Biochemistry 40:651–656 [DOI] [PubMed]
- 25.Vincent KA, Tilley GJ, Quammie NC, Streeter I, Burgess BK, Cheesman MR, Armstrong FA (2003) Chem Commun 20:2590–2591 [DOI] [PubMed]
- 26.Watt GD, Reddy KRN (1994) J Inorg Biochem 53:281–294 [DOI]
- 27.Johnson MK (1998) Curr Opin Chem Biol 2:173–181 [DOI] [PubMed]
- 28.Rees DC (2002) Ann Rev Biochem 71:221–246 [DOI] [PubMed]
- 29.Guo ML, Sulc F, Ribbe MW, Farmer PJ, Burgess BK (2002) J Am Chem Soc 124:12100–12101 [DOI] [PubMed]
- 30.Lowery TJ, Wilson PE, Zhang B, Bunker J, Harrison RG, Nyborg AC, Thiriot D, Watt GD (2006) Proc Natl Acad Sci USA 103:17131–17136 [DOI] [PMC free article] [PubMed]
- 31.Yoo SJ, Angove HC, Burgess BK, Hendrich MP, Münck E (1999) J Am Chem Soc 121:2534–2545 [DOI]
- 32.Yoo SJ, Meyer J, Achim C, Peterson J, Hendrich MP, Münck E (2000) J Biol Inorg Chem 5:475–487 [DOI] [PubMed]
- 33.Noodleman L, Lovell T, Liu TQ, Himo F, Torres RA (2002) Cur Opin Chem Biol 6:259–273 [DOI] [PubMed]
- 34.Torres RA, Lovell T, Noodleman L, Case DA (2003) J Am Chem Soc 125:1923–1936 [DOI] [PubMed]
- 35.Gaffney BJ, Silverstone HJ (1993) In: Berliner LJ, Reuben J (eds) EMR of paramagnetic molecules (biological magnetic resonance), vol 13. Plenum, New York
- 36.Hanson GR, Gates KE, Noble CJ, Griffin M, Mitchell A, Benson S (2004) J Inorg Biochem 98:903–916 [DOI] [PubMed]
- 37.Trautwein AX, Bill E, Bominaar EL, Winkler H (1991) Struct Bond 78:1–95
- 38.Klees AG, Linder D, Buckel W (1992) Arch Microbiol 158:294–301 [DOI] [PubMed]
- 39.Mabbs FE, Collison D (1992) Electron paramagnetic resonance of d transition metal compounds. Elsevier, Amsterdam
- 40.Hagen WR (1982) Biochim Biophys Acta 708:82–98
- 41.Hendrich MP, Debrunner PG (1989) Biophys J 56:489–506 [DOI] [PMC free article] [PubMed]
- 42.Bencini A, Gatteschi D (1990) EPR of exchange coupled systems. Springer, Berlin
- 43.Schulz CE, Debrunner PG (1976) J Phys (Paris) Colloq 37:153
- 44.Yoo SJ, Meyer J, Münck E (1999) J Am Chem Soc 121:10450–10451 [DOI]
- 45.Strubl R (1938) Collect Czech Chem Commun 10:475–492
- 46.Holm RH, Kennepohl P, Solomon EI (1996) Chem Rev 96:2239–2314 [DOI] [PubMed]
- 47.Spiro TG (1982) Iron sulfur proteins. Wiley-Interscience, New York
- 48.Cammack R (1973) Biochem Biophys Res Commun 54:548–554 [DOI] [PubMed]
- 49.Rao PV, Holm RH (2004) Chem Rev 104:527–559 [DOI] [PubMed]
- 50.Scott TA, Berlinguette CP, Holm RH, Zhou HC (2005) Proc Natl Acad Sci USA 102:9741–9744 [DOI] [PMC free article] [PubMed]
- 51.Eaton WA, Lovenberg W (1973) In: Lovenberg W (ed) Iron–sulfur proteins, vol 2. Academic, New York, pp 131–162
- 52.Georgiadis MM, Komiya H, Chakrabarti P, Woo D, Kornuc JJ, Rees DC (1992) Science 257:1653–1659 [DOI] [PubMed]
- 53.Lindahl PA, Day EP, Kent TA, Orme-Johnson WH, Münck E (1985) J Biol Chem 260:11160–11173 [PubMed]
- 54.Kim J, Lu Y, Buckel W (2007) C R Chim 10:742–747
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Below is the link to the electronic supplementary material.
Comparison of the fit model for the zero-field Mössbauer spectra of Ti(III) citrate-treated activator with one and two Lorentzian doublets for the all-ferrous cluster. (PDF 374 kb)