

Abstract
Applications of tandem mass spectrometry (MS/MS) techniques coupled with high-performance liquid chromatography (HPLC) in the identification and determination of phase I and phase II drug metabolites are reviewed with an emphasis on recent papers published predominantly within the last 6 years (2002–2007) reporting the employment of atmospheric pressure ionization techniques as the most promising approach for a sensitive detection, positive identification and quantitation of metabolites in complex biological matrices. This review is devoted to in vitro and in vivo drug biotransformation in humans and animals. The first step preceding an HPLC-MS bioanalysis consists in the choice of suitable sample preparation procedures (biomatrix sampling, homogenization, internal standard addition, deproteination, centrifugation, extraction). The subsequent step is the right optimization of chromatographic conditions providing the required separation selectivity, analysis time and also good compatibility with the MS detection. This is usually not accessible without the employment of the parent drug and synthesized or isolated chemical standards of expected phase I and sometimes also phase II metabolites. The incorporation of additional detectors (photodiode-array UV, fluorescence, polarimetric and others) between the HPLC and MS instruments can result in valuable analytical information supplementing MS results. The relation among the structural changes caused by metabolic reactions and corresponding shifts in the retention behavior in reversed-phase systems is discussed as supporting information for identification of the metabolite. The first and basic step in the interpretation of mass spectra is always the molecular weight (MW) determination based on the presence of protonated molecules [M+H]+ and sometimes adducts with ammonium or alkali-metal ions, observed in the positive-ion full-scan mass spectra. The MW determination can be confirmed by the [M-H]- ion for metabolites providing a signal in negative-ion mass spectra. MS/MS is a worthy tool for further structural characterization because of the occurrence of characteristic fragment ions, either MSn analysis for studying the fragmentation patterns using trap-based analyzers or high mass accuracy measurements for elemental composition determination using time of flight based or Fourier transform mass analyzers. The correlation between typical functional groups found in phase I and phase II drug metabolites and corresponding neutral losses is generalized and illustrated for selected examples. The choice of a suitable ionization technique and polarity mode in relation to the metabolite structure is discussed as well.
Keyword: Metabolite, Drug, Xenobiotic, High-performance liquid chromatography–mass spectrometry, Mass spectrometry, Atmospheric pressure ionization
Introduction
A wide variety of manufactured and natural chemicals such as drugs, industrial chemicals, pollutants, pesticides, pyrolytic products from cooked food, secondary plant metabolites (polyphenols, flavonoides, glycosides, terpenes, steroids, alkaloids, antibiotics) and toxins from bacteria, molds, plants and animals can influence the vital functions of an organism and they represent serious risks for its homeostasis, integrity and health. These categories of chemical compounds are usually denoted as xenobiotics, foreign compounds or exogenous compounds. The ability of the organism to cope with exposure of xenobiotics is dependent on the existence of various protective and defensive mechanisms leading to the consecutive detoxication connected usually with an increase of the hydrophilicity via metabolism and with the excretion of modified xenobiotics from the organism.
This review is focused on the methodical approaches in drug bioanalysis. Drugs are extensively employed xenobiotics used for the prevention, diagnosis or therapy of various diseases and pathological states. For the possibility to monitor the movement of drugs in various body compartments, their conversion in tissues and organs and their excretion from the organism, suitable bioanalytical methods have to be developed and validated. The development and validation of bioanalytical methods involve sample preparation steps followed by the separation of the drug and its metabolites from interfering endogenous compounds and the detection of analytes by various detection principles. Every experienced bioanalyst should be familiar with the fate of the drug after its administration into the body and the disposition of the organism with the drug molecule during the movement among various body compartments. This information is very important for the planning of required absorption–distribution–metabolism–excretion (ADME) studies.
From the chemical structure and physicochemical properties of the drug, the bioanalyst should be able to forecast (1) the probable behavior of the drug during its transfer through various biological membranes and among various body compartments and (2) the expected chemical structures of drug metabolites formed during phase I and phase II biotransformation. This knowledge may help during the selection of suitable sample preparation steps. In the subsequent instrumental analysis, the selection of a suitable liquid-phase separation technique and detection principle (photodiode-array, fluorescence, polarimetric, mass-spectrometric detection) should be performed.
Pharmacological and xenobiochemical notes
The drug is administered in necessary doses usually repeatedly into the organism via parenteral (intravenous, intramuscular, subcutaneous, intraperitoneal), enteral (oral, sublingual, rectal) or inhalation routes [1, 2]. Transdermal administration, when the drug is dosed on the skin together with an enhancer of transdermal penetration, is sometimes also employed for systemic drug delivery [3]. Further drug movement among the individual compartments of the organism and its interaction with the macromolecules (transporters, receptors, enzymes, etc.) can be divided into four phases: absorption, distribution, metabolism and excretion. Detailed information about the fate of drugs in an organism is available in many monographs [1, 2, 4, 5]. In this text only the basic facts needed for the bioanalyst are discussed.
The absorption is the transfer of a drug from the site of administration to the distribution compartment (bloodstream). As the preparation of a sample from blood and its derivatives (plasma, serum) is relatively simple, the identification and determination of the drug and its metabolites in blood (plasma, serum) is often employed in pharmacokinetic studies (including bioequivalence studies). The actual concentration of the drug (active metabolite) should range between subtherapeutic and toxic concentrations. Therapeutic drug monitoring employs bioanalytical methods for the individual optimization of therapeutic dosing in patients with different ability to absorb, metabolize and excrete the administered drug (patients with intestinal, hepatic and/or renal malfunction). Bioequivalence studies compare the pharmacokinetic parameters of two drug forms (test vs. reference) administered sequentially to volunteers and are also based on the instrumental determination of the drug or its active metabolite(s) [6, 7].
The drug distribution is the process by which the drug leaves the bloodstream and enters the interstitium (extracellular fluid) or the tissue cells, including target tissues for the drug. The receptors located in target tissues interact with the drug molecules and trigger the corresponding pharmacodynamical (therapeutic) effect. If drug analysis in target tissue is needed, the tissue sample has to be homogenized in a suitable medium before further sample preparation steps.
The drug administered into the organism interacts not only with above mentioned receptors but also with other macromolecules, such as enzymes and nucleic acids. During the drug circulation in the organism, its chemical structure is usually enzymatically modified [4, 5]. The sense of this drug metabolism (biotransformation) consists in the increase of drug hydrophilicity in order to facilitate its excretion from the organism, predominantly via the uropoetic system and the gastrointestinal tract. The modification of the chemical structure of the parent drug is catalyzed by various drug-metabolizing enzymes [8–11], which are localized preponderantly in liver, but enzyme activities were also found in lung, intestinum, kidney, brain and blood. When the drug is administered orally, it may be absorbed from the gastrointestinal tract by either passive diffusion or active transport via carrier proteins. Being absorbed across the gastrointestinal tract, the drug must traverse the portal system and undergoes the so-called first-pass metabolism before entering the systemic circulation [1, 4, 5]. Hence, the bioavailability of drugs, which are rapidly metabolized by the liver or intestinal enzymes, is relatively low, because the active drugs are changed to more hydrophilic and usually inactive metabolites. Some of these metabolites are released into the systemic circulation and excreted via the uropoetic system; some of them are transported from liver to bile and via the gastrointestinal tract are excreted from the organism. The enterohepatic recycling may prolong the pharmacological effect of certain drugs and their metabolites [12]. Urine, bile, intestinal content and feces contain large amounts of drug metabolites. From the view of bioanalysts, these biomatrices are most important sources of interesting chemical structures formed during the drug biotransformation [13–17].
The reactions catalyzed by xenobiotic biotransforming enzymes can be divided into two groups called phase I and phase II reactions. Phase I reactions involve oxidation [8], reduction [9–11], hydrolysis [18, 19] and some other less frequent reactions, for example, chiral inversion [20, 21]. The oxidation, reduction and hydrolysis reactions expose or introduce a reactive functional group (–OH, –SH, –NH2, –NH-, –COOH) in the molecule of parent drug. These phase I metabolites, but also the parent drug possessing one or more of the above mentioned functional groups, may significantly increase their hydrophilicity during phase II biotransformation reactions by the conjugation of a xenobiotic molecule (drug or its metabolites) with an eobiotic (endogenous) molecule (glucuronic acid, amino acid, etc). Two of the phase II reactions (acylation, methylation) cause increased lipophilicity, but on the other hand the detoxication of an irritant group in the molecule of the xenobiotic compound, for example, 4-aminophenol → N-acetyl-4-aminophenol (i.e., paracetamol), sulfane contained in flatus → dimethyl sulfide. Most of these reactions (glucuronidation, sulfation, conjugation with glutathione, GSH, amino acids) lead to a large increase in xenobiotic hydrophilicity; hence, they greatly promote the excretion of the drug from the body.
Sample preparation
A representative biomatrix sampling is the first important step of an effective bioanalysis. The fluid samples (blood, urine, bile) are mostly well accessible and treatable; the solid samples (feces, tissues) have to be homogenized (Potter-Elvehjem homogenizer or ultrasonic disintegrator) in a suitable medium (for example, phosphate buffer). As early as possible in the assay, a suitable amount of the chosen internal standard should be added to account for losses and errors introduced throughout the sample preparation process. Analytes contained in the homogenized samples have to be separated from endogenous macromolecules in the biomatrix and transferred into a solution containing the analytes studied and only a minimized amount of endogenous compounds. Many processes (filtration, centrifugation, derivatization, solvent evaporation, solvent pH adjustment) can be employed at this stage of bioanalysis in order to increase the recovery of analytes during the final extraction.
The presence of any phase II metabolite in the biomatrix can be preliminarily verified using xenobiochemical experiments with β-glucuronidase and/or arylsulfatase [14, 17, 22–25]. These two enzymes are able to quite selectively and quantitatively cleave the ether or ester bond between glucuronic or sulfuric acid and the xenobiotic part of the phase II drug metabolite under the back-formation of the phase I drug metabolite. Comparative high-performance liquid chromatography (HPLC) analyses of the sample incubated without an enzyme and the same sample after enzymatic treatment using β-glucuronidase and/or arylsulfatase afford qualitative and quantitative information about phase I and phase II drug metabolites in the biological sample [14].
The derivatization is a chemical modification of the analyte(s) in the sample leading to the stabilization of labile molecule(s) before the instrumental analysis [26], to increase the lipophilicity and extraction yield [27], to mask functional group(s) causing separation problems during the chromatography (e.g., esterification of carboxylic acids) or to introduce a chromophore (fluorophore) into the molecule of the analyte to improve spectrophotometric detection of a weakly absorbing analyte (a detection-oriented derivatization [28]). When HPLC–mass spectrometry (MS) analyses are used, only the first three arguments for the derivatization are plausible, but additionally the derivatization can lead to the characteristic fragmentation applicable, for example, for distinguishing positional isomers [29].
The extraction of the drug, its metabolites and the internal standard is usually the final part of the sample preparation process, enabling the preconcentration of analytes into a small volume before the instrumental analysis. In the drug bioanalyses, the following types of extraction procedures are mostly employed [30]: liquid–liquid extraction [31], solid-phase extraction [32–34], solid-phase microextraction [35–37], supercritical fluid extraction [38] and specific extraction using molecularly imprinted polymers [39].
Bioanalysis based on HPLC-MS measurements combines the separation efficiency of the chromatographic columns with the detection power of MS. In general, MS is considered to be one of the most universal tools for the identification and determination of xenobiotics. However, MS detection must be preceded by separation techniques (e.g., HPLC, capillary electrophoresis or gas chromatography after the derivation) for the structural discrimination of positional isomers and stereoisomers based on different retention times of positional isomers [40] or stereoisomers [41], or additionally by polarimetric or circular-dichroism detectors based on the measurement of the changed direction of plane-polarized light when passing through the individual concentration zones of both enantiomers [41]. Figure 1 shows the polarimetric detection (Fig. 1b) of two enantiomeric metabolites formed by the reduction of the keto group to a secondary alcohol in comparison with UV detection (Fig. 1a). The potential of a photodiode-array UV detector should not be underestimated, because it can, in some cases, differentiate the positional isomers, e.g., the position of O-acetylation is identified on the basis of the UV spectra [17]. When metabolic products have a strong change in the chromophoric system, UV spectra enable one to distinguish different chromophoric groups of metabolites [40], e.g., dimeflurone has a carbonyl group in the conjugation with three aromatic rings. When this carbonyl is reduced to alcohol, the conjugation system and the corresponding UV spectra are significantly changed (Fig. 2). The hyphenation approach to the bioanalysis (the combination of HPLC with photodiode-array UV, fluorescence, polarimetry, NMR and MS as detection techniques) offers the most comprehensive analytical information involving the chromatographic behavior of individual analytes under study and the information from individual detectors. These data are usually sufficient for the identification of the chemical structures of unknown metabolites and for their determination in the extract from the biomatrix together with the parent compound.
Fig. 1.

Chromatogram of reduced S- and R-enantiomeric metabolites of flubendazole using a UV detection at 250 nm and b polarimetric detection at 430 nm. (Adapted with permission from [41])
Fig. 2.

Comparison of UV spectra of a the parent drug and related metabolites with a keto group conjugated with three aromatic rings and b metabolites with a keto group reduced to hydroxyl. (Adapted with permission from [40])
High-performance liquid chromatography
The structural changes caused by metabolic reactions obviously have appropriate effects on the chromatographic behavior of metabolites (Tables 1 and 2). In most cases, the metabolic changes lead to an increased polarity of the metabolites and therefore decreased retention in reversed-phase (RP) HPLC systems in relation to the parent drug. The polarity increase and simultaneous retention decrease effects may be especially strong for phase II metabolites, for example, sulfates, glucuronides and other polar conjugates (Table 2). The separation of the parent drug and polar metabolites in one chromatographic run usually requires the use of gradient elution and often the addition of ionic modifiers into the mobile phase to increase the retention of polar conjugates [30]. Common HPLC methods for the analysis of drugs and their metabolites without MS detection frequently rely on the use of nonvolatile buffers and additives, such as phosphate buffers and other inorganic additives. These nonvolatile additives cannot be recommended for HPLC-MS coupling owing to the possible contamination of the ion source and ion optics, and also strong ion-suppression effects [42, 43]. The orthogonal geometry of the ion source can somewhat reduce the contamination and suppression effects [42], but anyway it is better to substitute nonvolatile additives by more volatile analogs providing in most cases comparable chromatographic performance. The concentration of additives should be kept as low as possible, maintaining the required retention selectivity as one aspect, but avoiding the contamination and ion-suppression effects as another point of view. Inorganic acids in the mobile phase should be replaced by organic acids (formic or acetic acid) with typical concentrations lower than 0.1% [44, 45], but sometimes higher [46, 47]. For an alkaline environment, ammonium hydroxide in a similar concentration range can be used with HPLC columns with an extended stability at alkaline pH [18, 48]. Typical volatile salts in HPLC-MS are ammonium acetate or formate with a common concentration range from 2 to 10 mmol/l [42, 49–52], rarely higher [30, 53]. Trifluoroacetic acid is a favorite ionic additive in bioassays [54, 55], but it may cause serious ion-suppression effects in HPLC-MS. If ion-pairing HPLC is needed for the successful separation, then dialkylammonium or trialkylammonium acetates or formates [42] are recommended for negatively charged analytes and perfluorated carboxylic acids [43] for positively charged analytes. For HPLC-MS analysis of chiral drugs or metabolites formed with a new chiral center (e.g., keto reduction to alcohol [40, 56]), a chiral chromatographic column should be used for proper analytical characterization of metabolites [41, 57]. The typical column packings for chiral separations of metabolites are α-acid glycoprotein [58, 59], cellulose tris(3,5-dimethylphenylcarbamate) [41], vancomycin [60], etc. Polarimetric detection is an obvious detection choice for the chiral separation, because it can distinguish enantiomers, unlike MS detection [61].
Table 1.
Effects of common phase I metabolic reactions on the mass-spectrometric behavior and retention in comparison with the parent drug
| Nominal mass shift (ΔDa) | Metabolic reaction (elemental composition change) | Exact mass shift (mDa) | Examples of relative retention shifta | References |
|---|---|---|---|---|
| – | Azo reduction to amines (R1N = NR2 to R1NH2 + R2NH2) | – | – | [145] |
| –14xb | Hydrolysis of esters to carboxylic acid (-CxH2x) | –15.7x | – | [106] |
| –90 | Reductive debenzylation (-C7H6) | –47.0 | 0.74 [134] | [134] |
| –78 | Reductive debromination (-Br+H) | +89.5 | – | [113] |
| –74 | Oxidative debenzylation (-C7H6+O) | –52.0 | – | [180] |
| –68 | Loss of CF3 (-CF3+H) | +12.6 | – | [107] |
| –62 | Oxidative debromination (-Br+OH) | +84.4 | 0.39 [17] | [17] |
| –56 | Debutylation (-C4H8) | –62.6 | – | [107] |
| –45 | Hydrolysis of nitrate to alcohol (-NO2+H) | +14.9 | – | [113] |
| Reductive loss of nitro group (-NO2+H) | +14.9 | 1.37 [46] | [46, 106] | |
| –44 | Decarboxylation (-CO2) | +10.2 | – | [106] |
| –42 | Depropylation (-C3H6) | –47.0 | 0.73—isopropyl [131]; 0.85, 1.09—propyl [24] | [24, 131] |
| –36 | Loss of HCl (-HCl) | +23.3 | – | [148] |
| –34 | Reductive dechlorination (-Cl+H) | +39.0 | – | [148] |
| –30 | Nitro reduction to amine (-O2+H2) | +25.8 | 0.75 [144] | [144] |
| –28 | Deethylation (-C2H4) | –31.3 | 0.87 [76] | [76] |
| –25 | Reductive loss of nitrile group (-CN+H) | +4.8 | 0.88 [45] | [45, 106] |
| –18 | Alcohol dehydration (-H2O) | +10.6 | 2.08c [17] | [17] |
| Oxidative dechlorination (-Cl+OH) | +33.9 | 0.73 [146] | [146] | |
| Reductive defluorination (-F+H) | +9.4 | – | [147] | |
| –16 | Desulfuration (-S+O) | +22.8 | 0.54 [68] | [68] |
| Reduction of sulfoxide to thioether (-O) | +5.1 | – | [113] | |
| Reduction of hydroxylamine to amine (-O) | +5.1 | – | [113] | |
| –14 | O-demethylation (-CH2) | –15.7 | 0.88, 0.93 [19]; 0.56, 0.64 [40]; 0.59 [129]; 0.87, 0.88, 0.90 [130] | [19, 25, 40, 46, 108, 129–131] |
| N-demethylation (-CH2) | –15.7 | 0.82 [52]; 0.84 [135]; 0.87 [51]; 0.88 [19]; 0.90 [40]; 0.92 [133]; 0.95 [130]; 0.99 [45] | [19, 40, 45, 47, 50–52, 130, 133, 135, 136] | |
| S-demethylation (-CH2) | –15.7 | – | [132] | |
| –6 | Aromatization of saturated ring (-H6) | –47.0 | – | [152] |
| –2 | Alcohol oxidation to ketone/aldehyde (-H2) | –15.7 | 1.01 [83] | [83, 106] |
| Ring formation (-H2) | –15.7 | 1.20 [112] | [112] | |
| Oxidative defluorination (-F+OH) | +4.3 | – | [77, 147] | |
| –1 | Oxidative deamination to ketone/aldehyde (-NH3+O) | –31.6 | – | [142] |
| +1 | Oxidative deamination to alcohol (-NH+O) | –16.0 | – | [113] |
| Hydrolysis of amide to carboxyl (-NH+O) | –16.0 | – | [143] | |
| +2 | Ketone/aldehyde reduction to alcohol (+H2) | +15.7 | 0.72 [56]; 0.70 [40] | [33, 40, 56, 126] |
| Hydrogenation (+H2) | +15.7 | 1.13 [124] | [17, 124] | |
| Ring opening (+H2) | +15.7 | 0.59 [149] | [47, 149–151] | |
| +14 | Methyl/methylene oxidation to aldehyde/ketone (+O-H2) | –20.7 | 0.92, 1.02 [102]; 1.05 [45] | [45, 102] |
| Alcohol oxidation to carboxylic acid (+O-H2) | –20.7 | – | [106] | |
| Hydroxylation and cyclization (+O-H2) | –20.7 | – | [106] | |
| +16 | Hydroxylation (+O) | –5.1 | 0.38 [51]; 0.40 [131]; 0.46 [137]; 0.46, 0.50, 0.62 [124]; 0.51, 0.73, 0.82, 0.89, 0.96 [76]; 0.56 [40]; 0.58 [135]; 0.60, 0.62 [112]; 0.61 [45]; 0.69 [130]; 0.80 [52]; 0.82 [138]; 0.85 [102] | [23, 24, 40, 45, 47, 51, 52, 76, 83, 102, 108, 112, 124, 130, 131, 135–139] |
| Epoxidation (+O) | –5.1 | 0.56 [124]; 0.72 [140]; 0.89 [77] | [77, 124, 140, 141] | |
| N-oxidation (+O) | –5.1 | 0.93 [40]; 1.04 [45]; 1.09 [76] | [40, 45, 51, 76, 131, 136] | |
| S-oxidation of thioether to sulfoxide or sulfoxide to sulfone (+O) | –5.1 | 0.34 (S-SO) [141]; 0.82 (S-SO) [138]; 1.02 (SO-SO2) [143] | [138, 141, 143] | |
| Aldehyde oxidation to carboxyl (+O) | –5.1 | – | [126] | |
| Oxidation and ring formation (+O) | –5.1 | 0.46, 0.51 [137]; 0.53, 0.64 [80] | [80, 137] | |
| +18 | Ring opening by water addition (+H2O) | +10.6 | 1.10 [18] | [18, 19] |
| Hydrolysis of nitrile to amide (+H2O) | +10.6 | – | [45] | |
| +30 | Methyl oxidation to carboxylic acid (+O2-H2) | –25.8 | 0.24 [167]; 0.34 [40] | [40, 50, 167] |
| +32 | Dihydroxylation (+O2) | –10.2 | 0.49 [54]; 0.39 [124]; 0.80, 0.81 [160]; 0.84 [83]; 0.63, 0.67, 0.71, 0.79, 0.94 [76] | [54, 76, 83, 124, 160] |
| S-oxidation of thioether to sulfone (+O2) | –10.2 | 0.79 [138] | [138] | |
| +34 | Epoxidation and hydration (+H2O2) | +5.5 | 0.39 [124]; 0.47 [140]; 0.51, 0.68 [141] | [124, 140, 141] |
| +48 | Trihydroxylation (+O3) | –15.3 | – | [113] |
| S-oxidation of thiol to sulfonic acid (+O3) | –15.3 | – | [113] |
aThe parent drug has a relative retention shift (RRS) of 1.00.
bThe value of x corresponds to the length of the alkyl chain.
cRRS is related to another metabolite.
Table 2.
Effects of common phase II metabolic reactions on the mass-spectrometric behavior and retention in comparison with the parent drug
| Nominal mass shift (ΔDa) | Conjugation reaction (elemental composition change) | Drug functional group | Exact mass shift (mDa) | Examples of relative retention shifta | References |
|---|---|---|---|---|---|
| +14 | Methylation (+CH2) | NH2, OH, SH | +15.7 | – | [114] |
| +28 | Formyl conjugation (+CO) | NH2, OH | –5.1 | 1.59 [27] | [27] |
| +42 | Acetyl conjugation (+C2H2O) | NH2, NHNH2, SO2NH2, OH | +10.6 | 1.24 [17], 1.11, 1.81 [27] | [17, 27] |
| +56 | Propionyl conjugation (+C3H4O) | NH2, OH | +26.2 | 2.39, 3.17 [27] | [27] |
| +57 | Glycine conjugation (+C2H3ON) | COOH | +21.5 | 0.70 [164] | [125, 164] |
| +70 | Butyryl conjugation (+C4H6O) | NH2, OH | +41.9 | 6.50 [27] | [27] |
| +79 | Phosphorylation (+PO3) | OH | –41.5 | – | [74] |
| +80 | Sulfation (+SO3) | NH2, SO2NH2, OH | –43.2 | 0.41 [129]; 0.49 [135]; 0.69, 0.92 [160]; 0.78 [83]; 0.79 [51]; 0.81 [52]; 1.13, 1.14 [154]; 1.27, 1.37 [23] | [23, 25, 51, 52, 69, 74, 83, 93, 117, 129, 131, 134, 154, 158, 160] |
| +107 | Taurine conjugation (+C2H5O2NS) | COOH | +4.1 | – | [125, 165] |
| +119 | Cystein conjugation (+C3H5O2NS) | Epoxide, halide | +4.1 | 0.71 (-F+OH) [77] | [77, 117] |
| +129 | Glutamine conjugation (+C5H7O3N) | COOH | +42.6 | – | [117] |
| +144 | Carnitine conjugation (+C7H14O2N) | COOH | +102.5 | – | [181] |
| +160 | Disulfation (+S2O6) | NH2, OH | –86.4 | – | [120] |
| +161 | N-Acetylcystein conjugation (+C5H7O3NS) | Epoxide, halide | +14.7 | 0.83 (-F+OH) [77] | [77, 117, 165, 182] |
| +162 | Glucosylation (+C6H10O5) | OH, COOH | +52.8 | 0.55, 0.64 (cis), 0.79, 0.82, 0.90; (trans) [158]; 0.67 [76] | [76, 117, 130, 154, 158] |
| +176 | Hydroxyl O-glucuronidation (+C6H8O6) | OH | +32.1 | 0.41 [135]; 0.46 [53]; 0.63, 0.70 [157]; 0.65, 0.83 [55]; 0.67, 0.82, 0.86 [154]; 0.74 [51]; 0.78 [52]; 0.78, 0.96 [23]; 0.99, 1.01 [158] | [17, 23, 25, 29, 30, 33, 46, 51–53, 55, 69, 74, 94, 120, 129, 135, 154, 156–159, 183] |
| Acyl O-glucuronidation (+C6H8O6) | COOH | +32.1 | 0.65 [44] | [25, 44, 85, 125, 182, 183] | |
| N-glucuronidation (+C6H8O6) | NH2 | +32.1 | 0.84 [160] | [33, 46, 130, 160, 183] | |
| Quarternary N-glucuronidation (+C6H8O6) | NR3 | +32.1 | 0.31 [130]; 0.81 [53] | [33, 53, 130, 183] | |
| S-glucuronidation (+C6H8O6) | SH | +32.1 | – | [183, 184] | |
| C-glucuronidation (+C6H8O6) | (CO)2CRH | +32.1 | – | [153] | |
| +220 | Indirect carbamate glucuronidation of amines (+C7H8O8) | NH2+CO2 | +21.9 | 1.09 [24] | [24, 50, 117] |
| +306-X | Glutathione conjugation–halide substitution (-X+C10H16O6N3S) | Halide (X) | +76.0 -X | 0.72—fluoride [77] | [77] |
| +305 | Glutathione conjugation via epoxidation (+C10H15O6N3S) | Aromatic | +68.2 | – | [77, 101, 117, 123, 125–127, 161, 162] |
| +352 | Diglucuronidation (+C12H16O12) | OH, COOH,NH2 | +64.2 | – | [154] |
aThe parent drug has a RRS of 1.00.
The current trend in pharmaceutical analysis is the reduction of the analysis time and the increase in the sample throughput, without sacrificing the separation selectivity. Various approaches can be used to achieve these goals. The first possibility is shortening the column length together with reduction of the particle diameter. If the particle size is below 2 μm (sub-2-μm particles), then there is a substantial increase of the column backpressure, which may be technically solved by the development of instrumentation and columns surviving such high pressures, so-called ultraperformance liquid chromatography [49, 62–64]. Another important parameter is temperature, because separation at increased temperature reduces the analysis time and also decreases the column backpressure owing to the decreased viscosity at higher temperature [65]. In addition to C18 silica columns as the gold standard in RP-HPLC, other packing materials may be used as well, shorter alkyls C12 [54, 66] and C8 [67–69], a medium-polarity nitrile column [70] for polar analytes or longer alkyls such as C30 for hydrophobic drugs [34]. Hydrophilic interaction chromatography (HILIC) is based on the hydrophilic interactions between ionic analytes and zwitterionic groups covalently bonded to silica or polymer beads, e.g., –CH2N+ (CH3)2CH2CH2CH2SO3-, which provides a separation mechanism orthogonal to that of RP-HPLC [71]. The great potential of HILIC for polar phase II and also phase I metabolites has been demonstrated in several papers [55, 72, 73] and in many presentations at recent chromatographic conferences. Ionic analytes often have little or no retention in RP-HPLC and ionic mobile-phase additives are troublesome because of MS coupling, so HILIC seems to be the most promising direction for HPLC–tandem MS (MS/MS) analysis of ionic metabolites. Another approach for ionic metabolites is based on the combination of RP and weak anion-exchange separation mechanisms applied in HPLC-MS/MS [74]. An alternative approach to the architecture of chromatographic columns packed with fine particles is the use of monolithic columns [75]. Their high permeability allows the use of higher flow rates and therefore shorter chromatographic runs, as demonstrated for the separation of bupropion metabolites in 23 s [75] at a flow rate of 2 ml/min, which may require flow splitting in the HPLC-MS arrangement. In general, the different types of column packing and column type (particle vs. monoliths) offer different chromatographic selectivity and therefore the fine-tuning of complex separations is feasible. The role of careful optimization of separation conditions should not be overlooked even with MS/MS detection, because some structural isomers cannot be distinguished by MS, e.g., positional isomers and enantiomers.
Another issue in biological analysis is the limited sample amount, which directs attention to columns with reduced inner diameter, such as microcolumns or capillary HPLC columns [76, 77]. The most recent developments deal with the use of separation on a chip, which may be easily coupled with nanoelectrospray ionization [78] with extremely low sample and solvent consumption and enormous sensitivity.
High-performance liquid chromatography–tandem mass spectrometry
MS is considered the third most powerful spectral technique in the structural elucidation of unknown organic compounds, but it has some distinct advantages over X-ray diffraction and NMR spectroscopy in the field of bioanalytical assays. MS can be easily coupled with both gas-phase and liquid-phase separation techniques in the analysis of complex biological samples, it has an extremely high sensitivity, low sample consumption and required information may be obtained relatively easily by a proper selection of ionization technique and mass analyzer. For these reasons, HPLC-MS/MS plays a prominent role in the analysis of biotransformation products [79]. HPLC-NMR coupling suffers from low sensitivity, but if this obstacle can be overcome, then the combination of HPLC-NMR and HPLC-MS/MS generates superior data for full structure elucidation [80].
Ionization techniques
Owing to the large structural variety of all organic and bioorganic molecules, there is not one universal ionization technique suitable for all drugs and their metabolites, but several ionization techniques should be considered depending on the structure, compound polarity, molecular weight (MW), etc. In HPLC-MS/MS coupling, three atmospheric pressure ionization (API) techniques covers the whole range of compound polarities and MWs—electrospray ionization (ESI), atmospheric pressure chemical ionization (APCI) and atmospheric pressure photoionization (APPI). Moreover, the polarity mode can be chosen according to the acidic, neutral or basic character of the analytes. In some cases, the right choice of ionization technique and the polarity mode is not so obvious; then measurements in all available ionization modes should be the first step in the optimization.
The softest ionization technique, ESI, is the method of choice for polar to ionic compounds. This ionization technique has promoted a great boom not only in proteomics but also in drug metabolite identification, because it enables the soft ionization of phase II metabolites, providing reliable information on the MWs of these conjugates, unlike other ionization techniques. For parent drug and phase I metabolites with a lower polarity, APCI and APPI may provide better ionization efficiency and sensitivity [81, 82]. A better tolerance to salts and matrix effects has been reported for APCI [83] and APPI [84] in comparison with ESI. APPI has a similar application range as for APCI, but slightly extended towards nonpolar compounds [85]. APPI is still a relatively new and developing technique with a great potential in the analysis of drug metabolites. The characteristic ions are the same for all API techniques: the base peak of full-scan positive-ion mass spectra is usually the [M+H]+ ion often accompanied by less abundant alkali-metal adducts [M+Na]+ and [M+K]+ [14], while the negative-ion mode typically shows the peak of the deprotonated molecule [M-H]- as the base peak (most references in this review demonstrate such behavior). APPI shows more frequently the formation of radical molecular ions M+., unlike other API techniques. Depending on the mobile-phase composition, other adducts may be observed as well, for example, ammonium acetate additive favors the formation of [M+NH4]+ ions [14, 30]. Rarely, adducts with mobile-phase molecules are found, such as [M+H+ acetonitrile]+ or [M+H+ methanol]+ [86, 87]. The relative abundances of fragment ions in the full-scan spectra are usually low to negligible, but there are known exceptions with extensive fragmentation already in the full-scan spectra, especially for APCI [88].
Matrix-assisted laser/desorption ionization (MALDI) is the second most important ionization technique in proteomics, but its role in drug metabolite identification is not as high. The coupling with HPLC is usually performed in off-line mode, because it offers some advantages over on-line HPLC-MALDI-MS coupling. The effluent coming from the HPLC column is deposited on the MALDI plate using an xyz-positioning device deposited either as a continuous very narrow trace or as distinct droplets in certain time intervals [89]. The droplet deposition may affect the chromatographic resolution, so the continuous trace is preferred. The deposition on the MALDI target with embedded matrix allows one to decouple the chromatographic and mass-spectrometric analyses. An important application of MALDI is the imaging analysis of whole tissue, which is useful for the characterization of distribution of metabolites in individual organs [90, 91].
The discovery of desorption ESI (DESI) [92] should be mentioned among new developments in ionization techniques with potential impact in drug metabolite identification [93, 94]. The principle of DESI is based on the solvent spraying through the stainless steel capillary with the applied voltage, but the sample is not dissolved in the eluent but is placed in front of the sprayed charged liquid, for example, the slice of whole organ tissue or skin. This technique does not require any sample preparation and may be used directly for metabolite imaging of whole tissues or a living organism to monitor where the metabolites are located [95]. Desorption interfaces are also available for other API techniques, such as APCI [96], APPI [97] and the new ionization technique of direct analysis in real time (DART) [98].
The whole review is devoted to the mass-spectrometric analysis of ionized molecules, but in some cases also atomic MS can provide useful information. The hyphenation of HPLC with inductively coupled plasma MS can be used for metabolite detection of drugs containing metals (e.g., Pt, Au and Ru), halogens (I, Br and Cl) or sulfur [99, 100].
Mass analyzers
The function of a mass analyzer is the separation of ions according to their different mass-to-charge (m/z) ratios. The quality of mass separation is characterized by the degree to which close m/z values can be separated in the mass analyzer. The most practical definition of mass resolution (R) is expressed as RFWHM = m/z /Δm/z, where m/z is mass-to-charge value of the target peak and Δm/z is the full width at half maximum of this peak. Mass analyzers are classified into groups of low and high resolution. In general, low-resolution mass analyzers (e.g., quadrupoles and ion traps) have resolution in the low-thousands range, while high-resolution analyzers should provide a minimum resolution of 15,000 for time of flight (TOF) based analyzers [42, 71, 101] or over 50,000 for Fourier transform mass analyzers [102–104]. The advantage of ion traps (both spherical and linear) is the possibility of MSn scan, which is useful for studying fragmentation paths [49, 105–108], as illustrated for the example of the fragmentation pattern determined on the basis of MSn spectra (Fig. 3). The use of high resolution is usually associated with another even more important parameter, mass accuracy, defined as 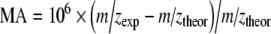 . High mass accuracy measurements should routinely provide values below 5 ppm with external calibration. Accurate masses enable one to determine the right elemental formula among different possibilities at a given nominal value or at least to significantly reduce the number of possible formulas [42]. The determination of the most probable elemental formula is performed in both MS and MS/MS modes, for molecular adducts and fragment ions as well. The benefit of reliable accurate mass measurements for the structural elucidation of unknown metabolites is extremely high, especially in the identification of unexpected or unusual metabolites.
. High mass accuracy measurements should routinely provide values below 5 ppm with external calibration. Accurate masses enable one to determine the right elemental formula among different possibilities at a given nominal value or at least to significantly reduce the number of possible formulas [42]. The determination of the most probable elemental formula is performed in both MS and MS/MS modes, for molecular adducts and fragment ions as well. The benefit of reliable accurate mass measurements for the structural elucidation of unknown metabolites is extremely high, especially in the identification of unexpected or unusual metabolites.
Fig. 3.

Radiochromatogram of monkey bile metabolites labeled with 14C. (Reprinted with permission from [24])
An important issue in accurate mass measurements is the right calibration of the mass scale and regular verification of the precision of the measured masses within the calibrated range [42, 102]. The best results should be obtained with internal calibration, because the calibrant and the analyte enter the mass analyzer at the same time, but on the other hand suppression effects or mass interferences with the calibrant masses may occur. To avoid interference with the chromatographic separation, the calibrant can be introduced after the column through a low-dead-volume T-piece or even better using the second independent sprayer. Robust instruments can provide comparable results even with external calibration on the condition that the calibration is performed as close as possible to the target analyte and no drift of mass scale is apparent. The simplest technical approach is the introduction of the calibrant in the void volume of the chromatographic system and immediately after the last chromatographic peak [109]. For longer runs, additional calibrant can be injected using a six-port valve and an infusion pump during the time when no compounds are eluted. This approach is superior for accurate mass measurements in the analysis of complex mixtures by HPLC-MS, but the critical requirement is the stability and robustness of the mass spectrometer. Other supporting information for the right elemental composition determination is the comparison of the experimental isotopic distribution with theoretically calculated patterns for all elemental compositions considered, expressed as a σ value [109, 110]. The use of an analog-to-digital converter is essential for that purpose, because a time-to-digital converter cannot provide such high precision of relative abundances of individual isotopic peaks [111].
The most widespread tandem mass analyzer providing high mass accuracy used in the metabolite identification is the hybrid double-quadrupole TOF instrument [19, 64, 77, 101, 112]. Other analyzers based on Fourier transform instruments exhibit even better mass accuracies, but at significantly higher instrument costs, such as ion cyclotron resonance (ICR) [104, 113] and Orbitrap as the newest types of mass analyzer [102, 103]. The advantage of ICR is significantly higher mass accuracy and resolution in comparison with all available mass analyzers (better than 1 ppm with the external calibration, achievable sub-ppm with the internal calibration), which often enables the spectral analysis without HPLC separation. However, its widespread is limited owing to high costs and also special technical demands for the installation and operation. In the past, double-focusing magnetic sector instruments had a leading role in this area, but nowadays the abovementioned mass analyzers offer clear advantages, such as more favorable price, smaller size and easier operation.
When the benefits of individual types of tandem mass analyzers for analysis of drug metabolites are compared, trap-based analyzers are preferably used for studying the fragmentation pathways, the triple-quadrupole analyzer is the most suitable choice for quantitation, while TOF-based analyzers or even better Fourier transform analyzers are superior for elemental composition determination because of high mass accuracy.
Metabolite structural changes compared with HPLC-MS/MS behavior
The majority of HPLC-MS/MS applications in metabolite identification are based on RP chromatographic systems. For ionic and ionizable compounds, HILIC separation is becoming more popular owing to the selectivity orthogonal to that of RP-HPLC [71]. In the following discussion, we focus only on the retention in RP-HPLC systems as the prevailing separation mode for drug metabolites. In agreement with the definition of metabolic processes of xenobiotics (see “Pharmacological and xenobiochemical notes”), all metabolites should have a higher polarity and lower retention in RP-HPLC in comparison with the parent drug, with only a few exceptions, such as acylation [27, 100], methylation [114] and some phase I metabolic reactions (Tables 1 and 2). All other metabolites should follow the assumption that their relative retention shift (RRS), 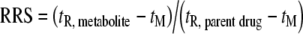 , should be in the interval (0, 1). It is not easy to provide a generalization of RRSs in relation to individual metabolic reactions, because various factors (e.g., mobile-phase composition, gradient steepness, column packing, positional isomerism, drug structure) can significantly affect the RRS, but regardless of these known limitations, Tables 1 and 2 show some distinct trends in the retention behavior of metabolites.
, should be in the interval (0, 1). It is not easy to provide a generalization of RRSs in relation to individual metabolic reactions, because various factors (e.g., mobile-phase composition, gradient steepness, column packing, positional isomerism, drug structure) can significantly affect the RRS, but regardless of these known limitations, Tables 1 and 2 show some distinct trends in the retention behavior of metabolites.
In the drug metabolite identification, the structure of the parent drug is always known; hence, the ionization and fragmentation behavior can be studied first with the parent drug and, if available, standards of phase I metabolites. Then, this knowledge is applied by analogy for the identification of unknown metabolites. Each metabolic reaction leads to a characteristic change in the MWs of the metabolites (Tables 1 and 2), which is the first indication for their identification. The mass shift is reflected both for nominal masses in Daltons and for exact masses in milliDaltons as well. Knowledge of the exact mass shift is applicable for setting very narrow mass windows in selected reaction monitoring (SRM) experiments, yielding significantly increased selectivity of the determination [113], or for the verification of proposed biotransformation pathways [113]. It is worthwhile to apply the so-called nitrogen rule which says that odd MW means an odd number of nitrogen atoms, while even MW means either an even number of nitrogen atoms or their absence. The formation of nitrogen-containing metabolites (e.g., adducts with amino acids, GSH) causes a characteristic change of the odd/even character of MWs. For other basic interpretation rules, reference to specialized literature for electron ionization [115] and soft ionization techniques [88, 116, 117] is recommended. It should be kept in mind that MS is mostly not applicable for distinguishing positional isomers (except for isomers with functional groups on different rings [29]), stereoisomers and enantiomers, so the support of other separation and spectral techniques is essential [117–119]. An established technique for in vivo metabolic studies is labeling with radioactive isotopes, such as 14C [24, 100, 120–122] or 3H [24, 122, 123], where radiocounting detection helps to positively identify metabolites arising from the parent drug in complex biological materials. Figure 4 shows a radiochromatogram of monkey bile after treatment with a drug against anxiety disorder labeled with 14C. Subsequently, drug metabolites detected by radiocounting detection are identified using HPLC-MS/MS and HPLC-NMR techniques [24].
Fig. 4.

Proposed metabolic pathways of AM-630 drug incubated with rat liver microsomes based on ion-trap MSn spectra. (Reprinted with permission from [150])
Tables 1 and 2 were compiled on the basis of references listed in this work and also our experiences and unpublished works. The intention of these tables is to provide a comprehensive list of common biotransformation reactions occurring in humans and animals. Metabolic reactions can occur multiple times or in combination with other reactions, but these reactions are mostly not included here for better clarity, with a few exceptions of important consecutive reactions, such as double hydroxylation [76, 124], epoxidation and GSH conjugation [101, 123, 125–127]. All metabolites are sorted according to their nominal mass shifts in comparison with the parent drug. Then the metabolic reactions are described with their exact mass shifts, RRSs and some selected references. The exact mass shift is the deviation of the atomic masses from the nominal values, for example, the exact mass of hydrogen is 1.007825, therefore the mass defect of hydrogen is +0.007825 Da = + 7.825 mDa (Table 3). If we consider that current TOF-based mass analyzers can routinely measure within an absolute mass error of ±1 mDa or mostly even better, the relatively high differences in mass defects of individual atoms (Table 3) and subsequently also metabolites (Tables 1 and 2) can be applied in metabolite identification. Another common tool in mass-spectrometric studies of fragmentation pathways is the use of standards labeled with stable isotopes [128]. In the case of biotransformation studies, the combination of a nonlabeled and labeled drug leads to twin peaks of metabolites with known mass difference, which is helpful for studying the fragmentation paths in tandem mass spectra [129].
Table 3.
Mass defects for common organic elements occurring in biotransformation reactions
| Element | Nominal atomic mass (Da) | Mass defect (mDa) |
|---|---|---|
| H | 1 | +7.825 |
| C | 12 | 0 |
| N | 14 | +3.074 |
| O | 16 | –5.085 |
| F | 19 | –1.597 |
| Si | 28 | –23.073 |
| P | 31 | –26.238 |
| S | 32 | –27.929 |
| Cl | 35 | –31.147 |
| Br | 79 | –81.662 |
| I | 127 | –95.532 |
Phase I metabolites
Concerning phase I metabolites, the only characteristic but nonspecific neutral loss is usually the loss of water for all metabolites containing hydroxyl functionality (typically for aliphatic alcohols, less common for phenols) often observed already in the full-scan mass spectra. Other less frequent losses are CO2 (for all carboxylic acids formed either by oxidation of alcohols and aldehydes or by the hydrolysis of esters), H2S (for thiols formed by S-dealkylation), SO3 and SO2 (for sulfonic acids formed by S-oxidation). Mostly, phase I metabolites have lower retention in RP-HPLC except for the following cases: denitration (RRS = 1.37 [46]), ring formation (RRS = 1.20 [112]), hydrogenation (RRS = 1.13 [124]) and hydration (RRS = 1.10 [18]). Some metabolites have retention very close to that of the parent drug, such as methyl oxidation to aldehyde (RRS = 0.92 and 1.02 [102], RRS = 1.05 [45]) and N-oxidation (RRS = 0.93 [40], RRS = 1.04 [45]), RRS = 1.09 [76]). Interestingly, some metabolites show a very narrow retention window for different drugs, positional isomers and chromatographic conditions, for example, RRSs of eight N-demethylated metabolites are within the range 0.82–0.99 (Table 1). On the other hand, the most widespread phase I reaction, hydroxylation, yields RRSs from 0.38 [51] to 0.96 [76] for 20 different metabolites, but most of them fit the region RRS = 0.60–0.85.
One of the most common phase I biotransformation reactions is demethylation or in general dealkylation (Table 1). Depending on the position of methyl/alkyl group, the uncovered polar group can be amine (N-demethylation), hydroxyl (O-demethylation) or thiol (S-demethylation). In the case of nitrogen, there is often double alkyl substitution; hence, two deakylations are necessary to uncover the amino group. When the demethylation occurs, new hydroxyl [19, 25, 40, 46, 108, 129–131] or thiol [132] groups can undergo fragmentation in MS/MS mode, yielding the typical neutral losses of H2O or H2S, respectively. In the case of the amino group formed by N-demethylation [133], the neutral loss of NH3 is not common. When the N,N-dimethyl group undergoes the first demethylation, the characteristic neutral loss is CH3NH2 (Δm/z 31) in comparison with CH3NHCH3 (Δm/z 45) for the parent drug, which can be used for differentiation between competitive O-demethylation [27]. In general, the dealkylation leads to the decrease of hydrophobicity and retention in RP-HPLC as well. The dealkylation on the carbon atom is less common, except for losses of bulky alkyls, such as tert-butyl [107] or benzyl [134].
The oxidation is, in general, the introduction of an oxygen atom by various reactions, such as hydroxylation (one or multiple times) [23, 24, 40, 45, 47, 51, 52, 76, 83, 102, 108, 112, 124, 130, 131, 135–139] and aliphatic and aromatic epoxidation [77, 124, 140, 141], which is often followed by hydrolysis leading to dihydrodiol formation [141]. Epoxides have the same elemental composition as hydroxylated metabolites and similar retention behavior, so their differentiation depends on the interpretation of their mass spectra and UV spectra for aromatic epoxides. The direct comparison of epoxide and hydroxylated metabolite in the same position under identical chromatographic conditions shows that the retention of epoxide is slightly lower [124, 140]. Epoxides are normally unstable intermediates, providing dihydrodiols by the subsequent hydration [141], but they may be detected for stabilized systems, such as polyaromatic compounds [77, 124, 140]. A well-known example of a stable epoxide is carbamazepine-10,11-epoxide [124, 140]. The characteristic neutral losses for dihydrodiols are two consecutive losses of water [141]. The oxidation of an already present functional group is represented by the following examples: alcohol oxidation to aldehyde/ketone [83, 106] and then to carboxylic acid [126], amine to hydroxylamine and then to a nitro group [107], oxidative deamination to aldehyde/ketone [142] or alcohol [113], thioether oxidation to sulfoxide [138, 141] and then to sulfone [143]. Oxidation reactions lead to a remarkable decrease of the relative retention in relation to that of the parent drug, with one interesting exception of N-oxide, where the retention is comparable [40] or even slightly higher [45, 76]. Reduction reactions are less frequent, but several biotransformation paths are known, for example, nitro reduction to amine [144], the reduction of azo bond functionality in organic dyes leading to two aromatic amines [145] and ketone/aldehyde reduction to secondary/primary alcohol [33, 40, 56, 126]. Positive-ion MS/MS spectra of N-oxides can show unusual loss of an OH. radical (Δm/z 17), while negative-ion spectra tend to show the formation [M+Cl]- adducts unlike other metabolites measured under the same experimental conditions [40]. Hydrolysis reactions have been reported for esters [106], nitrates [113], amides [143] and nitriles [45].
Organic halides can be degraded by oxidative (i.e., halogen is replaced by a hydroxyl group) [17, 77, 146, 147] or reductive (i.e., halogen is replaced by hydrogen) [113, 147, 148] dehalogenation, where the former is more frequent especially for biotransformation reactions or in general any reaction under aerobic conditions. In addition to these reactions, polyhalogenated xenobiotics (e.g., pesticides) can also provide stable metabolites formed by the loss of HX, for example, the loss of HCl from the banned pesticide DDT [148].
Metabolic reactions can also involve ring formation [112] or ring opening [47, 149–151], often associated with other oxidation and addition reactions. There are a wide variety of other phase I reactions, which are relatively rare, so only a few examples are listed in Table 1, such as decarboxylation of carboxylic acid [106], reductive loss of a nitrile group [45, 106], alcohol dehydration [17] and aromatization of a saturated ring [152].
Figure 5 illustrates the benefit of isotopic labeling for structural studies and fragmentation pattern recognition. Figure 5a shows MS2 and MS3 spectra of the nonlabeled drug 2-methylaminoethoxycarbonyl-4,4′-dimethoxy-5,6,5′,6′-dimethylenedioxybiphenyl-2′-carboxylic acid (DDB-S), while Fig. 5b shows the spectra for the d5-labeled drug. When the labeled part of the molecule is retained during the fragmentation, the fragment ion maintains the isotopic difference introduced by labeling. After the loss of the labeled moiety, the m/z values of both the labeled and the nonlabeled fragments are identical.
Fig. 5.

MS2 and MS3 spectra measured with an ion-trap analyzer for a nonlabeled and bd5-labeled drug 2-methylaminoethoxycarbonyl-4,4′-dimethoxy-5,6,5′,6′-dimethylenedioxybiphenyl-2′-carboxylic acid (DDB-S). (Reprinted with permission from [105])
Phase II metabolites
Glucuronidation is a major phase II metabolic pathway for vertebrates (except for the cat family). The site of glucuronidation (Fig. 6) is generally an electron-rich nucleophilic heteroatom (O, N, S), in rare cases activated carbon with suitable neighboring substitution, for example 1,3-dicarbonyl drugs sulfinpyrazone and phenylbutazone [153]. The second most important biotransformation path is the sulfate conjugation either with hydroxyl or amino groups. These two reactions can occur multiple times [154], if suitable functional groups are available in the drug structure. If one drug gives both sulfate and glucuronide conjugates, then glucuronide typically has a slightly lower retention then the corresponding sulfate in the same position [52]. Of course, this rule cannot be generally applied for conjugates in different positions, because the position of the conjugation has a comparable or an even stronger effect on the retention. The formation of polar phase II conjugates strongly affects the polarity, chromatographic retention, ionization and fragmentation behavior [117], because the most polar functional group in the molecule usually induces the primary fragmentation path. Phase II conjugates can be easily identified according to the shifts of the nominal masses, exact masses and also characteristic neutral losses (Table 2).
Fig. 6.

Different types of glucuronic acid conjugates: 1A hydroxy O-glucuronide, 1B acyl O-glucuronide, 2A amino N-glucuronide, 2B amide N-glucuronide, 2C sulfon amide N-glucuronide, 2D aliphatic quarternary N-glucuronide, 2E aromatic quarternary N-glucuronide, 2F indirect carbamoyl N-glucuronide, 3 thiol S-glucuronide, 4 C-glucuronide
HPLC methods for the separation of glucuronides have been reviewed recently [155]. Depending on the mobile-phase composition, the adduct formation of [M+NH4]+, [M+Na]+ and [M+K]+ can be observed in the positive-ion ESI mode in addition to protonated molecules [M+H]+, while the deprotonated molecule [M-H]- prevails in the negative-ion ESI mode [14, 94]. The most characteristic neutral loss for all types of glucuronides is the loss of anhydroglucuronic acid (Δm/z 176), which is observed in both polarity modes. The mass difference Δm/z 176 can be used in SRM for the quantitation of glucuronides or for the sensitive detection of glucuronides in complex biological mixtures using the neutral loss scan. Hydroxyl O-glucuronide may also provide the [M+H-193]+ fragment ion in addition to the loss of Δm/z 176 [156]. If the drug has more potential sites for glucuronidation, then more positional isomers [23, 55, 154, 157, 158] or multiple glucuronide conjugates [152] can be formed. The identification of the conjugation site by MS is a difficult task and the differentiation is mostly achieved only for conjugation sites on different aromatic rings. An interesting way for differentiating between hydroxyl and acyl glucuronides has been reported recently [25] based on the presence of the [M-H-CO2]- ion for hydroxyl glucuronides and the absence of this ion for acyl glucuronides (Fig. 7). However, it is not clear whether this case is specific or whether the rule can be generally applied owing to the absence of other references. Other attempts at the determination of the conjugation site are based on the chemical derivatization with 2-chloro-1-methylpyridinium iodide [29] or the formation of Co2+ adducts with the characteristic fragmentation [159]. In addition to the abovementioned ions, repetitive losses of water up to four molecules [14] can be observed in MSn spectra of glucuronides, but they do not have very high information content for the structural elucidation. If the concentration of the metabolite entering the mass spectrometer is too high, then the dimeric ions [2M+Na]+ and [2M+K]+ or even trimeric ions [3M+Na]+ and [3M+K]+ accompany common molecular adducts [14]. In general, positive-ion ESI spectra provide better results for the structural analysis of glucuronides, because MS/MS spectra show diagnostic ions unlike the negative-ion mode spectra, where the negative charge is retained on the glucuronide moiety, so the negative-ion spectra contain mainly deprotonated anhydroglucuronic acid at m/z 175 and subsequent losses of CO2 and H2O (m/z 113) and CO (m/z 85) [14, 117].
Fig. 7.

Negative-ion electrospray ionization tandem mass spectrometry spectra of isomeric O-glucuronides: a hydroxy O-glucuronide, b acyl O-glucuronide. (Adapted with permission from [25])
Typical neutral losses for sulfate conjugation are SO3 (Δm/z 80) and SO2 (Δm/z 64) [87, 154, 160], less frequently H2SO4 (Δm/z 98). The presence of the ionic sulfate group significantly induces the fragmentation already in the full-scan spectra, which can lead to reduced relative abundance of (de)protonated molecules. Sometimes, the radical ion [M-H-HSO3]-. may occur [23].
Acetylation and other less common acyl conjugations with formyl, propionyl and butyryl at the aromatic amino group lead to increased hydrophobic character of metabolites and hence significantly increased retention in RP-HPLC depending on the length of acyl chain (Table 2). Characteristic neutral losses for N-acylated metabolites are the following [27]: no neutral loss for formyl, CH2CO for acetyl, CH3CHCO for propionyl and CH3CH2CHCO for butyryl. O-acetylated metabolites also provide neutral losses of CH3COOH (Δm/z 60) [17] or corresponding losses of other acyls.
Phase II metabolites formed by methylation do not have any characteristic neutral loss, because the fragmentation of methyl is apparent only in the case of the absence of any other polar functional groups, for example, N-methylated aniline provides the neutral loss of methyl radical [117].
GSH concentration in cells is up to 10 mM; therefore, GSH conjugation is so frequent and important for biotransformation reactions. There are two basic mechanisms of GSH conjugation [77, 117, 123, 125, 161, 162]. The first mechanism is the substitution of aliphatic or aromatic halides by GSH. The mass shift of this substitution of halides depends on the type of halogen atom, generally expressed as -X+C10H16O6N3S. The second mechanism is GSH conjugation via epoxide formation, then the addition of GSH and the final dehydration resulting in the mass shift +C10H16O5N3S [117, 125, 161, 162]. The isolation and detection of reactive GSH metabolites is not a simple analytical task; therefore, an alternative approach for studying reactive GSH metabolites was suggested on the basis of on-line coupling of an electrochemical cell and tandem mass-spectrometric characterization of the products formed [163]. The mechanisms and fragmentation paths for GSH conjugates are discussed elsewhere in more detail [117]. The characteristic neutral loss is pyroglutamic acid (Δm/z 129) [101, 161, 162], but other losses may occur as well, such as glycine (Δm/z 75) [161, 162] and characteristic cleavages known from peptide chemistry.
The conjugation of the carboxyl functional group via a peptidic bond with amino acids is less common, but some reports can be found for glycine [124, 164], glutamine [117] and taurine [165]. Their identification is based on the characteristic shifts of nominal and exact masses (Table 2) and characteristic neutral losses in MS/MS spectra in a similar manner as known from proteomics. The clear sign of metabolites containing these amino acids is changed even/odd characters of the MW in accordance with the nitrogen rule. Characteristic neutral losses in the positive-ion ESI mode are the losses of free acids, HCOOH (Δm/z 46) and nonspecific loss of water (Δm/z 18), while in the negative-ion ESI mode two characteristic ions are known, the neutral loss of CO2 as for any carboxylic acid and the ion corresponding to deprotonated amino acid, such as m/z 74 for the deprotonated glycine molecule [14].
Phosphorylation is a relatively rare biotransformation path for xenobiotics [74]. The characteristic diagnostic ions for multiple reaction monitoring transitions are [PO2]- at m/z 63 and [PO3]- at m/z 79. Glucosylation is the most important reaction for plants and invertebrates, but its role in mammals is rather limited [74, 158].
Quantitation
The established method for the HPLC quantitation of drugs and their metabolites in biological samples is based on the use of tandem mass-spectrometric detection and the right choice of internal standards [166]. Nowadays, single-stage MS is not recommended for reliable bioanalytical quantitation, because it suffers from insufficient selectivity in comparison with MS/MS. Single-stage MS can only provide the selected ion monitoring record at a certain m/z value, for example the m/z value of [M+H]+ or [M-H]- ions, but the matrix or impurity interferences may occur at the same m/z value; however, in some specific cases of good chromatographic resolution and the absence of matrix interferences, selected ion monitoring may be considered as an alternative quantitative method. Tandem mass-spectrometric analyzers have an additional scan with utmost importance for the quantitative analysis in complex matrices called selected reaction monitoring (SRM) [54, 63, 68, 69, 75, 133, 157, 167]. The first mass analyzer isolates the selected precursor ion (e.g., [M+H]+ in positive-ion mode or [M-H]- in negative-ion mode) with a narrow isolation width, then the second step is the fragmentation of this ion by collision-induced dissociation in the second analyzer (i.e., collision cell) and finally only one abundant product ion with known m/z value is monitored by the third analyzer. A typical example of a tandem mass analyzer used for quantitation is a triple-quadrupole analyzer, where the first quadrupole isolates the precursor ion, the second quadrupole is used only for collision-induced dissociation but not for mass analysis, and the third quadrupole detects the product ions. Multiple reaction monitoring scan has the same meaning as SRM but for more fragmentation reactions in parallel [18, 58, 74, 83, 137, 138, 151, 168]. The typical neutral transitions are related to the metabolite type, for example, the neutral loss of SO3 (Δm/z 80) for sulfates, the anhydroglucuronide unit for glucuronides (Δm/z 176) and glucose for glucosides (Δm/z 162) [158].
Another important aspect is the appropriate choice of the internal standard. The best approach is the use of isotopically labeled analogs of target compounds (isotopomers). If such an internal standard labeled with stable isotopes is added to the biological sample before the sample preparation, the behavior of labeled and that of nonlabeled compounds will be identical in all common analytical procedures. Deuterated analogs have no observable differences in their chromatographic retention approximately up to six deuterium atoms [157], but a very small retention shift can occur for higher deuterium numbers, as illustrated for the example of d9–1-hydroxypyrene [168]. The only remarkable difference is found in their mass spectra, because the m/z values are shifted by the corresponding mass increment, which enables the parallel peak area integration of nonlabeled target drug (or metabolite) and labeled internal standard [74, 133, 167–169]. Another valuable application of deuterated analogs is the verification of proposed fragmentation pathways [105].
The internal standard used for spectrophotometric detection without MS should have a similar polarity, retention time sufficiently close to the compound(s) determined and exhibit similar behavior during the sample preparation procedures, e.g., comparable extraction recoveries. It is also desirable that the analytes and internal standards should have the same or a similar chromophore (fluorophore). From this point of view, the optimal internal standards are structural homologs or analogs of the analyte(s) under the study [28, 40, 41, 170].
In the development and validation of bioanalytical methods, chemical standards of drug metabolites are needed. These standards are sometimes accessible from commercial sources, or they have to be isolated from a biomatrix [16], biosynthesized using isolated or/and immobilized hepatocytes [117, 171–173] or synthesized [27, 40, 56, 61, 170, 174–176]. Many products of O-demethylation, N-demethylation, N-oxidation, carbonyl reduction, etc. are simply accessible by a modification of the parent drug structure [27, 40, 56, 170]. MS, NMR and HPLC purity control is required for each of the newly synthesized standards. Syntheses of the standards of phase II biotransformation products were also described; some of these reactions are very simple (N-acylation [27]), other reactions require an experienced synthesist [61, 174–176].
Concluding remarks and future perspectives
This review is devoted to the HPLC-MS/MS characterization of common phase I and phase II metabolites of xenobiotics. Tables 1 and 2 provide the most comprehensive overview of metabolic reactions occurring in human and animals reported so far in relation to their HPLC-MS/MS behavior. However, the list may not contain some rare metabolic reactions. The interpretation of mass spectra was discussed and some general conclusions were given for typical neutral losses associated with individual phase I and phase II metabolic reactions. The appropriate choices of ionization technique, polarity mode and the type of mass analyzer were considered as well.
Concerning the future perspectives, some trends are already notable at present. The available sample amount for metabolite analysis is typically extremely low; therefore, the use of nanotechnologies is desirable, e.g., capillary HPLC columns or chip-based separations coupled with on-line nanoelectrospray. Recently, several new ionization techniques have been introduced, such as desorption API techniques with potential in the spatial imaging of the distribution of metabolites in individual organs and tissues, which can provide better understanding of biotransformation processes. Fast technical developments in mass-spectrometric instrumentation are resulting in better sensitivity, higher resolution, better mass accuracy and also new types of mass analyzers and ionization techniques. A side effect of this growth is the extensive amount of analytical data, so manual interpretation is becoming a speed bottleneck for high-throughput analysis; therefore, sophisticated software algorithms for the prediction, search and positive identification of both expected and new metabolites are being developed and continuously improved [107, 177–179]. High-throughput analysis is becoming more important, so the use of ultrafast separations combining higher pressures, higher temperatures, faster flow rates, shorter columns, etc. is being established in common pharmaceutical analyses. Nowadays, the most powerful combination of high-tech analytical instrumentation for the structural elucidation of unknown metabolites is the coupling of high-resolution/high mass accuracy HPLC-MS/MS, HPLC-NMR and HPLC with radiocounting detection for metabolites arising from the drug labeled with the radioactive isotope, which can provide the maximum information content, but the investment and operational costs of such hypothetical ultrahyphenation are very high.
Acknowledgements
This work was supported by the grant projects MSM0021627502 and MSM0021620822 sponsored by the Ministry of Education, Youth and Sports of the Czech Republic and the project 203/08/1536 sponsored by the Czech Science Foundation.
References
- 1.Harvey RA, Champe PC (1992) Pharmacology. Lippincott, Philadelphia
- 2.Hardman JG, Limbird LE, Gilman AG (2001) Goodman & Gilman’s the pharmacological basis of therapeutics, 10th edn. McGraw-Hill, New York
- 3.Hrabálek A, Doležal P, Roman M, Macháček M, Šklubalová Z (1994) Pharmazie 49:325–328 [PubMed]
- 4.Parkinson A (2001) In: Klaassen CD (ed) Casarett & Doull’s toxicology—the basic science of poisons. McGraw-Hill, New York
- 5.Gibson GG, Skett P (2001) Introduction to drug metabolism, 3rd edn. Nelson Thornes, Cheltenham
- 6.Nobilis M, Kopecký J, Květina J, Chládek J, Svoboda Z, Voříšek V, Perlik F, Pour M, Kuneš J (2002) J Chromatogr A 949:11–22 [DOI] [PubMed]
- 7.Vybíralová Z, Nobilis M, Zoulová J, Květina J, Petr P (2005) J Pharm Biomed Anal 37:851–858 [DOI] [PubMed]
- 8.Anzenbacher P, Anzenbacherová E (2001) Cell Mol Life Sci 58:737–747 [DOI] [PMC free article] [PubMed]
- 9.Hyndman D, Bauman DR, Heredia VV, Penning TM (2002) Chem Biol Interact 143–144:621–631 [DOI] [PubMed]
- 10.Nordling E, Jörnvall H, Persson B (2002) Eur J Biochem 269:4267–4276 [DOI] [PubMed]
- 11.Kallberg Y, Oppermann U, Jörnvall H, Persson B (2002) Eur J Biochem 269:4409–4417 [DOI] [PubMed]
- 12.Roberts MS, Magnusson BM, Burczynski FJ, Weiss M (2002) Clin Pharmacokinet 41:751–790 [DOI] [PubMed]
- 13.Nobilis M, Kvasničková E, Šroler A, Hais IM (1991) Drug Metab Drug Interact 9:225–240 [DOI] [PubMed]
- 14.Nobilis M, Holčapek M, Kolářová L, Kopecký J, Kuneš M, Svoboda Z, Květina J (2004) J Chromatogr A 1031:229–236 [DOI] [PubMed]
- 15.Nobilis M, Anzenbacher P, Pastera J, Svoboda Z, Hrubý K, Květina J, Ubik K, Trejtnar F (1996) J Chromatogr B 681:143–151 [DOI] [PubMed]
- 16.Lyčka A, Jirman J, Nobilis M, Kvasničková E, Hais IM (1987) Magn Reson Chem 25:1054–1057
- 17.Nobilis M, Pour M, Pavlík J, Šenel P, Kuneš J, Vopršalová M, Kolářová L, Holčapek M (2007) J Chromatogr B 853:10–19 [DOI] [PubMed]
- 18.Vainchtein LD, Rosing H, Mirejovsky D, Huynh V, Lenaz L, Hillebrand MJX, Schellens JHM, Beijnen JH (2006) J Mass Spectrom 41:1268–1276 [DOI] [PubMed]
- 19.Liu Z, Floss HG, Cassady JM, Chan KK (2005) J Mass Spectrom 40:389–399 [DOI] [PubMed]
- 20.Lan SJ, Kripalani KJ, Dean AV, Egli P, Difazio LT, Schreiber EC (1976) Drug Metab Dispos 4:330–339 [PubMed]
- 21.Wsól V, Skálová L, Skotáková B (2004) Curr Drug Metab 5:517–533 [DOI] [PubMed]
- 22.Yilmazer M, Stevens JF, Buhler DR (2001) FEBS Lett 491:252–256 [DOI] [PubMed]
- 23.Nikolic D, Li Y, Chadwick LR, van Breemen RB (2006) Pharm Res 23:864–872 [DOI] [PMC free article] [PubMed]
- 24.Shaffer CL, Gunduz M, O’Connell TN, Obach RS, Yee S (2005) Drug Metab Dispos 33:1688–1699 [DOI] [PubMed]
- 25.Jaggi R, Addison RS, King AR, Suthers BD, Dickinson RG (2002) Drug Metab Dispos 30:161–166 [DOI] [PubMed]
- 26.Jack DB, Brechbühler S, Degen PH, Zbinden P, Riess W (1975) J Chromatogr A 115:87–92 [DOI] [PubMed]
- 27.Nobilis M, Vybíralová Z, Sládková K, Lísa M, Holčapek M, Květina J (2006) J Chromatogr A 1119:299–308 [DOI] [PubMed]
- 28.Nobilis M, Pour M, Kuneš J, Kopecký J, Květina J, Svoboda Z, Sládková K, Vortel J (2001) J Pharm Biomed Anal 24:937–946 [DOI] [PubMed]
- 29.Lampinen-Salomonsson M, Bondesson U, Petersson C, Hedeland M (2006) Rapid Commun Mass Spectrom 20:1429–1440 [DOI] [PubMed]
- 30.Samuel K, Yin W, Stearns RA, Tang YS, Chaudhary AG, Jewell JP, Lanza T Jr, Lin LS, Hagmann WK, Evans DC, Kumar S (2003) J Mass Spectrom 38:211–221 [DOI] [PubMed]
- 31.Pedersen-Bjergaard S, Rasmussen KE (2005) J Chromatogr B 817:3–12 [DOI] [PubMed]
- 32.Li KM, Rivory LP, Clarke SJ (2006) Curr Pharm Anal 2:95–102
- 33.Byrd GD, Ogden MW (2003) J Mass Spectrom 38:98–107 [DOI] [PubMed]
- 34.Moriwaki H, Watanabe A, Arakawa R, Tsujimoto Y, Shimizu M, Noda T, Warashina M, Tanaka M (2002) J Mass Spectrom 37:1152–1157 [DOI] [PubMed]
- 35.Flanagan RJ, Morgan PE, Spencer EP, Whelpton R (2006) Biomed Chromatogr 20:530–538 [DOI] [PubMed]
- 36.Ulrich S (2000) J Chromatogr A 902:167–194 [DOI] [PubMed]
- 37.Musteata FM, Pawliszyn J (2007) J Biochem Biophys Methods 70:181–193 [DOI] [PubMed]
- 38.Radcliffe C, Maguire K, Lockwood B (2000) J Biochem Biophys Methods 43:261–272 [DOI] [PubMed]
- 39.Theodoridis G, Konsta G, Bagia C (2004) J Chromatogr B 804:43–51 [DOI] [PubMed]
- 40.Císař P, Nobilis M, Vybíralová Z, Holčapek M, Kolářová L, Pour M, Kuneš J, Klimeš J (2005) J Pharm Biomed Anal 37:1059–1071 [DOI] [PubMed]
- 41.Nobilis M, Jira T, Lísa M, Holčapek M, Szotáková B, Lamka J, Skálová L (2007) J Chromatogr A 1149:112–120 [DOI] [PubMed]
- 42.Ferrer I, Thurman EM (2005) Anal Chem 77:3394–3400 [DOI] [PubMed]
- 43.Gustavsson SA, Samskog J, Markides KE, Langstrom B (2001) J Chromatogr A 937:41–47 [DOI] [PubMed]
- 44.Groff M, Riffel K, Song H, Lo MW (2006) J Chromatogr B 842:122–130 [DOI] [PubMed]
- 45.Mauriala T, Chauret N, Oballa R, Nicoll-Griffith DA, Bateman KP (2005) Rapid Commun Mass Spectrom 19:1984–1992 [DOI] [PubMed]
- 46.Chan W, Cui L, Xu G, Cai Z (2006) Rapid Commun Mass Spectrom 20:1755–1760 [DOI] [PubMed]
- 47.Weimann A, Sabroe M, Poulsen HE (2005) J Mass Spectrom 40:307–316 [DOI] [PubMed]
- 48.Stockvis E, Rosing H, Crul M, Rieser MJ, Heck AJR, Schellens JHM, Beijhen JH (2004) J Mass Spectrom 39:277–288 [DOI] [PubMed]
- 49.Dear GJ, James AD, Sarda S (2006) Rapid Commun Mass Spectrom 20:1351–1360 [DOI] [PubMed]
- 50.Link M, Hakala KS, Wsól V, Kostiainen R, Ketola RA (2006) J Mass Spectrom 41:1171–1178 [DOI] [PubMed]
- 51.Chen H, Chen Y, Du P, Han F, Wang H, Zhang H (2006) J Pharm Biomed Anal 40:142–150 [DOI] [PubMed]
- 52.Chen H, Chen Y, Wang H, Du P, Han F, Zhang H (2005) Talanta 67:984–991 [DOI] [PubMed]
- 53.Ogura K, Ishikawa Y, Kaku T, Nishiyama T, Ohnuma T, Muro K, Hiratsuka A (2006) Biochem Pharmacol 71:1358–1369 [DOI] [PubMed]
- 54.Gréen H, Vretenbrant K, Norlander B, Peterson C (2006) Rapid Commun Mass Spectrom 20:2183–2189 [DOI] [PubMed]
- 55.Bengtsson J, Jansson B, Hammarlund-Udenaes M (2005) Rapid Commun Mass Spectrom 19:2116–2122 [DOI] [PubMed]
- 56.Nobilis M, Kopecký J, Květina J, Svoboda Z, Pour M, Kuneš J, Holčapek M, Kolářová L (2003) J Pharm Biomed Anal 32:641–656 [DOI] [PubMed]
- 57.Chen YL, Akhtar S, Murai H, Kobayashi M (2005) Rapid Commun Mass Spectrom 19:2681–2688 [DOI] [PubMed]
- 58.Jansson B, Elsherbiny D, Simonsson USH (2006) Rapid Commun Mass Spectrom 20:463–472 [DOI] [PubMed]
- 59.Kelly T, Doble P, Dawson M (2005) J Chromatogr B 814:315–323 [DOI] [PubMed]
- 60.Liu W, Wang F, de Li H (2007) J Chromatogr B 850:183–189 [DOI] [PubMed]
- 61.Perrie JA, Harding JR, Holt DW, Johnston A, Meath P, Stachulski AV (2005) Org Lett 7:2591–2594 [DOI] [PubMed]
- 62.Plumb R, Castro-Perez J, Granger J, Beattie I, Joncour K, Wright A (2004) Rapid Commmun Mass Spectrom 18:2331–2337 [DOI] [PubMed]
- 63.Wang G, Hsieh Y, Cui X, Cheng KC, Korfmacher WA (2006) Rapid Commun Mass Spectrom 20:2215–2221 [DOI] [PubMed]
- 64.Castro-Perez J, Plumb R, Granger JH, Beattie I, Joncour K, Wright A (2005) Rapid Commun Mass Spectrom 19:843–848 [DOI] [PubMed]
- 65.Plumb RS, Rainville P, Smith BW, Johnson KA, Castro-Perez J, Wilson ID, Nicholson JK (2006) Anal Chem 78:7278–7283 [DOI] [PubMed]
- 66.Gasser A, Guse AH (2005) J Chromatogr B 821:181–187 [DOI] [PubMed]
- 67.Kyle PB, Adcock KG, Kramer RE, Baker RC (2005) Biomed Chromatogr 19:231–236 [DOI] [PubMed]
- 68.Williamson LN, Terry AV Jr., Bartlett MG (2006) Rapid Commun Mass Spectrom 20:2689–2695 [DOI] [PubMed]
- 69.Lampinen-Salomonsson M, Beckman E, Bondesson U, Hedeland M (2006) J Chromatogr B 833:245–256 [DOI] [PubMed]
- 70.Xu Y, Huang JC, Liu F, Gao S, Guo QX (2007) J Chromatogr B 852:101–107 [DOI] [PubMed]
- 71.SeQuant (2007) http://www.sequant.com/. Accessed 22 Nov 2007
- 72.Zhang X, Rauch A, Lee H, Xiao H, Rainer G, Logothetis NK (2007) Rapid Commun Mass Spectrom 21:3621–3628 [DOI] [PubMed]
- 73.Hsieh Y, Chen J (2005) Rapid Commun Mass Spectrom 19:3031–3036 [DOI] [PubMed]
- 74.Bicker W, Lämmerhofer M, Keller T, Schuhmacher R, Krska R, Lindner W (2006) Anal Chem 78:5884–5892 [DOI] [PubMed]
- 75.Borges V, Yang E, Dunn J, Henion J (2004) J Chromatogr B 804:277–287 [DOI] [PubMed]
- 76.Fandino AS, Nägele E, Perkins PD (2006) J Mass Spectrom 41:248–255 [DOI] [PubMed]
- 77.Wind M, Spickermann J, Schleimer M, Donzelli M, Gebhardt K, Sturm-Haurany R, Klauer D, Fullhardt P, Schmitt-Hoffmann A (2006) J Mass Spectrom 41:903–910 [DOI] [PubMed]
- 78.Staack RF, Varesio E, Hopfgartner G (2005) Rapid Commun Mass Spectrom 19:618–626 [DOI] [PubMed]
- 79.Kamel A, Prakash C (2006) Curr Drug Metab 7:837–852 [DOI] [PubMed]
- 80.Yilmazer M, Stevens JF, Deinzer ML, Buhler DR (2001) Drug Metab Dispos 29:223–231 [PubMed]
- 81.Sheen JF, Her GR (2004) Anal Bioanal Chem 380:891–897 [DOI] [PubMed]
- 82.Kantharaj E, Ehmer PB, Tuytelaars A, Vlaslaer AV, Mackie C, Gilissen RAHJ (2005) Rapid Commun Mass Spectrom 19:1069–1074 [DOI] [PubMed]
- 83.Schlüsener MP, Bester K (2005) Rapid Commun Mass Spectrom 19:3269–3278 [DOI] [PubMed]
- 84.Theron HB, van der Merwe MJ, Swart KJ, van der Westhuizen JH (2007) Rapid Commun Mass Spectrom 21:1680–1686 [DOI] [PubMed]
- 85.Cai Y, Kingery D, McConnell O, Bach AC II (2005) Rapid Commun Mass Spectrom 19:1717–1724 [DOI] [PubMed]
- 86.Holčapek M, Virelizier H, Chamot-Rooke J, Jandera P, Moulin C (1999) Anal Chem 71:2288–2293 [DOI] [PubMed]
- 87.Guan F, Uboh C, Soma L, Hess A, Luo Y, Tsang DS (2003) J Mass Spectrom 38:823–838 [DOI] [PubMed]
- 88.Holčapek M, Volná K, Ansorgová D (2007) Dyes Pigm 75:156–165
- 89.Foret F, Preisler J (2002) Proteomics 2:360–372 [DOI] [PubMed]
- 90.Khatib-Shahidi S, Andersson M, Herman JL, Gillespie TA, Caprioli RM (2006) Anal Chem 78:6448–6456 [DOI] [PubMed]
- 91.McDonnell LA, Heeren RMA (2007) Mass Spectrom Rev 26:606–643 [DOI] [PubMed]
- 92.Takats Z, Wiseman JM, Gologan B, Cooks RG (2004) Science 306:471–473 [DOI] [PubMed]
- 93.Williams JP, Nibbering NMM, Green BN, Patel VJ, Scrivens JH (2006) J Mass Spectrom 41:1277–1286 [DOI] [PubMed]
- 94.Kauppila TJ, Wiseman JM, Ketola RA, Kotiaho T, Cooks RG, Kostiainen R (2006) Rapid Commun Mass Spectrom 20:387–392 [DOI] [PubMed]
- 95.Ifa DR, Wiseman JM, Song Q, Cooks RG (2007) Int J Mass Spectrom (2007) 259:8–15
- 96.Williams JP, Patel VJ, Holland R, Scrivens JH (2006) Rapid Commun Mass Spectrom 20:1447–1456 [DOI] [PubMed]
- 97.Haapala M, Pól J, Saarela V, Arvola V, Kotiaho T, Ketola RA, Franssila S, Kauppila TJ, Kostiainen R (2007) Anal Chem 79:7867–7872 [DOI] [PubMed]
- 98.Petucci C, Diffendal J, Kaufman D, Mekonnen B, Terefenko G, Musselman B (2007) Anal Chem 79:5064–5070 [DOI] [PubMed]
- 99.Gammelgaard B, Jensen BP (2007) J Anal Atomic Spectrom 22:235–249
- 100.Duckett CJ, Lindon JC, Walker H, Abou-Shakra F, Wilson ID, Nicholson JK (2006) Xenobiotica 36:59–77 [DOI] [PubMed]
- 101.Castro-Perez J, Plumb R, Liang L, Yang E (2005) Rapid Commun Mass Spectrom 19:798–804 [DOI] [PubMed]
- 102.Peterman SM, Duczak N Jr, Kalgutkar AS, Lame ME, Soglia JR (2006) J Am Soc Mass Spectrom 17:363–375 [DOI] [PubMed]
- 103.Sanders M, Shipkova PA, Zhang HY, Warrack BM (2006) Curr Drug Metab 7:547–555 [DOI] [PubMed]
- 104.Godejohann M, Tseng LH, Braumann U, Fuchser J, Spraul M (2004) J Chromatrogr A 1058:191–196 [PubMed]
- 105.Yoo HH, Son J, Lee J, Kim NS, Shin M, Kang MJ, Kim DH (2006) Rapid Commun Mass Spectrom 20:1981–1988 [DOI] [PubMed]
- 106.Tozuka Z, Kaneko H, Shiraga T, Mitani Y, Beppu M, Terashita S, Kawamura A, Kagayama A (2003) J Mass Spectrom 38:793–808 [DOI] [PubMed]
- 107.Anari MR, Sanchez RI, Bakhtiar R, Franklin RB, Baillie TA (2004) Anal Chem 76:823–832 [DOI] [PubMed]
- 108.Lee J, Son J, Koh HY, Pae AN, Kim DH (2003) Anal Biochem 313:292–300 [DOI] [PubMed]
- 109.Ojanperä S, Pelander A, Pelzing M, Krebs I, Vuori E, Ojanperä I (2006) Rapid Commun Mass Spectrom 20:1161–1167 [DOI] [PubMed]
- 110.Jirásko R, Holčapek M, Kolářová L, Nádvornik M, Popkov A (2008) J Mass Spectrom. DOI http://dx.doi.org/10.1002/jms.1405 [DOI] [PubMed]
- 111.Čajka T, Hajšlová J (2007) LC-GC Eur 20:25–31
- 112.Nikolic D, Li Y, Chadwick LR, Grubjesic S, Schwab P, Metz P, van Breemen RB (2004) Drug Metab Dispos 32:272–279 [DOI] [PubMed]
- 113.Zhu M, Ma L, Zhang D, Ray K, Zhao W, Humphreys WG, Skiles G, Sanders M, Zhang H (2006) Drug Metab Dispos 34:1722–1733 [DOI] [PubMed]
- 114.Chang HC, Laly M, Prior RL, Badger TM (2004) J Food Drug Anal 12:161–166
- 115.McLafferty FW, Tureček F (1993) Interpretation of mass spectra. University Science Books, Mill Valley
- 116.Levsen K, Schiebel HM, Terlouw JK, Jobst KJ, Elend M, Preiß A, Thiele H, Ingendoh A (2007) J Mass Spectrom 42:1024–1044 [DOI] [PubMed]
- 117.Levsen K, Schiebel HM, Behnke B, Dötzer R, Dreher W, Elend M, Thiele H (2005) J Chromatogr A 1067:55–72 [DOI] [PubMed]
- 118.Farghali H, Hynie S (1997) In: Bickerstaff GF (ed) Methods in biotechnology: immobilization of enzymes and cells. Humana, Totowa
- 119.Prakash C, Shaffer CL, Nedderman A (2007) Mass Spectrom Rev 26:340–369 [DOI] [PubMed]
- 120.Mullen W, Hartley RC, Crozier A (2003) J Chromatogr A 1007:21–29 [DOI] [PubMed]
- 121.Perkins EJ, Cramer JW, Farid NA, Gadberry MG, Jackson DA, Mattiuz EL, O’Bannon DD, Weiss HJ, Wheeler WJ, Wood PG, Cassidy KC (2003) Drug Metab Dispos 31:1382–1390 [DOI] [PubMed]
- 122.Nassar AEF, Bjorde SM, Lee DY (2003) Anal Chem 75:785–790 [DOI] [PubMed]
- 123.Samuel K, Yin W, Stearns RA, Tang YS, Chaudhary AG, Jewell JP, Lanza T Jr, Lin LS, Hagmann WK, Evans DC, Kumar S (2003) J Mass Spectrom 38:211–221 [DOI] [PubMed]
- 124.Miao XS, Metcalfe CD (2003) Anal Chem 75:3731–3738 [DOI] [PubMed]
- 125.Ma S, Subramanian R (2006) J Mass Spectrom 41:1121–1139 [DOI] [PubMed]
- 126.Aldini G, Granata P, Orioli M, Santaniello E, Carini M (2003) J Mass Spectrom 38:1160–1168 [DOI] [PubMed]
- 127.Alvarez-Sanchez R, Montavon F, Hartung T, Pähler A (2006) Chem Res Toxicol 19:1106–1116 [DOI] [PubMed]
- 128.Zhang JY, Xu F, Breau AP (2004) J Mass Spectrom 39:295–30 [DOI] [PubMed]
- 129.Zhou L, Thakker DR, Voyksner RD, Anbazhagan M, Boykin DW, Hall JE, Tidwell RR (2004) J Mass Spectrom 39:351–360 [DOI] [PubMed]
- 130.Walles M, Thum T, Levsen K, Borlak J (2003) J Chromatogr B 798:265–274 [DOI] [PubMed]
- 131.Kamata T, Katagi M, Kamata HT, Miki A, Shima N, Zaitsu K, Nishikawa M, Tanaka E, Honda K, Tsuchihashi H (2006) Drug Metab Dispos 34:281–287 [DOI] [PubMed]
- 132.Baranczewski P, Stanczak A, Kautiainen A, Sandin P, Edlund PO (2006) Pharmacol Rep 58:341–352 [PubMed]
- 133.Guo B, Li C, Wang G, Chen L (2006) Rapid Commun Mass Spectrom 20:39–47 [DOI] [PubMed]
- 134.Lee J, La S, Ahn BR, Jeong TC, Kim DH (2004) Rapid Commun Mass Spectrom 18:1901–1910 [DOI] [PubMed]
- 135.Shima N, Tsutsumi H, Kamata T, Nishikawa M, Katagi M, Miki A, Tsuchihashi H (2006) J Chromatogr B 830:64–70 [DOI] [PubMed]
- 136.Gjerde J, Kisanga ER, Hauglid M, Holm PI, Mellgren G, Lien EA (2005) J Chromatogr A 1082:6–14 [DOI] [PubMed]
- 137.Guitton J, Cohen S, Tranchand B, Vignal B, Droz JP, Guillaumont M, Manchon M, Freyer G (2005) Rapid Commun Mass Spectrom 19:2419–2426 [DOI] [PubMed]
- 138.Mesplet N, Morin P, Ribet JP (2005) Rapid Commun Mass Spectrom 19:297–302 [DOI] [PubMed]
- 139.Berdelmeijer HA, Roelo ABGH, Hillebrand MJX, Beijnen JH, Schellens JHM, van Tellingen O (2005) Cancer Chemother Pharmacol 56:299–306 [DOI] [PubMed]
- 140.Breton H, Cociglio M, Bressolle F, Peyriere H, Blayac JP, Hillaire-Buys D (2005) J Chromatogr B 828:80–90 [DOI] [PubMed]
- 141.Wsól W, Szotáková B, Baliharová V, Šišpera L, Holčapek M, Kolářová L, Suchanová B, Kuchař M, Skálová L (2004) Collect Czech Chem Commun 69:689–702
- 142.Ensslin HK, Maurer HH, Gouzoulis E, Hermle L, Kovar KA (1996) Drug Metab Disp 24:813–820 [PubMed]
- 143.McKinney AR, Suann CJ, Stenhouse AM (2005) Rapid Commun Mass Spectrom 19:1217–1220 [DOI] [PubMed]
- 144.Sohda K, Minematsu T, Hashimoto T, Suzumura K, Funatsu M, Suzuki K, Imai H, Usui T, Kamimura H (2004) Chem Pharm Bull 52:1322–1325 [DOI] [PubMed]
- 145.Ansorgová D, Holčapek M, Jandera P (2003) J Sep Sci 26:1017–1027
- 146.Li AC, Gohdes MA, Shou WZ (2007) Rapid Commun Mass Spectrom 21:1421–1430 [DOI] [PubMed]
- 147.Pantuck EJ, Pantuck CB, Conney AH (1987) Anesthesiology 66:24–28 [DOI] [PubMed]
- 148.Kitamura S, Shimizu Y, Shiraga Y, Yoshida M, Sugihara K, Ohta S (2002) Drug Metab Dispos 30:113–118 [DOI] [PubMed]
- 149.Miao Z, Kamel A, Prakash C (2005) Drug Metab Dispos 33:879–883 [DOI] [PubMed]
- 150.Zhang Q, Ma P, Wang W, Cole RB, Wang G (2004) J Mass Spectrom 39:672–681 [DOI] [PubMed]
- 151.Segura M, Ortuno J, Farré M, Pacifici R, Pichini S, Joglar J, Segura J, de la Torre R (2003) Rapid Commun Mass Spectrom 17:1455–1461 [DOI] [PubMed]
- 152.Li QG, Humpel M (1992) Eur J Drug Metab Pharmacokinet 17:281–291 [DOI] [PubMed]
- 153.Kerdpin O, Elliot DJ, Mackenzie PI, Miners JO (2006) Drug Metab Disp 34:1950–1953 [DOI] [PubMed]
- 154.Mullen W, Boitier A, Stewart AJ, Crozier A (2004) J Chromatogr A 1058:163–168 [PubMed]
- 155.Kaushik R, Levine B, LaCourse WR (2006) Anal Chim Acta 556:255–266
- 156.Tevell A, Bondesson U, Törneke K, Hedeland M (2004) Rapid Commun Mass Spectrom 18:2267–2272 [DOI] [PubMed]
- 157.Murphy CM, Huestis MA (2005) J Mass Spectrom 40:1412–1416 [DOI] [PubMed]
- 158.Urpí-Sarda M, Jáuregui O, Lamuela-Raventós RM, Jaeger W, Miksits M, Covas MI, Andres-Lacueva C (2005) Anal Chem 77:3149–3155 [DOI] [PubMed]
- 159.Davis BD, Needs PW, Kroon PA, Brodbelt JS (2006) J Mass Spectrom 41:911–920 [DOI] [PubMed]
- 160.Chen C, Meng L, Ma X, Krausz KW, Pommier Y, Idle JR, Gonzalez FJ (2006) J Pharmacol Exp Ther 318:1330–1342 [DOI] [PMC free article] [PubMed]
- 161.Mutlib A, Lam W, Atherton J, Chen H, Galatsis P, Stolle W (2005) Rapid Commun Mass Spectrom 19:3482–3492 [DOI] [PubMed]
- 162.Yan Z, Maher N, Torres R, Caldwell GW, Huebert N (2005) Rapid Commun Mass Spectrom 19:3322–3330 [DOI] [PubMed]
- 163.Lohmann W, Karst U (2007) Anal Chem 79:6831–6839 [DOI] [PubMed]
- 164.Sidelmann UG, Bjornsdottir I, Shockcor JP, Hansen SH, Lindon JC, Nicholson JK (2001) J Pharm Biomed Anal 24:569–579 [DOI] [PubMed]
- 165.Kostiainen R, Kotiaho T, Kuuranne T, Auriola S (2003) J Mass Spectrom 38:357–372 [DOI] [PubMed]
- 166.Xu RNX, Fan LM, Rieser MJ, El-Shourbagy TA (2007) J Pharm Biomed Anal 44:342–355 [DOI] [PubMed]
- 167.Rodila RC, Kim JC, Ji QC, El-Shourbagy TA (2006) Rapid Commun Mass Spectrom 20:3067–3075 [DOI] [PubMed]
- 168.Pigini D, Cialdella AM, Faranda P, Tranfo G (2006) Rapid Commun Mass Spectrom 20:1013–1018 [DOI] [PubMed]
- 169.Duncan MW, Gale PJ, Yergey AL (2006) The principles of quantitative mass spectrometry. Rockpool Productions, Denver
- 170.Nobilis M, Květina J, Anzenbacher P, Vontor T, Zadák Z, Bláha V, Vlček J, Brátová M, Solichová D, Svoboda D (1998) Eur J Drug Metab Pharmacokinetic 23:287–294 [DOI] [PubMed]
- 171.Trejtnar F, Skálová L, Szotáková B, Wsól V (1999) Exp Toxic Pathol 51:432–435 [DOI] [PubMed]
- 172.Hynie S, Kameníková L, Farghali H (1997) In: Bickerstaff GF (ed) Methods in biotechnology: immobilization of enzymes and cells. Humana, Totowa
- 173.Farghali H (2000) In: Berry MN, Edwards AM (eds) The hepatocyte review. Kluwer, Dordrecht
- 174.Stachulski AV, Jenkins GN (1998) Nat Prod Rep 15:173–186 [DOI] [PubMed]
- 175.Stachulski AV (2001) Tetrahedron Lett 42:6611–6613
- 176.Harding JR, King CD, Perrie JA, Sinnott D, Stachulski AV (2005) Org Biomol Chem 3:1501–1507 [DOI] [PubMed]
- 177.Mortishire-Smith RJ, O’Connor D, Castro-Perez JM, Kirby J (2005) Rapid Commun Mass Spectrom 19:2659–2670 [DOI] [PubMed]
- 178.Idborg H, Edlund PO, Jacobsson SP (2004) Rapid Commun Mass Spectrom 18:994–954 [DOI] [PubMed]
- 179.Nassar AEF, Adams PE (2003) Curr Drug Metab 4:259–271 [DOI] [PubMed]
- 180.Ulgen M, Gorrod JW (1996) J Pharm Pharmacol 48:1320–1326 [DOI] [PubMed]
- 181.Olsen J, Li C, Bjornsdottir I, Sidenius U, Hansen SH, Benet LZ (2005) Chem Res Toxicol 18:1729–1736 [DOI] [PubMed]
- 182.Grillo MO, Knutson CG, Sanders PE, Waldon DJ, Hua F, Ware JA (2003) Drug Metab Dispos 31:1327–1336 [DOI] [PubMed]
- 183.Mullangi R, Bhamidipati RK, Srinivas NR (2005) Curr Pharm Anal 1:251–264
- 184.Ethell BT, Riedel J, Englert H, Jantz H, Oekonomopulos R, Burchell B (2003) Drug Metab Disp 31:1027–1034 [DOI] [PubMed]