

Abstract
To study the activation of macrophage and upregulation of costimulatory molecule of CD40 in lipopolysaccharide- (LPS-) induced acute lung injury (ALI) model, and to investigate the pathogenecy of ALI, mice were randomly divided into two groups. ALI model was created by injecting 0.2 mg/kg LPS in phosphate saline (PBS) in trachea. The pathologic changes of mice lungs were observed by HE staining at 24 and 48 hours after LPS treatment, then the alveolar septum damage, abnormal contraction, alveolar space hyperemia, and neutrophils or other inflammatory cells infiltration in the LPS group, but not in the control group, were observed. The expression of CD40 mRNA and CD40 protein molecules were higher in LPS group as compared to the control group by Northern blot and flow cytometry, respectively. Expression of Toll-like receptor-4 (TLR4) in activated macrophage (AMΦ) was higher in LPS group as compared to the control group by RT-PCR. The activation of NF-κB binding to NF-κB consensus oligos increased in LPS group by EMSA in macrophage. The concentrations of TNF-α, MIP-2, and IL-1β cytokines from bronchoalveolar lavage fluid (BALF) were increased significantly in LPS group as compared to the control group by ELISA. The activation of AM and upregulation of costimulatory molecule CD40 induced all kinds of inflammatory cytokines releasing, then led to ALI. Therefore, both of them played vital role in the process of development of ALI.
1. INTRODUCTION
It is well known that almost all of respiratory diseases entail acute lung injury (ALI); it is the end result of common pathways initiated by a variety of local or systemic insults leading to diffuse damage of the pulmonary parenchyma. Despite the accumulation of abundant information regarding the physiological and cellular basis of lung injury and increasing sophisticated intensive care, an improvement in prognosis has lagged behind. Therefore, the fatality rate of ALI is higher and still remains as the major cause of mortality in intensive care units (ICUs) [1]. It has become clear that there is not one mediator responsible for ALI, but rather a complex interplay including some inflammatory mediators. Lipopolysaccharide (LPS) is a glycolipid that constitutes the major portion of outmost membrane of gram-negative bacteria, and it is capable of inducing severe lung injury in Gram-negative bacteria sepsis and pneumonia, which are among the most common predisposing causes of ALI [2]. The interaction of the lipid A moiety of LPS with macrophages, especially activated alveolar macrophages (AMΦ) in the lung, appears to be especially important because subsequent cellular activation results in the release of inflammatory mediators and phenotypic changes [3, 4]. As inflammatory mediators, AMΦ could release systemically active proinflammatory cytokines and chemokines, including tumor-necrosis factor-α (TNF-α), Interleukin-1β (IL-1β), and macrophage inflammatory protein-2 (MIP-2) [5].
CD40, a costimulatory molecule for antigen presentation, is expressed by a wide variety of cells including macrophages. Aberrant expression of CD40 is associated with autoimmune inflammatory diseases. Interaction of Toll-like receptor-4 (TLR4) with the Gram-negative bacteria endotoxin LPS results in the induction of an array of immune response genes [6, 7]. As numerous recent studies have demonstrated, the CD40 ligation on APC is associated with the augmentation of inflammation as follow: secretion of cytokines TNF-α, IL-1β, and MIP-2. A great number of studies indicate that the CD40L on APC is related to inflammatory amplification. The interaction of CD40-CDL in the lung is a critical step to mediate the inflammation [8]. In fact, the mechanism of the activation of macrophage and upregulation of CD40 costimulatory molecule in ALI is undetermined. Because the natural history of many of these diseases is unknown, animal model studies have been undertaken to fill in the gaps and to provide important clues to their roles in ALI.
In this study, we describe that LPS is a strong inducer of CD40 expression in macrophages in ALI mice model, which occurs at the transcriptional level and involves the activation of the transcription factors nuclear factor-κB (NF-κB). LPS-induced CD40 expression involves a lot of cytokines such as TNF-α, IL-1β, and MIP-2.
2. MATERIALS AND METHODS
Reagents were obtained as follows: E.coil LPS was purchased from Sigma-Aldrich (St. Louis, Mo, USA). Enzyme-linked immunosorbent assay (ELISA) development kits for mouse TNF-α, IL-1β, and MIP-2 were obtained from Genzyme Techne Corporation (Minneapolis, Minn, USA). Anti-mouse TNF-α, MIP-2, and IL-1β signal clone antibiotics were from Sigma-Aldrich. Total cellular RNA extraction and purification kits were purchased from Roche Corporation (Basel, Switzerland). RT-PCR kits were obtained from Promage Corporation (Madison, Wis, USA). Rat anti-mouse PE-conjugated CD40 antibody (clone 3/23) and rat-PE IgG isotype control were from PtarMigen (San Diego, Calif, USA). Antibodies against CD40 and actin were from Santa Cruz Biotechnology (Santa Cruz, Calif, USA).
2.1. Induction of animal model
180 BALB/c wild-type (WT) mice (6-to-8 week old) from Shandong University animal experiment center (Shandong, China) were divided two groups named as LPS group (B group) and control group (A group), respectively. Mice were anesthetized with pentobarbital sodium followed by injecting 0.2 mg/kg LPS in phosphate saline (PBS) intratrachealy for group B and PBS alone for group A [9].
2.2. Collection and measurement of specimens
After mice were executed, a total of 2.5 mL brochoalveolar lavage fluid (BALF) was collected [10]. The trachea was cannulated, and the lungs were lavaged six times with PBS (0.5 mL each time). Total cell numbers were counted with a standard hemocytometer. After centrifugation, supernatants were stored −80°C for cytokine measure by ELISA, and cell pellets were used to prepare cytospins.
2.3. Cell culture
A smear of BAL cells was prepared with cytocentrifugation using acytospin 2 at 1000 rpm for 5 minutes and then stained with Giesma solution [11]. Cell differentiation was examined by counting at least 200 cells using hemocytologic criteria to classify the cells as neutrophils, eosinophils, lymphocytes, or macrophages. By morphologic estimation with Gimesa staining, it was confirmed that polled cells from each group consisted of >98% AMΦ for separate experiments. Cells were resuspended in RPMI-1640 medium supplemented with 2 mM L-glutamine, 100 U/mL penicillin, 100 μg/lstreptomycin, 0.25 μg/L amphotericcin B, and 10% fetal calf serum, which was used as a complete medium. After allowing the cells to adhere to plates for 3 hours, nonadjacent cells were removed with three washes. The remaining adhesive cells were used as AMΦ. Macrophages were about 1 × 105/mL in inverted microscope.
2.4. RNA analysis by RT-PCR
Total cellular RNA was extracted according to manufacture's instruction. TLR4 primers were designed by Gene Toll (616 bp) forward 5′TCCCACCACC GATTCACA3′, reverse 5′CAACC2CTTTCATTTCACA3′. PCR reaction conditions were as follow: 94°C denature 2 minutes, then 94°C denature 30 seconds, 58°C anneal 60 seconds, 68°C extend 2 minutes, after 40 cycles, 68°C extend 10 minutes. β-actin (200 bp) (5′TGGGTCAGAAGGACTCCTATGTG3′, reverse 5′CGTCCCAGTTGGTAACAATGC) was used as internal control for quantification by densitometry.
2.5. ELISA assay for TNF-α, MIP-2, and IL-1β
BALFs were collected as before, anti-mouse TNF-α, MIP-2, and IL-1β and polyclonal horseradish peroxidase-conjugated IgG were sequentially added as primary and secondary antibodies, respectively. The color was developed with o-phenylenediamine substrates, and read with universal microplate reader.
2.6. EMSA for NF-κB
Nuclear and cytoplasmic extracts were obtained from cells from two groups. Protein level was determined in all extracts using the Bio-Rad dye reagent assay (Hercules, Calif, USA). An equal amount (5 μg) of nuclear protein from each sample and NF-κB consensus oligonucleotide (5′-AGTTGAGGGGACTTTCCCAGGC-3′ (Promega) was used for EMSA following the manufacture's instructions as previously described [12].
2.7. Northern blot and immunofluorescence flow cytometry for CD40 mRNA and protein
Total cellular RNA was isolated at 24 and 48 hours after from two groups. Mouse CD40 and GAPAH probe were prepared as described previously [13, 14]. 20 μg of total RNA was hybridized with probes at 42°C overnight. The hybridized mixture was treated with RNaseA/T1 (1 : 2000) and analyzed by 5% denaturing polyacrylamide gel electrophoresis. Values for CD40 mRNA levels were normalized to GAPDH mRNA level for each experiment condition.
Cells were plated at 2 × 105 cells/well into 12 well plates after 48 hours, and then incubated with 100 μL of 2.4G2 hybridoma supernatant (which contains rat-anti-mouse Fc r R Ab) for blocking Fc r R. Cells were then incubated with 10 μg/mL PE-conjugated anti-mouse CD40 Ab, and analyzed on the FACStar (Becton Dickinson, Mountain View, Calif, USA) [15]. Nagative controls were incubated with IgG isotype-matched antibody. Fold induction of CD40 expression was calculated by dividing the value of mean fluorescence intensity (MFI) of regents treated samples by value of untreated samples.
2.8. Statistical analysis
Data were presented as means ±SD. Continuous variables were tested by analysis of t-test. The P-values are two-tailed and a P-value of less than .05 was considered statistically significant.
3. RESULTS
3.1. Pathologic changes of lung tissue
Compared with mice in control group, the lung tissue from LPS-treated mice demonstrated interalveolar septum collapse, abnormal constraction, alveolar space and interstitial congestion, and neutrophilic granulocyte infiltration (Figure 1).
Figure 1.

Histopathological examination. Three pictures of pathological sections showing (a) pathological appearance from control group, (b) pathological appearance from LPS group at 24 hours, and (c) pathological appearance from LPS group at 48 hours (HE*300).
3.2. TLR4 expresses in macrophages
The gene expression level of TLR4 in AMΦ detected by RT-PCR is higher in LPS group as compared to the controls (Figure 2).
Figure 2.

mRNA expression of TLR4 in AMΦ by RT-PCR. A representative agarose gel electrophoresis showing PCR amplification of TLR4 and β-actin from cultured AMΦ with control group and LPS group (at 24, 48 hours).
3.3. LPS induces NF-κB binding to the NF-κB consensus oligos
To investigate the role of LPS in regulation of transcription factor activity, NF-κB activity of macrophage in control and LPS groups were compared. LPS stimulation led to increased NF-κB binding to NF-κB consensus oligos as demonstrated by EMSA (Figure 3).
Figure 3.
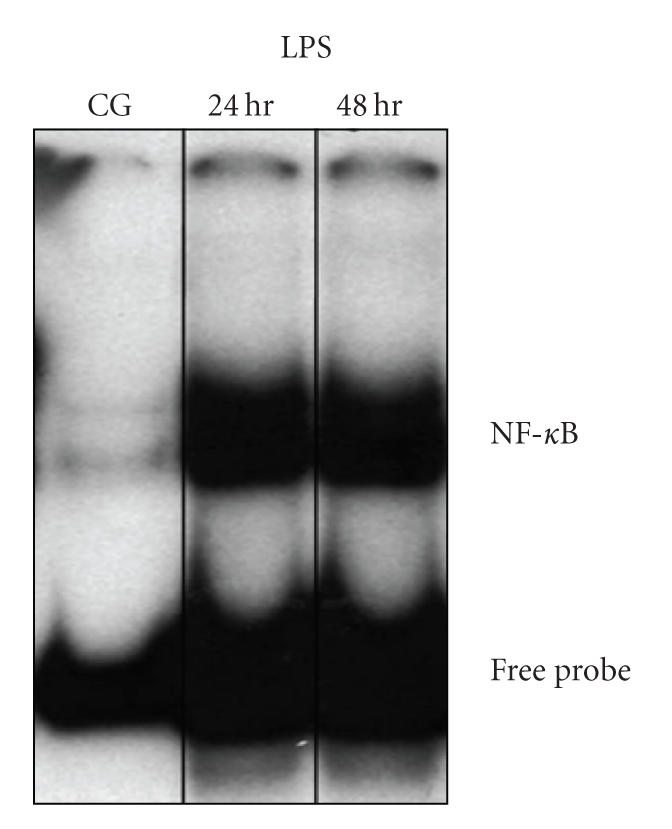
Activation of NF-κB by EMSA. NF-κB activity of macrophage in control and LPS groups (at 24, 48 hours).
3.4. The expression levels of CD40 mRNA and protein
The expression level of CD40 mRNA was determined by Northern blot as shown in Figure 4(a). LPS-induced CD40 mRNA expression increased in both 24 and 48 hours LPS group as compared to the control group. The expression level of CD40 protein determined by immunofluorescence flow cytometry increased after 48 hours in LPS group as compared to the control group (Figure 4(b)).
Figure 4.

mRNA and protein expression of CD40 in AMΦ by RT-PCR and flow cytometry, respectively, (a) a representative agarose gel electrophoresis showing PCR amplification of CD40 and β-actin from cultured AMΦ with control group and LPS group (at 24, 48 hours), (b) protein expression of CD40 in AMΦ with control group and LPS group.
3.5. The expression levels of TNF-α, MIP-2, and IL-1β in LPS-induced model
The inflammatory cytokine levels in BALF determined by ELISA demonstrated an increased expression of TNF-α, MIP-2, and IL-1β in LPS group as compared to the control group (Table 1, Figure 5).
Table 1.
Concentration of TNF-α, MIP-2, and IL-1β cytokines from BALF .
| Group | n | TNF-α (pg/mL) | IL-1β (pg/mL) | MIP-2 (pg/mL) |
|---|---|---|---|---|
| Control group | 10 | 61.02 ± 10.7 | 10.7 ± 2.7 | 84.5 ± 15.8 |
| LPS group | 10 | 115.0 ± 29.6* | 302.1 ± 17.3* | 524.9 ± 99.1* |
(*P < .05, compared with the control group).
Figure 5.

Concentration of TNF-α, MIP-2, and IL-1β cytokines from BALF: (a) concentration of TNF-α, (b) concentration of MIP-2, (c) concentration of IL-1β.
4. DISCUSSION
In this study, we demonstrated that the levels of inflammatory cytokines TNF-α, MIP-2, and IL-1β increased significantly in BALF in LPS-induced ALI mice model. Such cytokines have been studied more clearly than others in ALI, which have played critical role in inflammation development process. MIP-2, a functional analog of human IL-8, is an important mediator in the recruitment of neutrophils, and TNF-α acts locally to stimulate chemotaxis and activate neutrophils. These cytokines might result in the injury of the lung and eventually the development of ALI to some extent [16].
LPS can induce increased gene expressions in AMΦ, such as TLR4, CD40, TNF-α, MIP-2, and IL-1β, and activate some signal transduction pathways such as TLR4-mediated NF-κB activation, and CD40-CD40L interaction. These changes will result subsequently in releasing of large amount of inflammatory cytokines [17]. AMΦ was the first cell type in the primary immunity to kill bacterium after infection [18]. They are also the target cells to LPS. Organisms can release a large number of inflammatory and anti-inflammatory cytokines in endotoxemia [19]. TLR4 is a kind of transmembrane receptor on LPS target cell in immunity system [20]. It mediates the signal of LPS from outside to inside cell. From our experiments, we observed that TLR4 mRNA and TNF-α enhanced significantly in AMΦ of LPS-induced ALI. TLR4 is then regarded as the specific recognition receptor in the process of development ALI. The lung tissue was probably injured through increased secretion of TLR4 and strengthening its signal pathway. Therefore, TLR4 plays a vital role in a series of signals activation. LPS-TLR4 compound is able to activate many kinds of signals activation [21]: TLR4-induced NF-κB activation promotes the CD40-CD40L pathway as well as the CD40 self-activation, thus it causes the massive inflammation factors release and expanded inflammation response.
Previous reports demonstrated that the LPS-TLR4 interaction activated NF-κB directly. Subsequently, they all bind to the promoter of CD40, finally activated the CD40-CD40L interaction pathway and the CD40 self-activation, led to the release of inflammation factors and the anti-inflammation factors, which activated the acquired immunity. Therefore, the CD40 activation played a vital role in the process of development of ALI. In our study, we demonstrated that CD40 mRNA and the protein level increased in the macrophage in LPS group.
Large body of literatures demonstrated the expression of CD40L in the T lymphocytes and mast cells. Activation of CD40-CD40L pathway may promote the massive inflammation factors release, and the CD40-CD40L activation also indicates that T-cell dependence immunity already started [22, 23], which subsequently upregulated the other costimulatory molecules and the cell adhesion molecules to promote the massive preinflammation cell factor production and release, as well as inflammation cell differentiation. Our data demonstrated high concentrations of TNF-α, MIP-2, and IL-1β cell factors in BALF from LIP group, indicating that the CD40-CD40L pathway can enhance the inflammation response and gather massive inflammation cells, finally induces and aggravates the inflammation extent of lung injury.
5. CONCLUSIONS
Our data demonstrated that LPS-induced CD40 gene transcription, activated NF-κB transcription factors, increased their affinity and finally bound to the promoter of CD40, which led to the high expression of CD40 in the acute lung injury. Studying the mechanism of LPS-induced, TLR4-mediated in vitro CD40 transcription allowed us to provide the experimental evidences of the important role of CD40 in the development of LPS-induced ALI. We thus predict that blocking the expression of TLR4 or the inhibition of expression of CD40 through interruption of its transcription regulation complexes will be attractive targets to the treatment of LPS-induced ALI.
ACKNOWLEDGMENT
The authors are thankful to Yan Ding, Li Cong, and Yalun Zhang (School of Medicine, Shandong University) for their excellent technical assistance.
References
- 1.Ware LB, Matthay MA. The acute respiratory distress syndrome. The New England Journal of Medicine. 2000;342(18):1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 2.Fenton MJ, Golenbock DT. LPS-binding proteins and receptors. Journal of Leukocyte Biology. 1998;64(1):25–32. doi: 10.1002/jlb.64.1.25. [DOI] [PubMed] [Google Scholar]
- 3.Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406(6797):782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 4.Hoebe K, Du X, Georgel P, et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. 2003;424(6950):743–748. doi: 10.1038/nature01889. [DOI] [PubMed] [Google Scholar]
- 5.Lohmann-Matthes ML, Steinmuller C, Franke-Ullmann G. Pulmonary macrophages. European Respiratory Journal. 1994;7(9):1678–1689. [PubMed] [Google Scholar]
- 6.Imaizumi K, Kawabe T, Ichiyama S, et al. Enhancement of tumoricidal activity of alveolar macrophages via CD40-CD40 ligand interaction. American Journal of Physiology. 1999;277(1):L49–L57. doi: 10.1152/ajplung.1999.277.1.L49. [DOI] [PubMed] [Google Scholar]
- 7.Wiley JA, Geha R, Harmsen AG. Exogenous CD40 ligand induces a pulmonary inflammation response. Journal of Immunology. 1997;158(6):2932–2938. [PubMed] [Google Scholar]
- 8.van Kooten C, Banchereau J. CD40-CD40 ligand. Journal of Leukocyte Biology. 2000;67(1):2–17. doi: 10.1002/jlb.67.1.2. [DOI] [PubMed] [Google Scholar]
- 9.Nick JA, Young SK, Brown KK, et al. Role of p38 mitogen-activated protein kinase in a murine model of pulmonary inflammation. Journal of Immunology. 2000;164(4):2151–2159. doi: 10.4049/jimmunol.164.4.2151. [DOI] [PubMed] [Google Scholar]
- 10.Hashimoto N, Kawabe T, Imaizumi K, et al. CD40 plays a crucial role in lipopolysaccharide-induced acute lung injury. American Journal of Respiratory Cell and Molecular Biology. 2004;30(6):808–815. doi: 10.1165/rcmb.2003-0197OC. [DOI] [PubMed] [Google Scholar]
- 11.Johansson A, Lundborg M, Skold CM, et al. Functional, morphological, and phenotypical differences between rat alveolar and interstitial macrophages. American Journal of Respiratory Cell and Molecular Biology. 1997;16(5):582–588. doi: 10.1165/ajrcmb.16.5.9160840. [DOI] [PubMed] [Google Scholar]
- 12.Wesemann DR, Qin H, Kokorina N, Benveniste EN. TRADD interacts with STAT1-α and influences interferon-γ signaling. Nature Immunology. 2004;5(2):199–207. doi: 10.1038/ni1025. [DOI] [PubMed] [Google Scholar]
- 13.Nguyen VT, Benveniste EN. Involvement of STAT-1 and ets family members in interferon-γ induction of CD40 transcription in microglia/macrophages. Journal of Biological Chemistry. 2000;275(31):23674–23684. doi: 10.1074/jbc.M002482200. [DOI] [PubMed] [Google Scholar]
- 14.Wesemann DR, Dong Y, O'keefe GM, Nguyen VT, Benveniste EN. Suppressor of cytokine signaling 1 inhibits cytokine induction of CD40 expression in macrophages. Journal of Immunology. 2002;169(5):2354–2360. doi: 10.4049/jimmunol.169.5.2354. [DOI] [PubMed] [Google Scholar]
- 15.Fitzgerald KA, McWhirter SM, Faia KL, et al. IKKε and TBK1 are essential components of the IRF3 signaling pathway. Nature Immunology. 2003;4(5):491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 16.Tekamp-Olson P, Gallegos C, Bauer D, et al. Cloning and characterization of cDNAs for murine macrophage inflammatory protein 2 and its human homologues. Journal of Experimental Medicine. 1990;172(3):911–919. doi: 10.1084/jem.172.3.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kline JN. Eat dirt: CpG DNA and immunomodulation of asthma. Proceedings of the American Thoracic Society. 2007;4(3):283–288. doi: 10.1513/pats.200701-019AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hammermann R, Hirschmann J, Hey C, et al. Cationic proteins inhibit L-arginine uptake in rat alveolar macrophages and tracheal epithelial cells. Implications for nitric oxide synthesis. American Journal of Respiratory Cell and Molecular Biology. 1999;21(2):155–162. doi: 10.1165/ajrcmb.21.2.3574. [DOI] [PubMed] [Google Scholar]
- 19.Lim YS, Won T-B, Shim WS, et al. Induction of airway remodeling of nasal mucosa by repetitive allergen challenge in a murine model of allergic rhinitis. Annals of Allergy, Asthma and Immunology. 2007;98(1):22–31. doi: 10.1016/S1081-1206(10)60855-1. [DOI] [PubMed] [Google Scholar]
- 20.Hollingsworth JW, Whitehead GS, Lin KL, et al. TLR4 signaling attenuates ongoing allergic inflammation. Journal of Immunology. 2006;176(10):5856–5862. doi: 10.4049/jimmunol.176.10.5856. [DOI] [PubMed] [Google Scholar]
- 21.Wong CK, Cheung PFY, Ip WK, Lam CWK. Intracellular signaling mechanisms regulating toll-like receptor-mediated activation of eosinophils. American Journal of Respiratory Cell and Molecular Biology. 2007;37(1):85–96. doi: 10.1165/rcmb.2006-0457OC. [DOI] [PubMed] [Google Scholar]
- 22.Lei XF, Ohkawara Y, Stämpfli MR, et al. Disruption of antigen-induced inflammatory responses in CD40 ligand knockout mice. Journal of Clinical Investigation. 1998;101(6):1342–1353. doi: 10.1172/JCI1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Agalioti T, Lomvardas S, Parekh B, Yie J, Maniatis T, Thanos D. Ordered recruitment of chromatin modifying and general transcription factors to the IFN-β promoter. Cell. 2000;103(4):667–678. doi: 10.1016/s0092-8674(00)00169-0. [DOI] [PubMed] [Google Scholar]