

Abstract
Aim
To determine the risk of chronic obstructive pulmonary disease (COPD) associated with polymorphisms in the glutathione S-transferase (GST) M1, GST T1, and microsomal epoxide hydrolase (EPHX1) genes in a cohort of Slovak population.
Methods
Two hundred and seventeen patients with the diagnosis of COPD and 160 control subjects were enrolled in the study. Blood samples were collected from all subjects and the DNA from peripheral blood lymphocytes was used for subsequent genotyping assays, using polymerase chain reaction and restriction fragment-length polymorphism methods.
Results
In an unadjusted model, an increased risk for COPD was observed in subjects with EPHX1 His113-His113 genotype (odds ratio [OR], 2.32; 95% confidence interval [CI], 1.20-4.69; P = 0.008), compared with the carriers of the Tyr113 allele. However, after the adjustments for age, sex, and smoking status, the risk was not significant (adjusted OR, 1.79; 95% CI, 0.91-3.53; P = 0.093). In a combined analysis of gene polymorphisms, the genotype combination EPHX1 His113-His113/GSTM1 null significantly increased the risk of COPD in both, unadjusted (OR, 5.08; 95% CI, 1.70-20.43; P = 0.001) and adjusted model (OR, 4.87; 95% CI, 1.57-15.13; P = 0.006).
Conclusion
Although none of the tested gene polymorphisms was significantly related to an increased risk of COPD alone, our results suggest that the homozygous exon 3 mutant variant of EPHX1 gene in the combination with GSTM1 null genotype is a significant predictor of increased susceptibility to COPD in the Slovak population. The findings of the present study emphasize the importance of detoxifying and antioxidant pathways in the pathogenesis of COPD.
Chronic obstructive pulmonary disease (COPD) represents a major public health care problem worldwide due to its increasing prevalence, morbidity, and mortality (1). Generally, COPD is characterized by progressive and only partially reversible airflow limitation (2). Although cigarette smoking is the most important risk factor for COPD, only 20%-30% of chronic smokers develop severe impairment of lung function associated with COPD (3). Besides smoking, other environmental and genetic factors and gene-environment interactions influence the development of COPD (4).
Severe α-1-antitrypsin deficiency is a well established genetic risk factor for COPD that has provided a basis for the protease-antiprotease hypothesis in the pathogenesis of COPD (5,6). Other candidate genes that might play a role in the development of COPD are involved in endogenous protease/antiprotease imbalance, inflammatory processes, metabolism of mutagens and carcinogens in tobacco smoke, and in mucocilliary clearance (7). Interindividual differences in the polymorphisms of enzymes metabolizing the xenobiotic substances and free radicals contained in the cigarette smoke may play a role in the individual susceptibility to the decrease in lung functions in smokers (8).
Microsomal epoxide hydrolase (EPHX1) is generally considered to be a protective enzyme involved in the defense from oxidative damage (9,10). Two common polymorphic sites in the EPHX1 gene that influence the enzyme activity can be detected (11). An exon 3 thymine-to-cytosine mutation changes Tyr residue 113 to His, thus reducing the enzyme activity by about 50%. The second mutation, an adenine-to-guanine transition in exon 4 of the gene, changes His residue 139 to Arg and results in the production of EPHX1 with the activity increased by about 25% (11). The combination of these polymorphisms leads to a formation of several functional phenotypes of EPHX1. The slow metabolizing type of EPHX1 was associated with emphysema and COPD (9). In another study, an association of slow metabolizing EPHX1 phenotype with an accelerated deterioration of lung function in smokers was observed (12). In addition, several studies conducted in different populations have suggested that the EPHX1 genotype may influence individual susceptibility to COPD (9,13-15). Nevertheless, other investigators failed to confirm an association between the EPHX1 gene polymorphisms and COPD (16-18).
Glutathione S-transferases (GST) play a role in the detoxification of carcinogenic compounds contained in cigarette smoke and in the antioxidant protection (19,20). Recently, the GSTM1 and the GSTT1 gene polymorphisms have been excessively studied with respect to their potential contribution to the risk of COPD (8,17,21,22). The deficiency in the activity of GSTM1 and GSTT1 enzymes is caused by the inherited homozygous absence of the GSTM1 or GSTT1 gene, respectively (ie, GSTM1 null or GSTT1 null genotype). Previously, the homozygous GSTM1 null genotype has been associated with lung cancer (23), emphysema (21), and reductions in the lung function in Caucasian smokers with non-small-cell lung cancer (22). However, another study conducted in Koreans found no differences in the frequencies of polymorphic genotypes of GSTM1 and GSTT1 genes between patients with COPD and healthy smokers (17).
Since current data on the potential associations between an increased COPD risk and genes encoding the enzymes metabolizing xenobiotic substances are inconsistent, the aim of our study was to analyze the relation between COPD and gene polymorphisms of EPHX1, GSTM1, and GSTT1 genes in a sample of Slovak population.
Participants and methods
Participants
Patients with the diagnosis of COPD stage I to IV according to the American Thoracic Society/European Respiratory Society guidelines (1,2) referred to an outpatient respiratory clinic in a tertiary university hospital in Slovakia were consecutively recruited in the study in the period 2004-2007. The presence of fixed airflow obstruction was confirmed by spirometry in all patients with postbronchodilator Tiffeneau ratio <0.7 and negative bronchodilator reversibility test results (1,2). Exclusion criteria were respiratory disorders other than COPD, such as interstitial lung disease, bronchiolitis obliterans, diffuse bronchiectasis, lung cancer, tuberculosis, previous medical records of bronchial asthma, thoracic surgery in the past, and/or recent pulmonary infiltrate or pleural effusion on chest x-ray. Patients with pulmonary embolism, overt heart failure, malignancy, systemic autoimmune disorders, infectious diseases, recent surgery, severe endocrine, hepatic or renal diseases, and patients unable to perform spirometry to confirm the diagnosis were also excluded. Healthy age-matched subjects without previous medical history of respiratory disorders and without any present respiratory symptoms, seen by their general practitioner for a regular check-up, were recruited consecutively as controls. All COPD patients and controls were individuals from the general population of Eastern Slovakia. The study was approved by the institutional ethics committee and all subjects gave their written informed consent.
Pulmonary function tests
In patients, pulmonary function tests were performed with a bodyplethysmograph (Jaeger, Höchberg, Germany) 20 minutes before and after 400 μg dose of inhaled salbutamol (Ventolin; GlaxoSmithKline, Greenford, Middlesex, UK). Short-acting inhaled drugs were not used 6 hours before the testing, and long-acting bronchodilator therapy was stopped for 12 hours before the test. Bodyplethysmograph was calibrated before use. All pulmonary function testing was performed according to the European Respiratory Society standards, with the patients in the sitting position and the same technician performing the test in order to ensure the consistency (24). Three technically acceptable measurements were performed in each patient before and after salbutamol inhalation; the highest value was included in the analyses.
Genotyping assays
Blood (4 mL) for genotyping was drawn into an EDTA-tube by routine venipuncture and stored at -20°C until DNA extraction was carried out. Genomic DNA was isolated from the separated peripheral blood lymphocytes using Wizard Genomic DNA Purification Kit (#A1125, Promega Corp., Madison, WI, USA).
Two separate polymerase chain reactions (PCR) were used to detect two mutations in the EPHX1 gene (9). The primer pairs EPO1 (5′-GATCGATAAGTTCCGTTTCACC-3′) and EPo2 (5′-ATCCTTAGTCTTGAAGTGAGGAT-3′) were used for exon 3 variants. The primer pairs EPO3 (5′-ACATCCACTTCATCCACGT-3′) and EPO4 (5′-ATGCCTCTGAGAAGCCAT-3′) were used to test mutation in exon 4. PCR was carried out on MJ Research thermal cycler (Minicycler PTC-150, MJ Research Inc., Watertown, MA, USA). For exon 3, the PCR conditions were as follows: after 5 minutes of pretreatment at 94°C, the reaction mixture was subjected to 47 cycles at 94°C for 1 minute, at 46°C for 70 seconds, and at 72°C for 70 seconds; for exon 4, the initial pretreatment at 94°C for 5 minutes was followed by 38 cycles at 94°C for 1 minute, at 56°C for 1 minute, and at 72°C for 1 minute. Final extension was carried out at 72°C for 10 minutes. The resulting DNA fragments were digested with EcoRV (exon 3) and RsaI (exon 4) (37°C, overnight). Negative controls were included in every PCR run. The fragments were separated on 3% agarose gel after enzyme digestion. The gels were stained with ethidium bromide and transilluminated with the UV light (Figure 1).
Figure 1.
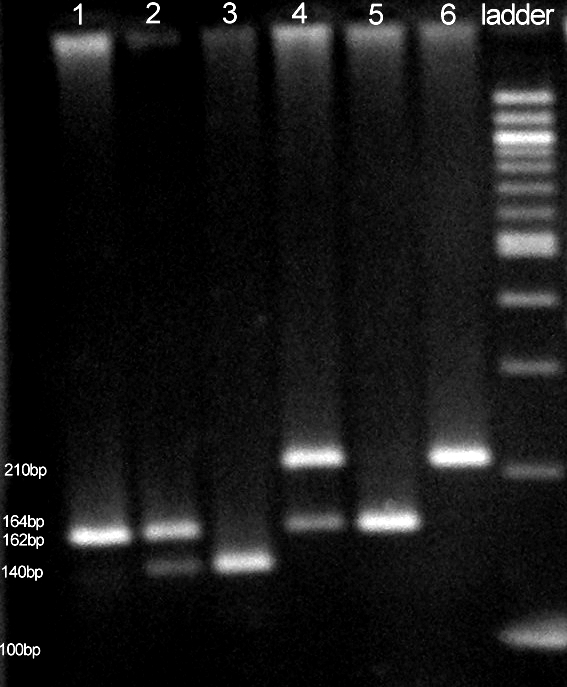
Polymerase chain reaction analysis of two polymorphic loci in epoxide hydrolase 1 (EPHX1) gene. Lanes 1-3 – EPHX1 exon 3 polymorphism: 1. His/His; 2. Tyr/His; 3. Tyr/Tyr. Lanes 4-6 – EPHX1 exon 4 polymorphism: 4. His/Arg; 5. Arg/Arg; 6. His/His. Lane 7 – 100 bp ladder (#N3231S, New England Biolabs, Ipswitch, USA).
The plus/minus (null/non-null) polymorphism of the GSTM1 and GSTT1 genes were determined according to the PCR-based method of Arand et al (25), where the simultaneous amplification of GSTM1 and GSTT1 genomic fragments in the same reaction was performed. The resulting PCR fragments were separated by electrophoresis on 3% agarose gel, followed by ethidium bromide staining and directly visualized by UV detection. The primer pairs for this multiplex PCR were the following: 5′-GAACTCCCTGAAAAGCTAAAGC-3′ and 5′-GTTGGGCTCAAATATACGGTGG-3′ for GSTM1; 5′-TTCCTTACTGGTCCTCACATCTC-3′ and 5′-TCACCGGATCATGGCCAGCA-3′ for GSTT1; and 5′-GCCCTCTGCTAACAAGTCCTAC-3′ and 5′-GCCCTAAAAAGAAAATCGCCAATC-3′ for albumin. Multiplex PCR analysis was carried out in MJ Research thermal cycler under the following conditions: initial pretreatment at 95°C for 2 minutes, followed by 30 cycles at 94°C for 1 minute, at 64°C for 1 minute, and at 72°C for 1 minute. Final extension was carried out at 72°C for 5 minutes. The involvement of GSTM1 deficiency was concluded from the absence of the specific 215 base pair (bp) fragment and GSTT1 deficiency from the absence of 480 bp fragment. Albumin gene fragment used as internal positive control resulted in constant 350 bp band in all samples (Figure 2). Subjects with heterozygous (+/0) and non-null homozygous (+/+) GSTM1 or GSTT1 genotypes were pooled together and formed the non-null genotype group, in agreement with previous studies (22), indicating that individuals with GSTM1 heterozygous and non-null homozygous genotypes have similar GST expression level in the liver (26). To test the consistency of the genotyping and to detect possible misgenotyping, 20 random samples from patients with COPD and 20 samples from the control group were retested. All the results of retested samples matched the previous ones.
Figure 2.
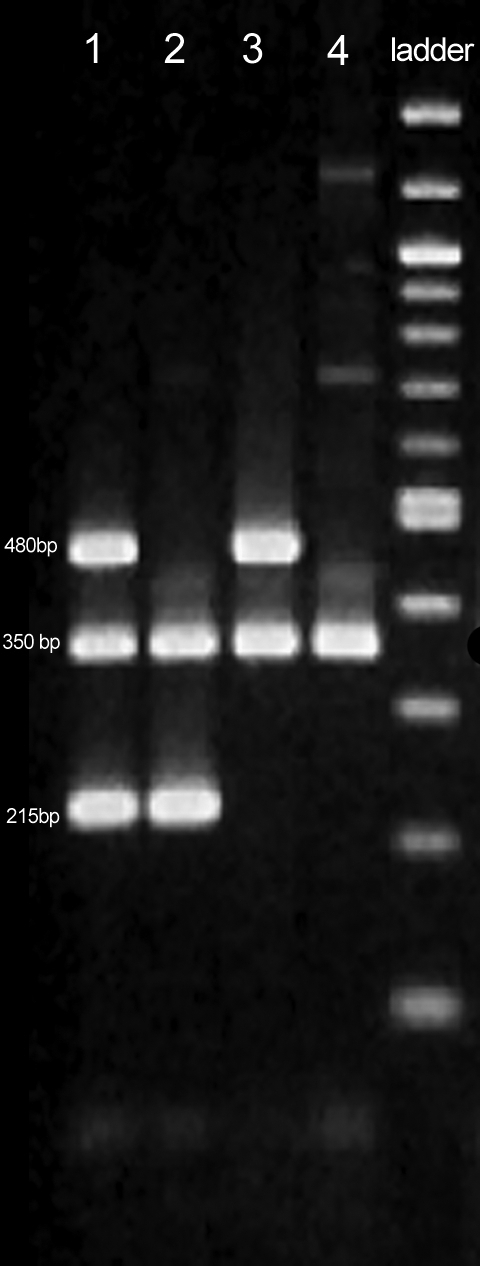
Multiplex polymerase chain reaction analysis of glutathione S-transferase M1 (GSTM1) and glutathione S-transferase T1 (GSTT1) polymorphisms. Lane 1 – GSTM1 non-null/GSTT1 non-null. Lane 2 – GSTM1 non-null/GSTT1 null. Lane 3 – GSTM1 null/GSTT1 non-null. Lane 4 – GSTM1 null/GSTT1 null genotype. Lane 5 – 100 bp ladder (#N3231S, New England Biolabs, Ipswitch, USA).
Statistical analysis
Demographic data and spirometric parameters are presented as means ± standard deviation. Mann-Whitney U-tests were used to compare the means and χ2 tests to compare the proportions of categorical variables in cases and controls. The distributions of genotypes for each polymorphic site in the EPHX1 gene were tested to match the Hardy-Weinberg heredity equilibrium by χ2 tests.
Crude odds ratios (OR) and 95% confidence intervals (CI) were calculated using Gart’s method (27) to examine the association between the GSTM1, GSTT1, and EPHX1 genotypes, and the risk of COPD. The recessive model was used, in which homozygous mutant genotype was compared with the remaining genotypes within the tested gene. In addition, logistic regression was used to determine the odds ratios adjusted for potential confounding factors (age, sex, and smoking status). All tests were performed using Statistical Package for the Social Sciences, version 14.0 (SPSS Inc., Chicago, IL, USA). P<0.05 was considered significant.
Results
A total of 377 Caucasians (217 patients with COPD and 160 control subjects) were enrolled in the study. No differences were observed between control subjects and COPD patients in mean age (63 ± 18 vs 67 ± 11 years; P = 0.09). The proportion of women was higher (46% vs 25%; P<0.001) and the proportion of smokers was lower (39% vs 78%; P<0.001) in control group than in patients with COPD. Mean smoking history in controls was 5 ± 18 pack-years. Patients with COPD had a 27 ± 25 pack-years history of smoking; their pulmonary function tests showed predicted mean post-bronchodilator forced expiratory volume in one second (FEV1) of 49 ± 21%, and predicted mean forced vital capacity (FVC) of 70 ± 22%, with mean FEV1/FVC ratio 57 ± 15%.
Genotype distributions of GSTM1, GSTT1, and EPHX1 gene polymorphisms in the studied population are reported in Table 1. Genotype distributions for exon 3 and exon 4 polymorphisms in the EPHX1 gene were in the Hardy-Weinberg equilibrium (P = 0.994 and P = 0.696, respectively).
Table 1.
Genotype distribution of glutathione S-transferase M1 (GSTM1), glutathione S-transferase T1 (GSTT1), and epoxide hydrolase 1 (EPHX1) in patients with chronic obstructive pulmonary disease and healthy controls in a sample of Slovak population*
| Genotype | No. (%) of |
OR (95% CI) for homozygous mutation | P | OR adjusted (95% CI) for homozygous mutation† | P | |
|---|---|---|---|---|---|---|
| COPD cases | controls | |||||
| GSTM1: | ||||||
| non-null genotype | 88 (40.6) | 75 (46.9) | ||||
| null genotype | 129 (59.4) | 85 (53.1) | 1.29 (0.84-1.99) | 0.221 | 1.34 (0.85-2.12) | 0.208 |
| GSTT1: | ||||||
| non-null genotype | 178 (82.0) | 129 (80.6) | ||||
| null genotype | 39 (18.0) | 31 (19.4) | 0.91 (0.52-1.60) | 0.729 | 0.91 (0.51-1.64) | 0.766 |
| EPHX1 exon 3 codon 113: | ||||||
| Tyr/Tyr | 105 (48.4) | 78 (48.8) | ||||
| Tyr/His | 70 (32.3) | 67 (41.9) | ||||
| His/His | 42 (19.3) | 15 (9.3) | 2.32 (1.20-4.69) | 0.008 | 1.79 (0.91-3.53) | 0.093 |
| EPHX1 exon 4 codon 139: | ||||||
| His/His | 140 (64.5) | 91 (56.9) | ||||
| His/Arg | 69 (31.8) | 63 (39.4) | ||||
| Arg/Arg | 8 (3.7) | 6 (3.7) | 0.98 (0.29-3.51) | 0.974 | 0.61 (0.20-1.90) | 0.393 |
*Abbreviations: COPD – chronic obstructive pulmonary disease; OR – odds ratio; CI – confidence interval.
†Adjusted for potential confounding factors (age, sex, and smoking status).
In an unadjusted model, the EPHX1 His113-His113 genotype at exon 3 was associated with an increased COPD risk (OR, 2.32; 95% CI, 1.20-4.69; P = 0.008). However, this association did not remain significant after the adjustments for age, sex, and smoking status (OR, 1.79; 95% CI, 0.91-3.53; P = 0.093) (Table 1). In contrast, no association with COPD risk, expressed both as crude and adjusted odds ratios, was observed in exon 4 codon 139 polymorphic site of the EPHX1 gene. Furthermore, no significant associations were observed between the GSTM1 or GSTT1 genotypes and the risk of COPD (Table 1).
To assess whether the combinations of the EPHX1, GSTM1, and GSTT1 genotype polymorphisms are associated with an increased risk for the development of COPD, the GSTM1, GSTT1, and EPHX1 gene polymorphisms were analyzed in combinations. Table 2 shows the distributions of combined genotypes of GSTM1, GSTT1, and EPHX1 genes in cases and controls. In crude combined analysis of gene polymorphisms for the EPHX1, GSTM1, and GSTT1 genes, the combined EPHX1 His113-His113/GSTM1 null genotype was associated with significantly increased risk of COPD (OR, 5.08; 95% CI, 1.70-20.43; P = 0.001). After adjustments for age, sex, and smoking status, the relationship between the combined EPHX1 His113-His113/GSTM1 null genotype and the increased risk for COPD remained significant (OR, 4.87; 95% CI, 1.57-15.13; P = 0.006).
Table 2.
Distribution of combined genotypes of glutathione S-transferase M1 (GSTM1), glutathione S-transferase T1 (GSTT1), and epoxide hydrolase 1 (EPHX1) genes in patients with chronic obstructive pulmonary disease (COPD) and healthy controls in a sample of Slovak population
| Genotype | No. of patients/controls |
||||||
|---|---|---|---|---|---|---|---|
|
EPHX1 exon4 |
GSTM1 |
GSTT1 |
|||||
| His/His | His/Arg | Arg/Arg | non-null genotype | null genotype | non-null genotype | null genotype | |
| EPHX1 exon3: | |||||||
| Tyr/Tyr | 68/44 | 34/34 | 3/0 | 40/33 | 65/45 | 84/61 | 21/17 |
| Tyr/His | 47/38 | 21/25 | 2/4 | 31/31 | 39/36 | 58/53 | 12/14 |
| His/His | 25/9 | 14/4 | 3/2 | 17/11 | 25/4 | 36/15 | 6/0 |
| GSTT1: | |||||||
| Non-null genotype | 118/73 | 53/51 | 7/5 | 75/64 | 103/65 | ||
| Null genotype | 22/18 | 16/12 | 1/1 | 13/11 | 26/20 | ||
| GSTM1: | |||||||
| Non-null genotype | 55/44 | 27/27 | 6/4 | ||||
| Null genotype | 85/47 | 42/36 | 2/2 | ||||
| Combined GSTM1 null and EPHX1 exon 3 His/His | 5.08 (1.70-20.43)* | 4.87 (1.57-15.13)† | |||||
*Odds ratio (95% confidence interval); P = 0.001.
†Adjusted odds ratio (95% confidence interval); P = 0.006.
Discussion
The combination of His113-His113 EPHX1 polymorphism at codon 113 with the GSTM1 null/non-null gene polymorphism significantly increased the risk of COPD in a sample of Slovak population, whereas none of the studied polymorphisms alone predicted the risk.
The only well-established genetic cause of COPD, the α-1-antitrypsin deficiency, is rare (6). A number of studies have been performed to find other susceptibility genes of COPD, and a variety of approaches have been used to identify them. Familial aggregation study on 5003 subjects participating in the Framingham Study suggested polygenic inheritance of obstructive ventilatory impairment, with the genetic component consisting of several genes of a small effect each rather than of a single major gene (28). In a genome-wide scan of pulmonary function measures in the National Heart, Lung, and Blood Institute Family Heart Study, FEV1 and FVC were suggestively linked to regions on the chromosome 18 (29). In addition, many studies were performed to search for potential associations between a given gene polymorphism and susceptibility to COPD (7,30-32). Up to now, more than 25 different candidate genes have been investigated for their potential role in lung function impairment in smokers (7,30-32).
The evidence of a linkage between the candidate genes and COPD and its associated phenotypes is seldom definitive and is often conflicting (33). The development and progression of COPD involves multiple genes, gene-gene, and gene-environment interactions (4). In general, although association studies have successfully identified various significant genetic associations to the reduction in lung function in smokers (7,9,12-15,21,22,31), the inconsistency and lack of replication has been a major flaw in these studies. In addition, in some studies the distribution of genotypes deviated from Hardy-Weinberg equilibrium (9,14,17). Moreover, COPD is a heterogeneous disease, and its definition has been changing over the last decade (1,2). As a result, genetic association studies in patients with COPD use different phenotype definition of the disease. In our study, the current definition of COPD according to GOLD guidelines (2) was used, and the cohort of subjects under investigation was ethnically well defined.
One of the possible approaches to understanding the influence of genetic factors on COPD, as well as their mutual interactions, is to assess the combined genotypes (34) in an effort to explain gene-gene interactions. Current study suggest that the genotype combination of two genes involved in metabolism of xenobiotics appears as a significant risk factor for the susceptibility to COPD.
Since genetic polymorphisms in xenobiotic enzymes may play a role in individual susceptibility to oxidant-related lung disease, several studies assessed the associations between COPD and candidate genes of enzymes involved in the metabolism of mutagens and carcinogens originating in cigarette smoke. Epoxide derivatives of cigarette-smoke components may be the cause of some of the lung damage characteristic of these diseases. Indeed, Smith and Harrison (9) were the first to report a strong association between the innate slow EPHX1 activity (Tyr113His polymorphism of EPHX1 gene) and COPD. These findings were strengthened by further studies in Taiwanese (14) and Caucasian (15,35) populations. Nevertheless, since some authors failed to confirm these initial results (16-18), Brogger et al (36) performed a meta-analysis of 16 studies on the associations between COPD and several candidate gene polymorphisms. This meta-analysis supported the role of EPHX1 in the etiology of COPD (36).
Although there has been some early evidence that the GSTM1 null genotype might be related to severe bronchitis in heavy smokers (37) and to the reduction in lung function and emphysema in smokers with bronchogenic carcinoma (21,22), Hersh et al (35) did not find any significant association between the GSTM1 genotype and COPD. Similarly, Yim et al (17) did not find any associations between genetic polymorphisms in GSTM1 and GSTT1 genes and the development of COPD in Koreans. In agreement with these findings, the results of our study could not provide any support for the possible relationship between the GSTM1 and GSTT1 gene polymorphisms and COPD in the Slovak population.
The most important finding of our study is that the combination of GSTM1 null genotype and EPHX1 His113-His113 genotype significantly increased the risk of COPD. GSTM1 null genotype is related to the total lack of the GSTM1 enzyme activity, whereas the EPHX1 His 113 allele is conferred to be slow (11). Taken together, patients with the particular EPHX1 His113-His113/GSTM1 null genotype combination might possess a decreased ability to detoxify carcinogenic compounds contained in cigarette smoke and an impaired antioxidant protection, resulting in an increased risk for the development of COPD. Indeed, Cheng et al (14) reported in a Taiwanese population an increased odds ratio for COPD when two genotypes of at least one slow EPHX1 exon 3 allele and GSTM1 null were combined. To our knowledge, our study is the first to identify an increased risk for the development of COPD in the carriers of the combined GSTM1 null and slow EPHX1 genotype in the Slovak population.
Several limitations might have influenced our results. First, the size of the studied population was moderate, thus affecting the potential to detect and replicate the formerly reported associations. Indeed, Brogger et al (36) have recently shown that the genetic associations tend to vary in small studies. The second potential source of bias in our study relates to the genotyping method that was originally described by Smith and Harrison (9). In an association study on the relationship between EPHX1 gene polymorphism and emphysema in a Japanese cohort, the frequency of His113, a possible emphysema susceptibility allele of the EPHX1 gene, was overestimated when DNA samples were genotyped in a conventional way (38). This pitfall for PCR-based association studies on EPHX1 gene polymorphisms results from the interference of the nearby single nucleotide polymorphism (Lys119) with the primer hybridization, and therefore, the distribution of polymorphisms should be tested for Hardy-Weinberg equilibrium (38). In our study, the distributions of EPHX1 gene polymorphisms were in Hardy-Weinberg equilibrium. Therefore, our results are likely unaffected by the potential bias resulting from the conventional way of genotyping EPHX1 gene. Finally, a limitation common to most studies with similar design as ours is that they only try to find the association in the genes previously known to be related to a given disease. A different approach, ie, the genome-wide scanning should be employed to find a novel genetic link to COPD.
In conclusion, in our study the combined EPHX1 His113-His113/GSTM1 null genotype was associated with significantly increased risk of COPD in Slovaks. Since simple association studies give only limited and often contradictory results, the real challenge is to understand the role of multiple genetic and environmental factors, as well as gene-gene and gene-environment interactions in the pathogenesis of lung impairment in COPD.
Acknowledgments
The authors thank Terezia Hudáková for her help with DNA isolation and the laboratory work. This study was supported by operating grant VEGA 1/2312/05 and VEGA 1/4231/07 of the Ministry of Education and grant 2005/5-FNLPKE-01 of the Ministry of Health, Slovakia.
References
- 1.Celli BR, MacNee W, ATS/ERS Task Force. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 2004;23:932–46. doi: 10.1183/09031936.04.00014304. [DOI] [PubMed] [Google Scholar]
- 2.Rabe KF, Hurd S, Anzueto A, Barnes PJ, Buist SA, Calverley P, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2007;176:532–55. doi: 10.1164/rccm.200703-456SO. [DOI] [PubMed] [Google Scholar]
- 3.Lokke A, Lange P, Scharling H, Fabricius P, Vestbo J. Developing COPD: a 25 year follow up study of the general population. Thorax. 2006;61:935–9. doi: 10.1136/thx.2006.062802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sandford AJ, Silverman EK. Chronic obstructive pulmonary disease. Susceptibility factors for COPD the genotype-environment interaction. Thorax. 2002;57:736–41. doi: 10.1136/thorax.57.8.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Janoff A. Elastases and emphysema. Current assessment of the protease-antiprotease hypothesis. Am Rev Respir Dis. 1985;132:417–33. doi: 10.1164/arrd.1985.132.2.417. [DOI] [PubMed] [Google Scholar]
- 6.Janus ED, Phillips NT, Carrell RW. Smoking, lung function, and alpha 1-antitrypsin deficiency. Lancet. 1985;1:152–4. doi: 10.1016/S0140-6736(85)91916-6. [DOI] [PubMed] [Google Scholar]
- 7.Molfino NA. Genetics of COPD. Chest. 2004;125:1929–40. doi: 10.1378/chest.125.5.1929. [DOI] [PubMed] [Google Scholar]
- 8.He JQ, Ruan J, Connett JE, Anthonisen NR, Pare PD, Sandford AJ. Antioxidant gene polymorphisms and susceptibility to a rapid decline in lung function in smokers. Am J Respir Crit Care Med. 2002;166:323–8. doi: 10.1164/rccm.2111059. [DOI] [PubMed] [Google Scholar]
- 9.Smith CA, Harrison DJ. Association between polymorphism in gene for microsomal epoxide hydrolase and susceptibility to emphysema. Lancet. 1997;350:630–3. doi: 10.1016/S0140-6736(96)08061-0. [DOI] [PubMed] [Google Scholar]
- 10.Harrison DJ, Hubbard AL, MacMillan J, Wyllie AH, Smith CA. Microsomal epoxide hydrolase gene polymorphism and susceptibility to colon cancer. Br J Cancer. 1999;79:168–71. doi: 10.1038/sj.bjc.6690028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hassett C, Aicher L, Sidhu JS, Omiecinski CJ. Human microsomal epoxide hydrolase: genetic polymorphism and functional expression in vitro of amino acid variants. Hum Mol Genet. 1994;3:421–8. doi: 10.1093/hmg/3.3.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sandford AJ, Chagani T, Weir TD, Connett JE, Anthonisen NR, Pare PD. Susceptibility genes for rapid decline of lung function in the lung health study. Am J Respir Crit Care Med. 2001;163:469–73. doi: 10.1164/ajrccm.163.2.2006158. . REMOVED HYPERLINK FIELD. [DOI] [PubMed] [Google Scholar]
- 13.Park JY, Chen L, Wadhwa N, Tockman MS. Polymorphisms for microsomal epoxide hydrolase and genetic susceptibility to COPD. Int J Mol Med. 2005;15:443–8. [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng SL, Yu CJ, Chen CJ, Yang PC. Genetic polymorphism of epoxide hydrolase and glutathione S-transferase in COPD. Eur Respir J. 2004;23:818–24. doi: 10.1183/09031936.04.00104904. [DOI] [PubMed] [Google Scholar]
- 15.Rodriguez F, Jardi R, Costa X, Juan D, Galimany R, Vidal R, et al. Detection of polymorphisms at exons 3 (Tyr113→His) and 4 (His139→Arg) of the microsomal epoxide hydrolase gene using fluorescence PCR method combined with melting curves analysis. Anal Biochem. 2002;308:120–6. doi: 10.1016/S0003-2697(02)00219-1. [DOI] [PubMed] [Google Scholar]
- 16.Takeyabu K, Yamaguchi E, Suzuki I, Nishimura M, Hizawa N, Kamakami Y. Gene polymorphism for microsomal epoxide hydrolase and susceptibility to emphysema in a Japanese population. Eur Respir J. 2000;15:891–4. doi: 10.1034/j.1399-3003.2000.15e13.x. [DOI] [PubMed] [Google Scholar]
- 17.Yim JJ, Park GY, Lee CT, Kim YW, Han SK, Shim YS, et al. Genetic susceptibility to chronic obstructive pulmonary disease in Koreans: combined analysis of polymorphic genotypes for microsomal epoxide hydrolase and glutathione S-transferase M1 and T1. Thorax. 2000;55:121–5. doi: 10.1136/thorax.55.2.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matheson MC, Raven J, Walters EH, Abramson MJ, Ellis JA. Microsomal epoxide hydrolase is not associated with COPD in a community-based sample. Hum Biol. 2006;78:705–17. doi: 10.1353/hub.2007.0015. [DOI] [PubMed] [Google Scholar]
- 19.Ketterer B, Harris JM, Talaska G, Meyer DJ, Pemble SE, Taylor JB, et al. The human glutathione S-transferase supergene family, its polymorphism, and its effects on susceptibility to lung cancer. Environ Health Perspect. 1992;98:87–94. doi: 10.2307/3431252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mannervik B, Danielson UH. Glutathione transferases – structure and catalytic activity. CRC Crit Rev Biochem. 1988;23:283–337. doi: 10.3109/10409238809088226. [DOI] [PubMed] [Google Scholar]
- 21.Harrison DJ, Cantlay AM, Rae F, Lamb D, Smith CA. Frequency of glutathione S-transferase M1 deletion in smokers with emphysema and lung cancer. Hum Exp Toxicol. 1997;16:356–60. doi: 10.1177/096032719701600703. [DOI] [PubMed] [Google Scholar]
- 22.Tkacova R, Salagovic J, Ceripkova M, Tkac I, Stubna J, Kalina I. Glutathione S-transferase M1 gene polymorphism is related to COPD in patients with non-small-cell lung cancer. Wien Klin Wochenschr. 2004;116:131–4. doi: 10.1007/BF03040750. [DOI] [PubMed] [Google Scholar]
- 23.Schneider J, Bernges U, Philipp M, Woitowitz HJ. GSTM1, GSTT1, and GSTP1 polymorphism and lung cancer risk in relation to tobacco smoking. Cancer Lett. 2004;208:65–74. doi: 10.1016/j.canlet.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 24.Miller MR, Hankinson J, Brusasco V, Burgos F, Casaburi R, Coates A, et al. Standardisation of spirometry. Eur Respir J. 2005;26:319–38. doi: 10.1183/09031936.05.00034805. [DOI] [PubMed] [Google Scholar]
- 25.Arand M, Muhlbauer R, Hengstler J, Jager E, Fuchs J, Winkler L, et al. A multiplex polymerase chain reaction protocol for the simultaneous analysis of the glutathione S-transferase GSTM1 and GSTT1 polymorphisms. Anal Biochem. 1996;236:184–6. doi: 10.1006/abio.1996.0153. [DOI] [PubMed] [Google Scholar]
- 26.Bell DA, Taylor JA, Paulson DF, Robertson CN, Mohler JL, Lucier GW. Genetic risk and carcinogen exposure: a common inherited defect of the carcinogen-metabolism gene glutathione S-transferase M1 (GSTM1) that increases susceptibility to bladder cancer. J Natl Cancer Inst. 1993;85:1159–64. doi: 10.1093/jnci/85.14.1159. [DOI] [PubMed] [Google Scholar]
- 27.Gart JJ. Point and interval estimation of the common odds ratio in the combination of 2 × 2 tables with fixed marginals. Biometrika. 1970;57:471–5. [Google Scholar]
- 28.Givelber RJ, Couropmitree NN, Gottlieb DJ, Evans JC, Levy D, Myers RH, et al. Segregation analysis of pulmonary function among families in the Framingham Study. Am J Respir Crit Care Med. 1998;157:1445–51. doi: 10.1164/ajrccm.157.5.9704021. [DOI] [PubMed] [Google Scholar]
- 29.Wilk JB, DeStefano AL, Arnett DK, Rich SS, Djousse L, Crapo RO, et al. A genome-wide scan of pulmonary function measures in the National Heart, Lung, and Blood Institute Family Heart Study. Am J Respir Crit Care Med. 2003;167:1528–33. doi: 10.1164/rccm.200207-755OC. [DOI] [PubMed] [Google Scholar]
- 30.Lomas DA, Silverman EK. The genetics of chronic obstructive pulmonary disease. Respir Res. 2001;2:20–6. doi: 10.1186/rr34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Joos L, He JQ, Shepherdson MB, Connett JE, Anthonisen NR, Pare PD, et al. The role of matrix metalloproteinase polymorphisms in the rate of decline in lung function. Hum Mol Genet. 2002;11:569–76. doi: 10.1093/hmg/11.5.569. [DOI] [PubMed] [Google Scholar]
- 32.Hersh CP, Demeo DL, Lazarus R, Celedon JC, Raby BA, Benditt JO, et al. Genetic association analysis of functional impairment in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;173:977–84. doi: 10.1164/rccm.200509-1452OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Silverman EK. Progress in chronic obstructive pulmonary disease genetics. Proc Am Thorac Soc. 2006;3:405–8. doi: 10.1513/pats.200603-092AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Habalova V, Salagovic J, Kalina I, Stubna J. Combined analysis of polymorphisms in glutathione S-transferase M1 and microsomal epoxide hydrolase in lung cancer patients. Neoplasma. 2004;51:352–7. [PubMed] [Google Scholar]
- 35.Hersh CP, Demeo DL, Lange C, Litonjua AA, Reilly JJ, Kwiatkowski D, et al. Attempted replication of reported chronic obstructive pulmonary disease candidate gene associations. Am J Respir Cell Mol Biol. 2005;33:71–8. doi: 10.1165/rcmb.2005-0073OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brogger J, Steen VM, Eiken HG, Gulsvik A, Bakke P. Genetic association between COPD and polymorphisms in TNF, ADRB2 and EPHX1. Eur Respir J. 2006;27:682–8. doi: 10.1183/09031936.06.00057005. [DOI] [PubMed] [Google Scholar]
- 37.Baranova H, Perriot J, Albuisson E, Ivaschenko T, Baranov VS, Hemery B, et al. Peculiarities of the GSTM1 0/0 genotype in French heavy smokers with various types of chronic bronchitis. Hum Genet. 1997;99:822–6. doi: 10.1007/s004390050455. [DOI] [PubMed] [Google Scholar]
- 38.Keicho N, Emi M, Kajita M, Matsushita I, Nakata K, Azuma A, et al. Overestimated frequency of a possible emphysema-susceptibility allele when microsomal epoxide hydrolase is genotyped by the conventional polymerase chain reaction-based method. J Hum Genet. 2001;46:96–8. doi: 10.1007/s100380170116. [DOI] [PubMed] [Google Scholar]