

Abstract
The tumor suppressor protein p53 is a redox active transcription factor that organizes and directs cellular responses in the face of a variety of stresses that lead to genomic instability. One of the most important questions in the study of p53 is how selective transactivation of certain p53 target genes is achieved. Reactive oxygen species (ROS), generated by cells as products or byproducts, can function either as signaling molecules or as cellular toxicants. Cellular generation of ROS is central to redox signaling. Recent studies have revealed that each cellular concentration and distribution of p53 has a distinct cellular function, and that ROS act as both an up-stream signal that triggers p53 activation and as a downstream factor that mediates apoptosis. Here, we examine the newly discovered role of p53 in regulating cellular ROS generation and how ROS modulate selective transactivation of p53 target genes. The focus is on interlinks between ROS and p53.
Keywords: p53, ROS, Redox signaling, Oxidative stress, Antioxidant, Mitochondrial translocation, Redox modification, Selection of p53 target gene
Introduction
Aerobic organisms are continually subjected to reactive oxygen species (ROS), derivatives of O2 that are generated as products or byproducts by a plethora of enzymatic reactions in life processes. Superoxide radical (O2•-) is the primary derivative of O2 and it gives rise to hydrogen peroxide (H2O2), alkoxyl/peroxyl radical (RO•/ROO•), and peroxynitrite (ONOOH/ONOO−). Reactions between ROS and redox active amino acid residues (e.g., cysteine) in transcription factors (e.g., AP-1, NF-κB, and HIF-1) and enzymes (e.g., protein tyrosine phosphatases) can modulate the activities of these proteins [1, 2]. Nature has integrated such redox reactions into a variety of signaling pathways to regulate life processes [3–6], where ROS are recruited as second messengers [6,7]. Cellular processes in which ROS are involved range from proliferation to growth arrest or senescence and cell death [4,5]. Cellular generation of ROS is central to redox signaling. Site of generation, spatial distribution, pulse concentration and temporal duration are important parameters of ROS in governing target-specific transduction of redox signals [5, 7, 8], and thus are subject to strict cellular scrutiny. ROS-generating enzymes are compartmentalized [7] and tightly controlled at both the genetic and activity levels [9, 10]; moreover, concentrations of ROS are exquisitely balanced by non-enzymatic antioxidants (e.g., α-tocopherol and glutathione [GSH]) and antioxidant enzymes (e.g., superoxide dismutase [SOD] and catalase) [7]. Correspondingly, antioxidant systems are also specifically organized. For example, cellular thiol/disulfide systems form three distinct redox circuitries (GSH/GSSH, Trx1 [-SH2/-SS-], and cysteine/cystine) to mediate differential responses to physical and toxicological redox stimuli [11, 12]. When the balance between oxidants and antioxidants tips towards the oxidant side, or when a disruption of redox signaling and control occurs, oxidative stress ensues [11], causing damage to bio-molecules such as DNA, proteins and lipids through oxidative modification and contributing to the pathogenesis of human diseases [3] and cytotoxicity of chemotherapy [13].
Tumor suppressor protein p53 occupies a pivotal position in maintaining genomic integrity [14]. In response to cellular stresses that lead to DNA damage, wild-type p53 orchestrates transcriptions of numerous genes and directs cells either to cell cycle arrest, senescence, or apoptosis via differential activation of target genes [15], preventing the propagation of damaged DNA [16]. One of the most important questions in the study of p53 is how p53 determines a specific cellular outcome (e. g., selecting cell cycle arrest between senescence and apoptosis) via selectively regulating certain groups of target genes. Current knowledge shows that various effectors, including proteins and even non-coding RNAs (e.g., myc [17], hCAS/CSE1L [18], Hzf [19], and miR-34 [20]), can play a role in selective transactivation of p53 target genes that leads to different cellular outcomes.
Given that both ROS and p53 participate in multiple cellular processes, there should be interactions between ROS and p53 and intersections between their signaling pathways. A microarray analysis of H2O2-treated human cells identified one-third of the 48 highly H2O2-reponsive genes as targets of p53 [21]. Though it is generally recognized that oxidative stress is associated with p53-dependent cell cycle arrest, DNA repair, and apoptosis, a clear understanding of the mechanisms of the interactions between ROS and p53 is still elusive. In our summary of recent advances in the study of interactions between p53 and ROS, we focus on two main questions in the relationship between ROS and p53: namely, how cellular levels and distribution of p53 influence ROS generation and how ROS modulate selective transactivation of p53 target genes. Since numerous mutations of p53 exist, in this discussion p53 is referred to as wild-type p53, unless otherwise specified.
Cellular levels of p53 and ROS
In unstressed mammalian cells, p53 has a short half-life and is normally maintained at low levels by continuous ubiquitylation catalyzed by Mdm2 [22], COP1 (constitutively photomorphogenic 1) [23], and Pirh2 (p53-induced protein with a RING-H2 domain) [24], and subsequent degradation by 26S proteasome. Current data show that hyper-physiological and physiological levels of p53 exert different effects on cellular redox status either through directly regulating the expressions of pro-oxidant and antioxidant genes or through modulating the cellular metabolic pathways. Consequently, cellular ROS can be modulated by p53 in a number of ways (depicted in Figure 1).
Figure 1.
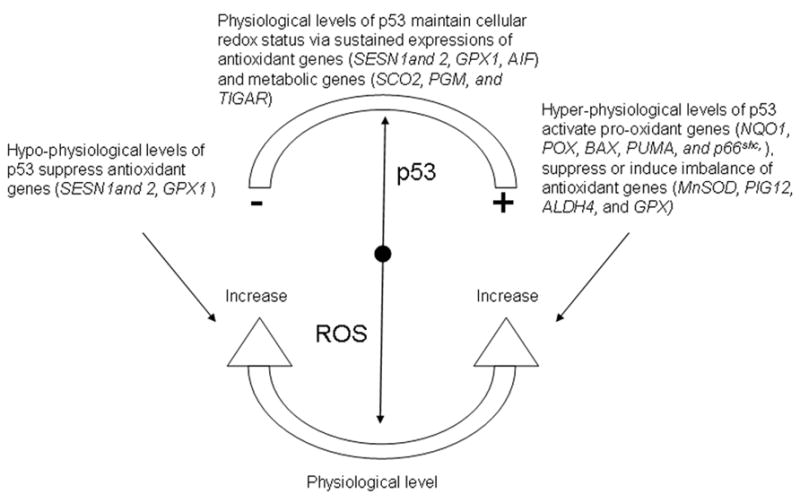
Diagram depicting how cellular levels of p53 regulate cellular ROS generation.
Hyper-physiological levels of p53 and ROS
Polyak et al discovered that overexpression of p53 transactivates a series of p53-induced genes (PIGs) and many of these PIGs encode redox active proteins including two ROS-generating enzymes, NQO1(quinone oxidoreductase, PIG3) [25] and proline oxidase (POX, PIG6) [26]. Upregulation of these pro-oxidant enzymes leads to oxidative stress and consequently to apoptosis [25, 26]. This was the first clear connection between p53 and ROS generation. More candidates have been added to the list of p53-induced pro-oxidant genes, which include BAX, PUMA, and p66Shc. BAX and PUMA can induce uncoupling of mitochondria, resulting in ROS being generated from a less efficient electron transport chain (ETC) [27, 28]. A downstream target of p53 [29], p66Shc predominantly exists in cytoplasm and is translocated into mitochondria with the help of prolyl isomerase 1 (Pin1) and mitochondrial heat shock protein 70 (mtHsp 70) [30]. Confronted in the mitochondria with pro-apoptotic stimulation, p66shc oxidizes cytochrome c, producing H2O2, which promotes the opening of the mitochondrial permeability transition pore and triggers apoptosis [31].
Suppression of antioxidant genes by p53 is an alternative way to increase cellular ROS, conferring oxidative stress. MnSOD (manganese superoxide dismutase) has been shown to be suppressed at the promoter level by p53 activation [32] or overexpression [33]. Surprisingly, even though some antioxidant genes, e.g., PIG12 (microsomal glutathione transferase homologue) [25] and ALDH4 (aldehyde dehydrogenase 4) [34], are concomitantly up-regulated upon p53 overexpression, they are not able to reverse the apoptotic process and seem more like an adaptive response to p53-induced oxidative stress. More surprisingly, oxidative stress can result from the imbalanced induction of antioxidant enzymes by p53. In human lymphoblast cell line TK6, Hussain et al. found that overexpression of p53 increases cellular levels of MnSOD and glutathione peroxidase (GPX), but does not alter the level of catalase [35]. The authors concluded that the oxidative stress is due to increased generation of H2O2 by the elevated levels of MnSOD and possibly inadequate removal by catalase. However, the actual mechanism can be complex [36], with glutathione and NADPH possibly being involved [12, 35].
Basal levels of p53 and ROS
One of the most important developments in p53 research concerns the biological function of physiological levels of p53. In contrast to the pro-oxidant function of hyper-physiological levels of p53, physical levels of p53 have a subtle but vital function in containing ROS at non-toxic levels through transactivation of antioxidant genes [27, 37, 38]. At physical levels, p53 is required to maintain a normal basal transcription of antioxidant genes, SESN1 (mammalian sestrin homolog), SESN2, and GPX1 (glutathione peroxidase-1) [27]. Interestingly, AIF (apoptosis-inducing factor), a pro-apoptosis protein by definition, has paradoxically been found to work as an antioxidant enzyme under physiological conditions regulated by basal levels of p53 [38]. Suppression of p53 results in a significant decrease in the basal transcriptions of SESN1, SESN2, and GPX1 without affecting the expression of pro-oxidant genes BAX, NQO1, and PUMA [27]. This leads to an increase of ROS and subsequently to oxidative damage of DNA, while restoring physical levels of p53 up-regulates the antioxidant enzymes and decreases the levels of ROS. Regulating antioxidant defense against physiological levels of ROS may be one of the tumor suppressing mechanisms of p53. This mechanism seems to be general, for its validity has been seen in multiple normal and carcinoma human cell lines as well as in p53-knockout mice [27].
In addition to transactivation of antioxidant genes, physiological levels of p53 are also indispensable to the expression of key metabolic enzymes, exerting an intricate function in balancing energy metabolism among mitochondrial respiration, glycolysis and the pentose phosphate shunt. Mitochondrial respiration is the major cellular process that produces ROS. Under normal conditions, it is estimated that approximately 1–2% of the electrons leak out of ETC, forming ROS [39]. Conceivably, the regulation of energy metabolism simultaneously controls ROS formation. Constitutive levels of p53 are coupled to normal mitochondrial respiration through its target gene, SCO2 (synthesis of cytochrome c oxidase 2), which has a critical function in maintaining the cytochrome c oxidase complex, the major site of O2 utilization [40]. Mutations of p53 in cancer cells result in defective mitochondrial respiration, forcing cancer cells to use glycolysis for energy (Warburg effect), together with a reduced level of ROS generation [41], suggesting that constitutive levels of p53 sustain the basal level of ROS generated from mitochondrial ETC. In addition to SCO2, phosphoglycerate mutase (PGM), a glycolytic enzyme encoded by a p53 target gene, is another site where physiological levels of p53 regulate energy metabolism. Cell type-specific p53-induced regulation of PGM results in distinct patterns of ROS generation. In mouse embryo fibroblasts, p53 represses the expression of PGM, thus inhibiting the glycolytic pathway and promoting respiration and ROS formation [42]. In muscle cells, PGM seems to be positively regulated by p53, favoring energy production via glycolysis and reduced ROS formation [43]. TIGAR (TP53-induced glycolysis and apoptosis regulator) is another p53 target gene. TIGAR inhibits glycolysis via lowering the intracellular levels of fructose-2,6-bisphosphate (Fru-2,6-P2), a substrate that promotes glycolysis by activation of 6-phospho-1-kinase, a key enzyme of the glycolytic pathway [44]. As a consequence, TIGAR directs glucose into the pentose phosphate pathway that produces NADPH, a cellular reducing power that is utilized to reduce GSSG back to GSH [44]. p53 couples energy metabolism and ROS formation by modulating the transcription of target genes that control the fluxes through mitochondrial respiration, glycolysis or the pentose phosphate shunt.
In order for p53 to effectively regulate the basal levels of ROS, a redox active regulation loop should exist to serve as a channel for crosstalk between the basal levels of cellular ROS and p53 and to keep the fluctuations of ROS and p53 within physiological ranges [45]. Preliminary evidence suggests that two redox proteins, redox factor-1 (Ref-1) and thioredoxin (TRX) reductase (TRR), reciprocally regulate the basal level and activity of p53, with Ref-1 as the stabilizing factor and TRR the destabilizing factor [46]. More in-depth investigations are needed to further elucidate the signaling mechanism between the basal levels of cellular ROS and p53.
Mitochondrial translocation of p53 and ROS
Subcellular localization is an important way to control the function of p53 [47]. Traditionally, studies on p53 localization have been focused on nuclear import and export [48]. Recent investigations of mitochondrial translocation of p53 have added a new direction to this field [49–55]. In all stresses, including oxidative stress, a fraction of cellular p53 (~ 2%) traffics to mitochondria and initiates apoptosis [49, 53]. This stress-induced mitochondrial migration occurs before nuclear import and transactivation of pro-apoptotic genes [51, 54], indicating that the pro-apoptosis function of p53 is independent of its transactivation function. Interestingly, there are two waves of p53 mitochondrial migration [52], which are carried out by different species of p53 [52, 56]. The first wave of mitochondrial trafficking seems to be executed by monoubiquitylated p53 [56] with the help of mtHsp 70 [49]. Zhao et al have found that, once in the mitochondria, p53 binds to and inhibits MnSOD, playing a direct role in promoting ROS formation and eventually in apoptosis [51]. The second wave of mitochondrial trafficking is carried out by fragments of p53 generated by caspases after the onset of apoptosis, further augmenting mitochondrial membrane depolarization and ROS generation [52].
Oxidative stress and apoptosis are not the only outcomes of p53 mitochondrial trafficking [53]. Upon mild oxidative stress, the biological consequences of p53 mitochondrial localization may provide protection to the mitochondrial genome [57, 58], which consists of a circular mitochondrial DNA (mtDNA) and is susceptible to oxidative damage owing to its proximity to the site of ROS generation and its lack of introns and histones [59]. In mitochondrion, polymerase γ is one of the key curators of mtDNA. It has been found that p53 complexes with mtDNA and polymerase γ in mitochondria and enhances DNA replication [57]. In light of the discoveries that at basal levels p53 has an antioxidant role and that hyper-physiological levels of p53 are pro-oxidant, we are curious whether there is a concentration threshold of mitochondria-localized p53 at which the pro-apoptotic function and genome curator function of p53 is differentiated. In addition, as preliminary data have indicated that mitochondrial migration of p53 is under redox modulation [58], it would be important to determine whether ROS play a signaling role in this process. Further investigations are also needed to determine whether mitochondrial targeted antioxidants will reverse the apoptotic effect of mitochondrial localized p53.
Redox regulation of p53
Numerous investigations have correlated oxidative stress with different p53-directed cell fates, such as cell cycle arrest, DNA repair, and apoptosis. For example, excess generation of ROS in mitochondria resulting from treatments with chemotherapeutic agents leads to apoptosis [13, 60], while oxidative stress in the nucleus directs cells to p53-dependent DNA repair [61]. Distinct response patterns suggest that multiple pathways exist that integrate redox and p53 signaling, converting various redox signals into the selection of certain categories of p53 target genes that determine the final fate of the cells. Pharmacological studies on cisplatin and ginkgo bilobalide have shown that chemotherapeutics-induced ROS increase C-myc [62, 63]. Elevated levels of C-myc suppress p53- transactivation of p21Cip1, inhibiting cell cycle arrest, but do not affect p53-transactivation of the pro-apoptosis gene PUMA, thus leading cells into apoptosis [17]. In pathogenic bacterium Pseudomonas aeruginosa induced cell death, azurin, a copper-containing redox protein excreted by Pseudomonas aeruginosa binds to p53 and transactivates pro-apoptosis protein Bax, leading to apoptosis [64, 65]. Currently, knowledge about how ROS regulate p53 target gene selection is still in its infancy. More in-depth research is needed to understand the cause and effect relationship.
Redox modification of p53
In addition to the interactions between ROS and p53 through signaling networks, direct effects of ROS may also affect the fate of p53. The stability and activity of p53 are subject to diverse covalent post-translational modifications such as ubiquitylation [66], phosphorylation [67], acetylation [68], neddylation [69], sumoylation [70], and methylation [71]. Ubiquitylation is involved in p53 degradation [22–24] and mitochondrial trafficking [56]. As for sumoylation, it is still controversial whether it activates or represses p53 activity [72]. Phosphorylation, methylation, and acetylation of p53 generally lead to its stabilization, accumulation, and activation [67, 73, 74]. Among these post-translational modifications, ROS have been implicated in the phosphorylation of p53 mediated via protein kinases, including p38α MARK (mitogen activated protein kinase) [75], ATM (ataxia-telangiectasia mutated protein) [76], and ERK (extracelluar signal-regulated kinases) [77]. However, activation of these protein kinases, which are common downstream effectors of signaling pathways that respond to DNA damage, is not necessarily ROS specific. Genotoxic stresses in addition to ROS, e.g., UV light, share the same signaling pathways [67, 76, 78].
p53 itself is redox active due to the presence of cysteines (Cys) that contain redox sensitive thiol groups (-SH) [79] (Fig. 2). In human p53, there are two clusters of cysteines in the DNA-binding domain, which are essential to the specific binding of p53 to its consensus sequence [80]. Cys 176, 238, and 242, along with histidine 179, consist of a binding site for Zn2+ [79]. Mutation of these Zn2+-ligands diminishes the sequence-specific DNA binding of p53 [81]. Cys 124, 135, 141, 182 and 277 are located in the loop-sheet-helix region of the proximal DNA-binding domain of p53. They constitute a structural platform for redox modulation. Theoretically, there are multiple possible structures of oxidized thiol groups in proteins, including sulphenic acid (-SOH), disulfide (-S-S-), sulphenamide (-SNR1R2), sulphinic acid (-SO2H), and sulphonic acid (-SO3H) [82]. It has been observed that treating p53 with oxidizing reagents abolishes its DNA-binding activity. Two recent studies identified the sites and structural details of p53 oxidation [83, 84]. GSH was found to be attached to either Cys124 or 141, and to 182 of p53 via disulfide bond after oxidant treatment, decreasing the DNA-binding activity of p53, which could be reversed by antioxidants [83,84]. Similar effects of S-glutathionylation of the conserved cysteines, which were observed in the study of p53, were also observed for AP-1 [85], NF-κB [86], and Pax-8 [87]. S-glutathionylation of p53 occurs both in vitro and in vivo, and is regulated by the ratio of GSH/GSSH [83]. As a major cellular redox buffer, total GSH (including both GSH and GSSG) exists in high concentrations in cytosol (1–11 mM), nuclei (3–15 mM), and mitochondria (5–11 mM) [3]. Fluctuations in the ratio of GSH/GSSG, either on the local or on the global level, can serve as the driving force for S-glutathionylation. Previous studies showed that redox proteins TRX, TRR, and APE/Ref-1 affect p53 activity [46, 61, 88, 89]. Identification of protein effectors involved in the redox modification process will provide mechanistic insights into how redox modification of p53 is channeled to the fluctuations of organelle or cellular redox status.
Figure 2.
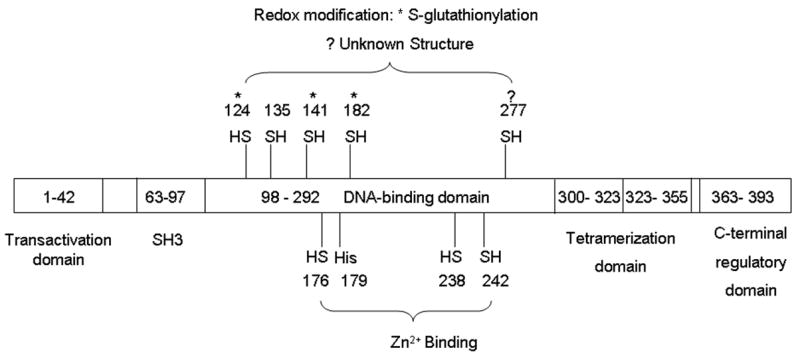
Redox modifications of functionally important amino acid residues on p53. Cys 176, 238, and 242 and His 179 form the Zn2+-binding site. Among the potential redox sensitive cysteines, it has been found that Cys 124, 141, and 182 can form a disulfide bond with GSH. Oxidation of Cys 277 decreases p53 binding to GADD45, but does not affect its binding to p21CIP1, even though the structural nature of this redox modification is unknown.
Bezek et al previously reported that oxidation of Cys 277 decreases p53 binding to GADD45 but not to p21CIP1 [90]. Although the structural nature of the redox modification on Cys 277 in p53 is still unknown and GADD45 and p21CIP1 are both involved in DNA repair and cell growth arrest [91], this discovery reveals a fascinating connection between ROS and p53 function; i.e., redox modification can be a potential mechanism for target gene selection. More work is needed to investigate whether, in addition to disulfide bond, other structures of the oxidation products of cysteines exist during p53 oxidation.
Different post-translational modifications can coexist on one molecule, as has been observed in p53 phosphorylation and acetylation [67], both of which generally stabilize p53. S-glutathionylation of p53 coexists with phosphorylation [83]. However, how S-glutathionylation or, in general, how redox modification affects p53 stability is unknown, though researchers have noticed cellular accumulation of oxidized p53 [46]. The major turnover pathway for p53 is through ubiquitylation and subsequent 26S proteasome degradation. In p53, Cys124, 141, 182 and 277 do not overlap with the ubiquitylation sites that are located on the C-terminus [83, 92, 93]. It is then unlikely that redox modification directly blocks the ubiquitylation sites. However, it cannot be excluded that redox modification might induce conformation changes that would affect the stability of p53. Future research is needed to address the effects of redox modification on p53 stability and whether different structures or sites of redox modifications convey differential binding affinity to the response elements in p53 target genes or the recruitment of p53 partners to its target genes, leading to target gene selection. Such information will add significantly to the rapidly growing number of fascinating reports on how p53 selects its target genes and how that impacts the various cellular responses of p53.
Conclusion
Even after almost three decades of intensive research, new discoveries of the biological roles of p53 continue to intrigue scientists from diverse fields. Connections between p53 and ROS provide a unique perspective to examine and appreciate their biological functions. Current data show that cellular concentration and subcellular localization are important to defining the functions of p53-mediated ROS generation. A redox sensitive protein, p53 is also under redox regulation, which determines cell fate via selection of p53 target genes. Other parameters, including cell type, source of stress, and intensity of stimuli, also determine the outcomes of the interaction between ROS and p53, and they may limit any generalized mechanistic explanation of how p53 and ROS interact. Hopefully, genome profiling techniques coupled to genetic modulation of p53 levels will prove to be effective tools to pinpoint the specific target effectors involved in ROS and p53 interaction [25, 27].
Acknowledgments
This work is supported by NIH grants CA 59797 and CA 94853.
Abbreviations
- ROS
Reactive oxygen species
- GSH
Glutathione
- COP1
Constitutively photomorphogenic 1
- Pirh2
p53-induced protein with a RING-H2 domain
- PIG
p53-induced gene
- NQO1
Quinone oxidoreductase
- POX
Proline oxidase
- Pin1
Prolyl isomerase 1
- ALDH4
Aldehyde dehydrogenase 4
- GPX
Glutathione peroxidase
- ETC
Electron transport chain
- SESN1
Mammalian sestrin homolog
- AIF
Apoptosis-inducing factor
- SCO2
Synthesis of cytochrome c oxidase 2
- PGM
phosphoglycerate mutase
- TIGAR
TP53-induced glycolysis and apoptosis regulator
- Fru-2
6-P2, Fructose-2,6-bisphosphate
- Ref-1
Redox factor-1
- TRX
Thioredoxin
- TRR
Thioredoxin reductase
- mtHsp70
Mitochondrial heat shock protein 70
- MnSOD
Manganese superoxide dismutase
- mtDNA
Mitochondrial DNA
- MAPK
Mitogen activated protein kinase
- ATM
Ataxia-telangiectasia mutated protein
- ERK
Extracelluar signal-regulated kinase
- Cys
Cysteine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 2.Salmeen A, Barford D. Functions and mechanisms of redox regulation of cysteine-based phosphatases. Antioxidants & Redox Signaling. 2005;7:560–577. doi: 10.1089/ars.2005.7.560. [DOI] [PubMed] [Google Scholar]
- 3.Valko M, Leibfritz D, Moncol J, Cronin MTD, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. International Journal of Biochemistry & Cell Biology. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 4.Martindale JL, Holbrook NJ. Cellular response to oxidative stress: Signaling for suicide and survival. Journal of Cellular Physiology. 2002;192:1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- 5.Menon SG, Goswami PC. A redox cycle within the cell cycle: Ring in the old with the new. Oncogene. 2007;26:1101–1109. doi: 10.1038/sj.onc.1209895. [DOI] [PubMed] [Google Scholar]
- 6.Takahashi A, Ohtani N, Yamakoshi K, Iida SI, Tahara H, Nakayama K, et al. Mitogenic signalling and the p16(ink4a)-rb pathway cooperate to enforce irreversible cellular senescence. Nature Cell Biology. 2006;8:1291–U1263. doi: 10.1038/ncb1491. [DOI] [PubMed] [Google Scholar]
- 7.Terada LS. Specificity in reactive oxidant signaling: Think globally, act locally. Journal of Cell Biology. 2006;174:615–623. doi: 10.1083/jcb.200605036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Forman HJ. Use and abuse of exogenous h2o2 in studies of signal transduction. Free Radic Biol Med. 2007;42:926–932. doi: 10.1016/j.freeradbiomed.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bokoch GM. Regulation of the human neutrophil nadph oxidase by the rac gtp-binding proteins. Current Opinion in Cell Biology. 1994;6:212–218. doi: 10.1016/0955-0674(94)90138-4. [DOI] [PubMed] [Google Scholar]
- 10.Sundaresan M, Yu ZX, Ferrans VJ, Sulciner DJ, Gutkind JS, Irani K, et al. Regulation of reactive-oxygen-species generation in fibroblasts by rac1. Biochemical Journal. 1996;318:379–382. doi: 10.1042/bj3180379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jones DP. Redefining oxidative stress. Antioxidants & Redox Signaling. 2006;8:1865–1879. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]
- 12.Jones DP. Disruption of mitochondrial redox circuitry in oxidative stress. Chemico-Biological Interactions. 2006;163:38–53. doi: 10.1016/j.cbi.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 13.Chen Y, Jungsuwadee P, Vore M, Butterfield DA, StClair DK. Collateral damage in cancer chemotherapy: Oxidative stress in nontargeted tissues. Molecular Interventions. 2007;7:147–156. doi: 10.1124/mi.7.3.6. [DOI] [PubMed] [Google Scholar]
- 14.Vousden KH, Lu X. Live or let die: The cell’s response to p53. Nat Rev Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 15.Gang Liu XC. Regulation of the p53 transcriptional activity. Journal of Cellular Biochemistry. 2006;97:448–458. doi: 10.1002/jcb.20700. [DOI] [PubMed] [Google Scholar]
- 16.Lim YP, Lim TT, Chan YL, Song ACM, Yeo BH, Vojtesek B, et al. The p53 knowledge base: An integrated information resource for p53 research. Oncogene. 2006;26:1517–1521. doi: 10.1038/sj.onc.1209952. [DOI] [PubMed] [Google Scholar]
- 17.Seoane J, Le H-V, Massague J. Myc suppression of the p21cip1 cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature. 2002;419:729–734. doi: 10.1038/nature01119. [DOI] [PubMed] [Google Scholar]
- 18.Tanaka T, Ohkubo S, Tatsuno I, Prives C. Hcas/cse1l associates with chromatin and regulates expression of select p53 target genes. Cell. 2007;130:638–650. doi: 10.1016/j.cell.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 19.Das S, Raj L, Zhao B, Kimura Y, Bernstein A, Aaronson SA, et al. Hzf determines cell survival upon genotoxic stress by modulating p53 transactivation. Cell. 2007;130:624–637. doi: 10.1016/j.cell.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He L, He X, Lowe SW, Hannon GJ. Micrornas join the p53 network [mdash] another piece in the tumour-suppression puzzle. Nat Rev Cancer. 2007;7:819–822. doi: 10.1038/nrc2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Desaint S, Luriau S, Aude JC, Rousselet G, Toledano MB. Mammalian antioxidant defenses are not inducible by H2O2. Journal of Biological Chemistry. 2004;279:31157–31163. doi: 10.1074/jbc.M401888200. [DOI] [PubMed] [Google Scholar]
- 22.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 23.Dornan D, Wertz I, Shimizu H, Arnott D, Frantz GD, Dowd P, et al. The ubiquitin ligase cop1 is a critical negative regulator of p53. Nature. 2004;429:86–92. doi: 10.1038/nature02514. [DOI] [PubMed] [Google Scholar]
- 24.Leng RP, Lin Y, Ma W, Wu H, Lemmers B, Chung S, et al. Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell. 2003;112:779–791. doi: 10.1016/s0092-8674(03)00193-4. [DOI] [PubMed] [Google Scholar]
- 25.Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389:300–305. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- 26.Rivera A, Maxwell SA. The p53-induced gene-6 (proline oxidase) mediates apoptosis through a calcineurin-dependent pathway. Journal of Biological Chemistry. 2005;280:29346–29354. doi: 10.1074/jbc.M504852200. [DOI] [PubMed] [Google Scholar]
- 27.Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005;11:1306–1313. doi: 10.1038/nm1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Z, Lu H, Shi H, Du Y, Yu J, Gu S, et al. Puma overexpression induces reactive oxygen species generation and proteasome-mediated stathmin degradation in colorectal cancer cells. Cancer Research. 2005;65:1647–1654. doi: 10.1158/0008-5472.CAN-04-1754. [DOI] [PubMed] [Google Scholar]
- 29.Trinei M, Giorgio M, Cicalese A, Barozzi S, Ventura A, Migliaccio E, et al. A p53-p66shc signalling pathway controls intracellular redox status, levels of oxidation-damaged DNA and oxidative stress-induced apoptosis. Oncogene. 2002;21:3872–3878. doi: 10.1038/sj.onc.1205513. [DOI] [PubMed] [Google Scholar]
- 30.Pinton P, Rimessi A, Marchi S, Orsini F, Migliaccio E, Giorgio M, et al. Protein kinase c beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66(shc) Science. 2007;315:659–663. doi: 10.1126/science.1135380. [DOI] [PubMed] [Google Scholar]
- 31.Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, et al. Electron transfer between cytochrome c and p66(shc) generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 2005;122:221–233. doi: 10.1016/j.cell.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 32.Drane P, Bravard A, Bouvard V, May E. Reciprocal down-regulation of p53 and sod2 gene expression--implication in p53 mediated apoptosis. Oncogene. 2001;20:430–439. doi: 10.1038/sj.onc.1204101. [DOI] [PubMed] [Google Scholar]
- 33.Dhar SK, Xu Y, Chen Y, StClair DK. Specificity protein 1-dependent p53-mediated suppression of human manganese superoxide dismutase gene expression. Journal of Biological Chemistry. 2006;281:21698–21709. doi: 10.1074/jbc.M601083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoon K-A, Nakamura Y, Arakawa H. Identification of aldh4 as a p53-inducible gene and its protective role in cellular stresses. J Hum Genet. 2004;49:134–140. doi: 10.1007/s10038-003-0122-3. [DOI] [PubMed] [Google Scholar]
- 35.Hussain SP, Amstad P, He P, Robles A, Lupold S, Kaneko I, et al. P53-induced up-regulation of mnsod and gpx but not catalase increases oxidative stress and apoptosis. Cancer Res. 2004;64:2350–2356. doi: 10.1158/0008-5472.can-2287-2. [DOI] [PubMed] [Google Scholar]
- 36.Liochev SI, Fridovich I. The effects of superoxide dismutase on H2O2 formation. Free Radical Biology and Medicine. 2007;42:1465–1469. doi: 10.1016/j.freeradbiomed.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 37.Tomko RJ, Jr, Bansal P, Lazo JS. Airing out an antioxidant role for the tumor suppressor p53. Mol Interv. 2006;6:23–25. doi: 10.1124/mi.6.1.5. [DOI] [PubMed] [Google Scholar]
- 38.Stambolsky P, Weisz L, Shats I, Klein Y, Goldfinger N, Oren M, et al. Regulation of aif expression by p53. Cell Death and Differentiation. 2006;13:2140–2149. doi: 10.1038/sj.cdd.4401965. [DOI] [PubMed] [Google Scholar]
- 39.Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiological Reviews. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 40.Matoba S, Kang J-G, Patino WD, Wragg A, Boehm M, Gavrilova O, et al. P53 regulates mitochondrial respiration. Science. 2006;312:1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- 41.Brand KA, Hermfisse U. Aerobic glycolysis by proliferating cells: A protective strategy against reactive oxygen species. The FASEB Journal. 1997;11:388–395. doi: 10.1096/fasebj.11.5.9141507. [DOI] [PubMed] [Google Scholar]
- 42.Kondoh H, Lleonart ME, Gil J, Wang J, Degan P, Peters G, et al. Glycolytic enzymes can modulate cellular life span. Cancer Research. 2005;65:177–185. [PubMed] [Google Scholar]
- 43.Ruiz-Lozano P, Hixon ML, Wagner MW, Flores AI, Ikawa S, Baldwin AS, Jr, et al. P53 is a transcriptional activator of the muscle-specific phosphoglycerate mutase gene and contributes in vivo to the control of its cardiac expression. Cell Growth & Differentiation. 1999;10:295–306. [PubMed] [Google Scholar]
- 44.Bensaad K, Tsuruta A, Selak MA, Vidal MNC, Nakano K, Bartrons R, et al. Tigar, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 45.Budanov AV, Sablina AA, Feinstein E, Koonin EV, Chumakov PM. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial ahpd. Science. 2004;304:596–600. doi: 10.1126/science.1095569. [DOI] [PubMed] [Google Scholar]
- 46.Seemann S, Hainaut P. Roles of thioredoxin reductase 1 and ape/ref-1 in the control of basal p53 stability and activity. Oncogene. 2005;24:3853–3863. doi: 10.1038/sj.onc.1208549. [DOI] [PubMed] [Google Scholar]
- 47.O’Brate A, Giannakakou P. The importance of p53 location: Nuclear or cytoplasmic zip code? Drug Resistance Updates. 2003;6:313–322. doi: 10.1016/j.drup.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 48.Vousden KH, Vande Woude GF. The ins and outs of p53. Nature Cell Biology. 2000;2:E178–E180. doi: 10.1038/35036427. [DOI] [PubMed] [Google Scholar]
- 49.Marchenko ND, Zaika A. Moll UMDeath signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J Biol Chem. 2000;275:16202–16212. doi: 10.1074/jbc.275.21.16202. [DOI] [PubMed] [Google Scholar]
- 50.Erster S, Mihara M, Kim RH, Petrenko O, Moll UM. In vivo mitochondrial p53 translocation triggers a rapid first wave of cell death in response to DNA damage that can precede p53 target gene activation. Mol Cell Biol. 2004;24:6728–6741. doi: 10.1128/MCB.24.15.6728-6741.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao Y, Chaiswing L, Velez JM, Batinic-Haberle I, Colburn NH, Oberley TD, et al. P53 translocation to mitochondria precedes its nuclear translocation and targets mitochondrial oxidative defense protein-manganese superoxide dismutase. Cancer Res. 2005;65:3745–3750. doi: 10.1158/0008-5472.CAN-04-3835. [DOI] [PubMed] [Google Scholar]
- 52.Sayan BS, Sayan AE, Knight RA, Melino G, Cohen GM. P53 is cleaved by caspases generating fragments localizing to mitochondria. J Biol Chem. 2006;281:13566–13573. doi: 10.1074/jbc.M512467200. [DOI] [PubMed] [Google Scholar]
- 53.Essmann F, Pohlmann S, Gillissen B, Daniel PT, Schulze-Osthoff K, Janicke RU. Irradiation-induced translocation of p53 to mitochondria in the absence of apoptosis. Journal of Biological Chemistry. 2005;280:37169–37177. doi: 10.1074/jbc.M502052200. [DOI] [PubMed] [Google Scholar]
- 54.Arima Y, Nitta M, Kuninaka S, Zhang D, Fujiwara T, Taya Y, et al. Transcriptional blockade induces p53-dependent apoptosis associated with translocation of p53 to mitochondria. Journal of Biological Chemistry. 2005;280:19166–19176. doi: 10.1074/jbc.M410691200. [DOI] [PubMed] [Google Scholar]
- 55.Yoshida Y, Izumi H, Torigoe T, Ishiguchi H, Itoh H, Kang DC, et al. P53 physically interacts with mitochondrial transcription factor a and differentially regulates binding to damaged DNA. Cancer Research. 2003;63:3729–3734. [PubMed] [Google Scholar]
- 56.Marchenko ND, Wolff S, Erster S, Becker K, Moll UM. Monoubiquitylation promotes mitochondrial p53 translocation. Embo Journal. 2007;26:923–934. doi: 10.1038/sj.emboj.7601560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Achanta G, Sasaki R, Feng L, Carew JS, Lu W, Pelicano H, et al. Novel role of p53 in maintaining mitochondrial genetic stability through interaction with DNA pol gamma. EMBO J. 2005;24:3482–3492. doi: 10.1038/sj.emboj.7600819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nithipongvanitch R, Ittarat W, Cole MP, Tangpong J, StClair DK, Oberley TD. Mitochondrial and nuclear p53 localization in cardiomyocytes: Redox modulation by doxorubicin (adriamycin)? Antioxidants & Redox Signaling. 2007;9:1001–1008. doi: 10.1089/ars.2007.1632. [DOI] [PubMed] [Google Scholar]
- 59.Wallace DC. Mitochondrial defects in cardiomyopathy and neuromuscular disease. American Heart Journal. 2000;139:S70–S85. doi: 10.1067/mhj.2000.103934. [DOI] [PubMed] [Google Scholar]
- 60.Hwang PM, Bunz F, Yu J, Rago C, Chan TA, Murphy MP, et al. Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nat Med. 2001;7:1111–1117. doi: 10.1038/nm1001-1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ueno M, Masutani H, Arai RJ, Yamaguchi A, Hirota K, Sakai T, et al. Thioredoxin-dependent redox regulation of p53-mediated p21 activation. Journal of Biological Chemistry. 1999;274:35809–35815. doi: 10.1074/jbc.274.50.35809. [DOI] [PubMed] [Google Scholar]
- 62.Zhou LJ, Zhu XZ. Reactive oxygen species-induced apoptosis in pc12 cells and protective effect of bilobalide. Journal of Pharmacology and Experimental Therapeutics. 2000;293:982–988. [PubMed] [Google Scholar]
- 63.Biroccio A, Benassi B, Amodei S, Gabellini C, Del Bufalo D, Zupi G. C-myc down-regulation increases susceptibility to cisplatin through reactive oxygen species-mediated apoptosis in m14 human melanoma cells. Molecular Pharmacology. 2001;60:174–182. doi: 10.1124/mol.60.1.174. [DOI] [PubMed] [Google Scholar]
- 64.Punj V, Bhattacharyya S, Saint-Dic D, Vasu C, Cunningham EA, Graves J, et al. Bacterial cupredoxin azurin as an inducer of apoptosis and regression in human breast cancer. Oncogene. 2004;23:2367–2378. doi: 10.1038/sj.onc.1207376. [DOI] [PubMed] [Google Scholar]
- 65.Yamada T, Hiraoka Y, Ikehata M, Kimbara K, Avner BS, Das Gupta TK, et al. Apoptosis or growth arrest: Modulation of tumor suppressor p53’s specificity by bacterial redox protein azurin. Proc Natl Acad Sci U S A. 2004;101:4770–4775. doi: 10.1073/pnas.0400899101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dai MS, Jin Y, Gallegos JR, Lu H. Balance of yin and yang: Ubiquitylation-mediated regulation of p53 and c-myc. Neoplasia. 2006;8:630–644. doi: 10.1593/neo.06334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bode AM, Dong ZG. Post-translational modification of p53 in tumorigenesis. Nature Reviews Cancer. 2004;4:793–805. doi: 10.1038/nrc1455. [DOI] [PubMed] [Google Scholar]
- 68.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 c-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 69.Xirodimas DP, Saville MK, Bourdon J-C, Hay RT, Lane DP. Mdm2-mediated nedd8 conjugation of p53 inhibits its transcriptional activity. Cell. 2004;118:83–97. doi: 10.1016/j.cell.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 70.Il Kim K, Baek SH. Sumoylation code in cancer development and metastasis. Molecules and Cells. 2006;22:247–253. [PubMed] [Google Scholar]
- 71.Chuikov S, Kurash JK, Wilson JR, Xiao B, Justin N, Ivanov GS, et al. Regulation of p53 activity through lysine methylation. Nature. 2004;432:353–360. doi: 10.1038/nature03117. [DOI] [PubMed] [Google Scholar]
- 72.Chen LH, Chen JD. Mdm2-arf complex regulates p53 sumoylation. Oncogene. 2003;22:5348–5357. doi: 10.1038/sj.onc.1206851. [DOI] [PubMed] [Google Scholar]
- 73.Lake AN, Bedford MT. Protein methylation and DNA repair. Mutation Research-Fundamental and Molecular Mechanisms of Mutagenesis. 2007;618:91–101. doi: 10.1016/j.mrfmmm.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 74.Ito A, Lai CH, Zhao X, Saito S, Hamilton MH, Appella E, et al. P300/cbp-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by mdm2. Embo Journal. 2001;20:1331–1340. doi: 10.1093/emboj/20.6.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bragado P, Armesilla A, Silva A, Porras A. Apoptosis by cisplatin requires p53 mediated p38α mapk activation through ros generation. Apoptosis. 2007;12:1733–1742. doi: 10.1007/s10495-007-0082-8. [DOI] [PubMed] [Google Scholar]
- 76.Kurz EU, Lees-Miller SP. DNA damage-induced activation of atm and atm-dependent signaling pathways. DNA Repair. 2004;3:889–900. doi: 10.1016/j.dnarep.2004.03.029. [DOI] [PubMed] [Google Scholar]
- 77.Persons DL, Yazlovitskaya EM, Pelling JC. Effect of extracellular signal-regulated kinase on p53 accumulation in response to cisplatin. 2000. pp. 35778–35785. [DOI] [PubMed] [Google Scholar]
- 78.Moiseeva O, Mallette FA, Mukhopadhyay UK, Moores A, Ferbeyre G. DNA damage signaling and p53-dependent senescence after prolonged beta-interferon stimulation. Molecular Biology of the Cell. 2006;17:1583–1592. doi: 10.1091/mbc.E05-09-0858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hainaut P, Mann K. Zinc binding and redox control of p53 structure and function. Antioxidants & Redox Signaling. 2001;3:611–623. doi: 10.1089/15230860152542961. [DOI] [PubMed] [Google Scholar]
- 80.Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science. 1994;265:346–355. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- 81.Rainwater R, Parks D, Anderson ME, Tegtmeyer P, Mann K. Role of cysteine residues in regulation of p53 function. Mol Cell Biol. 1995;15:3892–3903. doi: 10.1128/mcb.15.7.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.D’Autreaux B, Toledano MB. Ros as signalling molecules: Mechanisms that generate specificity in ros homeostasis. Nature Reviews Molecular Cell Biology. 2007;8:813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- 83.Velu CS, Niture SK, Doneanu CE, Pattabiraman N, Srivenugopal KS. Human p53 is inhibited by glutathionylation of cysteines present in the proximal DNA-binding domain during oxidative stress. Biochemistry. 2007;46:7765–7780. doi: 10.1021/bi700425y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sun XZ, Vinci C, Makmura L, Han SB, Tran D, Nguyen J, et al. Formation of disulfide bond in p53 correlates with inhibition of DNA binding and tetramerization. Antioxidants & Redox Signaling. 2003;5:655–665. doi: 10.1089/152308603770310338. [DOI] [PubMed] [Google Scholar]
- 85.Klatt P, Molina EP, De Lacoba MG, Padilla CA, Martinez-Galisteo E, Barcena JA, et al. Redox regulation of c-jun DNA binding by reversible s-glutathiolation. Faseb Journal. 1999;13:1481–1490. doi: 10.1096/fasebj.13.12.1481. [DOI] [PubMed] [Google Scholar]
- 86.Pineda-Molina E, Klatt P, Vazquez J, Marina A, de Lacoba MG, Perez-Sala D, et al. Glutathionylation of the p50 subunit of nf-kappa b: A mechanism for redox-induced inhibition of DNA binding. Biochemistry. 2001;40:14134–14142. doi: 10.1021/bi011459o. [DOI] [PubMed] [Google Scholar]
- 87.Cao X, Kambe F, Lu X, Kobayashi N, Ohmori S, Seo H. Glutathionylation of two cysteine residues in paired domain regulates DNA binding activity of pax-8. Journal of Biological Chemistry. 2005;280:25901–25906. doi: 10.1074/jbc.M411443200. [DOI] [PubMed] [Google Scholar]
- 88.Seo YR, Kelley MR, Smith ML. Selenomethionine regulation of p53 by a ref1-dependent redox mechanism. Proc Natl Acad Sci U S A. 2002;99:14548–14553. doi: 10.1073/pnas.212319799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hanson S, Kim E, Deppert W. Redox factor 1 (ref-1) enhances specific DNA binding of p53 by promoting p53 tetramerization. Oncogene. 2005;24:1641–1647. doi: 10.1038/sj.onc.1208351. [DOI] [PubMed] [Google Scholar]
- 90.Buzek J, Latonen L, Kurki S, Peltonen K, Laiho M. Redox state of tumor suppressor p53 regulates its sequence-specific DNA binding in DNA-damaged cells by cysteine 277. Nucleic Acids Research. 2002;30:2340–2348. doi: 10.1093/nar/30.11.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Giono LE, Manfredi JJ. The p53 tumor suppressor participates in multiple cell cycle checkpoints. Journal of Cellular Physiology. 2006;209:13–20. doi: 10.1002/jcp.20689. [DOI] [PubMed] [Google Scholar]
- 92.Nakamura S, Roth JA, Mukhopadhyay T. Multiple lysine mutations in the c-terminal domain of p53 interfere with mdm2-dependent protein degradation and ubiquitination. Molecular and Cellular Biology. 2000;20:9391–9398. doi: 10.1128/mcb.20.24.9391-9398.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gu J, Nie L, Wiederschain D, Yuan Z-M. Identification of p53 sequence elements that are required for mdm2-mediated nuclear export. Molecular and Cellular Biology. 2001;21:8533–8546. doi: 10.1128/MCB.21.24.8533-8546.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]