

Abstract
Cytochrome c oxidase catalyzes the reduction of oxygen to water that is accompanied by pumping of four protons across the mitochondrial or bacterial membrane. Triggered by the results of recent x-ray crystallographic analyses, published data concerning the coupling of individual electron transfer steps to proton pumping are reanalyzed: Conversion of the conventional oxoferryl intermediate F to the fully oxidized form O is connected to pumping of only one proton. Most likely one proton is already pumped during the double reduction of O, and only three protons during conversion of the “peroxy” forms P to O via the oxoferryl form F. Based on the available structural, spectroscopic, and mutagenesis data, a detailed mechanistic model, carefully considering electrostatic interactions, is presented. In this model, each of the four reductions of heme a during the catalytic cycle is coupled to the uptake of one proton via the D-pathway. These protons, but never more than two, are temporarily stored in the regions of the heme a and a3 propionates and are driven to the outside (“pumped”) by electrostatic repulsion from protons entering the active site during turnover. The first proton is pumped by uptake of one proton via the K-pathway during reduction, the second and third proton during the P → F transition when the D-pathway and the active site become directly connected, and the fourth one upon conversion of F to O. Atomic structures are assigned to each intermediate including F′ with an alternative route to O.
Cytochrome c oxidase (COX), located in the inner membrane of mitochondria and bacteria, is the terminal enzyme in the respiratory chains of many aerobic organisms. It catalyzes electron transfer from cytochrome c to molecular oxygen, reducing the latter to water. The reaction is accompanied by vectorial transport (“pumping”) of four protons across the membrane. Because cytochrome c donates its electron from the outside, and the protons consumed in water formation originate from the inside, a total of eight charges is translocated across the membrane.
The discovery that COX functions as a proton pump (1) has considerably increased the interest to understand this fundamental enzyme. COX (reviewed in refs. 2 and 3) contains four redox-active cofactors, namely two Cu atoms forming the so-called CuA, a low-spin heme a and a heme a3-CuB binuclear center. Cu A accepts the electrons from cytochrome c. Electrons are transferred via heme a to the binuclear center where oxygen reduction takes place.
During the catalytic cycle (see Fig. 1) uptake of one electron by the fully oxidized enzyme (O), leads to the formation of the one-electron reduced enzyme (E). Further electron transfer generates the two-electron reduced state (R). R can bind oxygen. Via compound A (5), the so-called peroxy state P is reached. P appears to exist in two forms, PM, a two-electron reduced one, and PR after the third reduction. In Fig. 1, then splitting of the O—O bond, uptake of two protons and formation of the first water molecule is indicated, generating the oxoferryl state F (6). Input of the fourth electron leads to the sequential uptake of two protons, formation of the second water molecule, and generation of the O-state, via the hydroxy intermediate H. Conventional P-states absorb at 607 nm, F-states at 580 nm.
Figure 1.

Model of a conventional catalytic cycle of COX (modified after ref. 4). Fe stands for heme a3: Cu, CuB; e−, electrons; Hi+, protons taken up from the inside of mitochondria or bacteria; Ho+, protons released to the outside. For details, see text.
Effects of the electric field across the mitochondrial membrane on the F:O ratio, and the extramitochondrial ATP/ADP ⋅ phosphate ratio on the P:O and F:O ratios by “reversed electron flow” led to the postulate (7) that the P → F and F → O transitions are coupled exclusively to proton pumping with a stoichiometry of two protons per transition. This proposal has found widespread acceptance. However, location of heme a in the middle of the dielectric barrier between the outer and inner membrane surfaces is crucial for the derived pumping stoichiometry (see below).
The fact that the P → F and F → O transitions as shown in Fig. 1 are accompanied by the formation of one water molecule each, is suggestive that water formation may somehow lead to proton pumping. Mitchell and Rich (8) have formulated an electroneutrality principle and postulated that reduction of the binuclear center causes the uptake of two protons to maintain electroneutrality. These protons are expelled to the periplasm through electrostatic repulsion by the protons required for water formation (9). One of the “histidine cycle” models (10, 11), postulated in parallel, also strictly obeys this electroneutrality principle. In the histidine cycle models, a CuB histidine ligand is the proton acceptor and cycles twice between the imidazolate, imidazole, and imidazolium states.
Surprisingly, Kitagawa and coworkers (12, 13) demonstrated that in the P-states (see Fig. 1) the O—O-bond is already broken and have suggested the P-states to be a hydrogen-bonded oxoferryl form. This suggestion raises the question whether water has been already formed during the transition to the P-state. If yes, all proton pump mechanisms, which use incoming protons needed for water formation to electrostatically repel protons taken up during reduction of the binuclear site, are in jeopardy because proton pumping is expected to occur only beyond the P-state.
The identification of the proton-accepting groups is of prime importance for elucidating the mechanism of proton pumping. We have used electrochemically induced redox-difference Fourier transform-IR spectroscopy (14, 15) to investigate this question. The results demonstrate the existence of reversible structural changes, which are absent in a subunit I-E278Q mutant. E278 stays, however, in its protonated (neutral) state (14). The equivalent residue in the related Escherichia coli cytochrome bo changes its IR absorption on release of CO from the fully reduced enzyme even at cryo-temperatures (16, 17). Pronounced conformational and/or protonation changes have been observed for at least two heme propionate side chains (15). In a theoretical approach, the electrostatic potential, the interaction energies of ionizable groups, and the titration curves of all protonable groups, in the recently determined two-subunit COX from Paracoccus denitrificans (18), have been calculated (19). The results reproduce well the observed proton uptake of 2.0 protons upon formation of the R-state (8). A putative CuB bound OH− (see below) is the most favored proton acceptor. There is a core cluster of strongly electrostatically interacting residues that includes the heme propionates and 13 other residues, which most likely accepts one proton. The contributions of individual residues are difficult to estimate, due to possible inhomogeneity of the dielectric medium in this cluster (see below), the inaccuracy of the coordinates, and the strong electrostatic interactions.
Constraints Imposed by Structural Data and Analysis of Mutants.
The recent structure determinations of COX from P. denitrificans (20, 18) and bovine heart mitochondria (21–23) reveal a surprisingly high similarity of the three core subunits I, II, and III. One potential pathway for proton transfer as shown in Fig. 2 leads from the conserved D124 under involvement of solvent molecules straight up to S192 and S193 and from there through a large presumably water-filled cavity to the conserved E278. The further pathway for protons is unclear. It may lead directly to the binuclear site via a temporarily established chain of water molecules (see below), or to ring D propionate of heme a3, either by direct contacts upon conformational changes of both acidic groups (20), or via unresolved intervening water molecules. Replacement of the residues corresponding to D124 by Asn or of E278 by Cys led to a similar phenotype (25, 26, 17); some residual turnover, however, uncoupled from proton pumping, was observed. Therefore, both residues are included in a so-called D-pathway of proton transfer. In the E278Q mutant enzymes the transition from the P-state to the subsequent states appears to be inhibited. Therefore, in this mutant, proton transfer from the D-pathway to the binuclear site may be blocked in this state.
Figure 2.

Structure of the core of COX. Helices in subunit I are shown in light blue and gray, helices of subunit II in green, β-strands in red. The cofactors (CuA, two red dots close to the β-strands), heme a (yellow, left porphyrin), heme a3 (yellow, right porphyrin), and CuB (red dot in front of heme a3) are also shown, as well as the key residues (red) D124, E278, and K354. The D-pathway of proton transfer leads straight up via D124 and then to E278. Proton transfer continues via the heme D propionate of heme a3 to the heme a propionate area (A in Fig. 3) or toward the other heme a3 propionate and Asp-399 (B in Fig. 3). A possible exit pathway is indicated. A connection from E278 to the binuclear site appears to be open during the PR → F transition. Protons taken up via the K-pathway might expel protons from the propionate areas taken up first via the D-pathway already during formation of the R-state. The figure was prepared by using the program setor (24).
A fact, apparently not discussed before, is the nearly symmetric location of both E278 and D124 with respect to hemes a and a3. In the P. denitrificans enzyme (18), the closest carboxyl oxygen of E278 is 12.3 Å (1 Å = 0.1 nm) away from the heme a3-Fe-atom and 12.8 Å from the heme a-Fe-atom. E278 may therefore sense the difference between the redox states of heme a and the binuclear site by electrostatic interactions and adopt different conformational states according to the difference. The Cα-atom of D124 is 35.6 Å away from the heme a Fe-atom and 35.1 Å away from the heme a3-Fe-atom. Reduction of both hemes therefore would equally well pull protons into the D-pathway. The proposal that a hydrogen-bonded chain of bound water molecules connects E278 (E242 in the bovine enzyme) to the binuclear site (17) is unlikely to be correct for the O- and R-states: First, the empty space between E278 and the binuclear site is much more narrow in the bacterial enzymes than in the bovine one; and second, the very same space may be part of the oxygen diffusion channel (27). A chain of bound water molecules would interfere seriously with oxygen diffusion to the active site. Third, the recent x-ray structure at 2.35 Å resolution of the reduced bovine enzyme, for which the existence of the water chain was postulated, does not provide any evidence for bound water molecules in this area (23).
K354 in the so-called K-pathway could receive protons either from S291 directly or from the conserved subunit II-E78 indirectly (19). It would transfer the proton via T351, the hydroxy group of the side chain of heme a3 to Y280, which is located very close to the active site. Y280 appears to be covalently crosslinked to the CuB-ligand H276 (23, 18). The significance of this crosslink is unclear. Because this histidine and the tyrosine maintain separate π-electron systems (see refs. 18 and 23), only a small influence on the pK can be expected. However, the existence of this crosslink indicates that during the first turnovers after assembly of the enzyme a radical is formed, which causes the crosslink. Formation of similar crosslinks by radical reactions is typical for peroxidase catalyzed reactions (28). K354 appears to be essential for the reduction of heme a3 (29), most likely by transferring a proton to a CuB-bound OH− (see below). Because the O-state of the K354M mutant enzymes reacts with H2O2 and undergoes a complete turnover to the O-state, it has been argued that the K-pathway delivers the first two protons needed during reduction of the binuclear site, whereas the six protons involved in the residual cycle use the D-pathway (29, 30). This contrasts the original proposal (20) that the D-pathway is used for pumped protons and the K-pathway is needed for protons for water formation. However, the hypothesis that the K-pathway is involved in the uptake of the first two protons only was challenged again (31) because in the analogous mutant enzyme from R. sphaeroides the KM for H2O2 is very high and its turnover very slow. It is therefore likely that the K-pathway is involved in the second half of the cycle also, but that the D-pathway can substitute in the second half of the cycle for a non-functional K-pathway, but that the D-pathway cannot deliver (at least one of the) protons normally taken up during the first part of the cycle.
Most interestingly, the entrance to the D-pathway seems to be surrounded by a proton-collecting antenna (30, 32). This pathway is rapidly protonated as expected for a high flux proton port, whereas the protonation of the K-pathway is a long continuous process. This behavior can be used to generate a simple pump mechanism. In such a mechanism first a proton is rapidly taken up via the D-pathway, and reaches a place near the outside, and later another proton enters the K-pathway, reaching a thermodynamically favored protonation site near the other proton but closer to the inside. Then electrostatic interactions would drive the proton first taken up to the outside. Such a mechanism of proton pumping is likely to be realized during the two-electron reduction of COX (see below).
Fig. 2 shows that both hemes are positioned relatively close to the outer membrane surface. The distance of the heme a iron to the outer surface is only ≈20 Å and to the inner surface ≈35 Å. Such a localization is hard to combine with the claim that heme a is located in the middle of the dielectric barrier. It could be correct if the dielectric between heme a and the inner membrane surface would be high but low between heme a and the outer membrane surface. Rather the opposite is found; the propionate side chains of both hemes are oriented toward the outside and reach into an area that contains many polar groups and water molecules (18), which easily could change the direction of their dipoles. For the evaluation of the kinetic electrometric measurements mentioned below, the exact localization of heme a with respect to the low membrane dielectric is of critical relevance. Particularly striking (and potentially of functional importance) is the observation that there are no sites for stably accepting protons below heme a, apart from E278, which, however, appears to be protonated in the oxidized and reduced states (33, 23, 19).
Analysis of Published Data and Proposal of a New Proton Pump Mechanism.
The claim that electron transfer from CuA to heme a occurs across 50% of the dielectric barrier is based on the observation that in mitochondria “the shift of apparent midpoint potential of cytochrome a and of cytochromes (a + a3) corresponds to about half the value of the membrane potential” (34). It therefore was concluded that the “environment (of heme a) might correspond to a region in the M phase about mid-way between the surfaces of the outer and inner aquous phase”. However, later work (35) showed that reduction of cytochrome a is accompanied by protonation from the mitochondrial matrix, with a pH-dependence of the midpoint redox potential of 23 mV/pH unit (35), corresponding to an uptake of 0.4 protons. These data are in excellent agreement with results by Mitchell and Rich (8), who showed that 2.4 ± 0.1 protons are taken up on reduction of COX, two of which are associated with the reduction of the binuclear site and 0.4 protons with heme a reduction. For the carbon monoxide inhibited enzyme used in the experiments by Hinkle and Mitchell (34), a pH dependence of 9 mV per pH-unit, corresponding to the uptake of 0.15 protons, was found (36). The shift of the heme a midpoint potential on the employed membrane potential was 43% for valinomycin-induced potassium diffusion potentials and 50% for uncoupler induced proton diffusion potentials. With these values, a relative location of heme a in the membrane dielectric of 33% and 41% is calculated.
Attemps to try to identify those steps in the catalytic cycle that are coupled to proton transfer showed surprisingly that the ratio of the F and O states in mitochondria upon “reversal of electron flow” by addition of ATP became nearly independent from the external pH above pH 7.2 (37). If the F → O-transition was coupled to pumping of two protons, the equation for this reaction would be
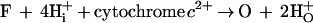 |
 |
and a strong dependence on external pH was expected. To explain this finding, the existence of a state, optically not distinguishable from O, called O′ (7), was postulated (37). O′ was placed between F and O, and proton pumping was postulated to occur between F and O′ (7). The reaction was thus split into two half reactions, one leading to O′ with electron transfer and proton pumping and the other from O′ to O with uptake of two protons for water formation.
For the investigation of the membrane potential dependence of the F:O ratio, only the first half reaction was considered. Such a procedure is inappropriate because the second half reaction also would come close to equilibrium. Because O′ and O are optically indistinguishable, the sum of both would be measured. The uptake of 2 H+ from the inside for water formation will contribute to the dependence of the F:O ratio on the electric field. The dependence of log [F]/[O] was found to be 26.7 mV per decade (7), corresponding to transport of 2.2 charges during the F → O transition. It was concluded that this value originates from one electron transported from cytochrome c to heme a across half of the dielectric barrier (0.5 charges) and 1.7 charges translocated by proton pumping. Assuming more correctly that an electron crosses 40% of the dielectric barrier, and considering proton uptake during the second half reaction, then 0.4 charges originate from electron transfer, 1.2 charges from proton uptake for water formation, and only 0.6 charges are left for proton pumping. In the mechanistic model presented below, only one proton is pumped, one proton is consumed, and one electron transferred, adding up to 2.0 charges per F → O transition. This model therefore agrees much better with the observed dependence of log [F]/[O] on the membrane potential. The situation may become more complicated, considering the possible existence of F′-states (38), also called CcO 580 (39), which have optical absorption properties like F-states, but possess one electron less than a true F-state. The dependence of P:F ratio and the F:O ratio on the ATP, ADP, and phosphate concentrations presented (7) cannot be quantitated because of the existence of multiple P- and F-states not known at the time when the experiment was performed. Nevertheless, the data might indicate that more ATP is synthesized upon the P → F than upon the F → O transition.
In a kinetic approach, Zaslavsky et al. (40) tried to determine the number of protons translocated upon injection of one electron into the F-state by using electrometric techniques. They calculated a number of 1.5 protons per F → O transition if the electron crossed 50% of the dielectric barrier and raised doubts about the localization of heme a in the middle of the dielectric barrier. They also discussed other reasons for their failure to observe a ratio of two. However, with a value of 41% for the fraction of the dielectric barrier between CuA to heme a, a value of 1.05, with 33% of 0.66 protons is calculated.
Taken together, the thermodynamic as well as the kinetic data suggest that the F → O transition is coupled to the pumping of one proton only. On the other hand, it is an undebated fact that 1 H+ is pumped per electron. Therefore, one proton must be pumped elsewhere. As outlined below, the structural data and the results of the analysis of mutants can be used to suggest a mechanism of proton pumping, which couples the pumping of one proton to the O → R transition.
The Mechanistic Model.
It is of crucial importance to start with the correct structure for the O-state. Unfortunately, x-ray structures at 2–3 Å resolution provide atomic models, but these are not based on electron densities of individual atoms. It is therefore necessary to use other information to derive atomic models especially around metals. The continuous electron density between the heme a3 iron and CuB in the P. denitrificans COX (18) can be interpreted by the presence of a water molecule bound to the iron atom and an OH− as a CuB-ligand. Evidence for a water or OH− as a CuB ligand has been obtained by EXAFS and ENDOR spectroscopy (41). A negatively charged ligand between both metals should exist to account for the strong antiferromagnetic coupling of the iron and the CuB. An OH− as an iron ligand can be excluded because heme a3 would become low spin. The positive charges at the irons and CuB2+ can be compensated in the following way: one positive charge of CuB2+ is neutralized by its OH− ligand, the residual charge of CuB2+ and the positive charges at the heme irons (formal charge +III, real charge +1) can be compensated by the heme propionates and D399 as indicated by the strong electrostatic coupling (19). The scheme in Fig. 3 therefore starts with an O-state consisting of the heme a3 iron, a water ligand, an OH− bound to CuB, and the covalently crosslinked Y280. In each state, the heme a iron appears on the left. A and B stand for two clusters of residues that could become protonated upon reduction of heme a (A) or the binuclear site (B). A and B are not individual residues; A should comprise at least the heme a propionates and B the heme a3 propionates and D399. Proton transfer between A and B should be possible through an extended hydrogen bonded network including several water molecules (18). A− and B− would receive the proton from the D-pathway residue E278 via the heme a3 ring D propionate (see above, Fig. 2). Direct proton transfer from BH to the binuclear site has to be excluded.
Figure 3.

Mechanistic model for proton pumping of COX by electrostatic repulsion. The left Fe symbol in each state [(O), (arO), (E), etc.] stands for the heme a-Fe-atom with +III or +II for its formal oxidation number. A− stands for a proton-accepting site near the heme a propionates. The Fe symbol more to the right stands for the heme a3 iron. A water molecule (H2O) may be bound to it in the O-state. An OH− bound to CuB is shown. B− stands for a common protonation site near the heme a3-CuB binuclear site. The reaction steps are given in brackets. The overall net charge at the heme a-Fe-site (without A) is indicated within the left circle; the net charge in the immediate vicinity of the binuclear site (including HY, without B) is shown within the right circle for each state. e− stands for electrons transferred from CuA, always to the heme a iron and always accompanied by uptake of one proton (HD+) via the D-pathway. Protons taken up via the K-pathway are shown as HK+, those via the D or the K-pathway as HD,K+. Proton pumping is indicated by a double arrow. Y• stands for the neutral radical of the H276-H280 crosslink, Y⊖ for the tyrosinate. (A) Regular catalytic cycle. (B) Alternative route by early, single protonation of the PM-state, leading via F′ to the true F-state. Then the cycle continues as in A. For further details, see text.
Step 1 is electron transfer to heme a, accompanied by proton uptake via the D-pathway, leading to protonation of A− and a still oxidized state of the binuclear site with heme a reduced (arO). Step 2 is nearly simultaneous transfer of the electron from heme a to CuB and of the proton from AH to B−, leading to the one electron reduced state E with the OH− still present as shown. Steps 1 and 2 together are fast. Although a protonation of OH− would be thermodynamically favored, the proton on B can partly stabilize OH−. The second electron transfer and proton uptake via the D-pathway (step 3) lead to formation of the protonated heme a reduced E-state (arE). The next step would be electron transfer from heme a to heme a3. This is problematic; the proton in the A position cannot follow because B is already occupied. In the A position the proton is too far away to sufficiently stabilize heme a3 reduction and OH−. Now a proton must be taken up via the K-pathway for protonation of OH−, to allow heme a3 reduction, in full agreement with the results obtained with the K354M mutants, in which this step is blocked. However, the additional positive charge taken up via the K-pathway must be compensated for by expulsion of a proton from AH or BH. This proton cannot go back, due to electrostatic repulsion by the incoming proton. This repulsion also would help to cross a possible barrier for proton transfer between the A, B-positions and the outer membrane surface.
As a result, step 4 constitutes the first proton pumping step. It leads to the reduced state R. In R the oxygen-binding site is no longer blocked; oxygen can be bound and “compound A” is formed. In step 6, internal proton and electron rearrangements occur leading to the PM-state. In agreement with recent results (12, 13), it is supposed to be a hydrogen-bonded oxoferryl state. The charge of CuB would be plus two; it is in a tetrahedral environment with the OH− being the H-bond donor to the oxoferryl oxygen atom. The missing electron and proton might be taken from HY, creating a neutral tyrosine radical, as proposed (18, 42, 43). The third electron uptake, accompanied by proton uptake and transfer (steps 7 and 8), leads to PR. In this step, protonation via the K-channel is not possible because Y280 does not possess a proton. This feature stabilizes the PM-state.
The electron most likely converts the neutral tyrosine radical into a tyrosinate. It must be noted that in the entire cycle PR is the only state where the immediate vicinity of the binuclear site is less positively charged (net charge zero) than the heme a iron (net charge +1). This inversion of the standard charge imbalance between the heme a3 iron and the binuclear site might be sensed by E278. It may undergo a conformational change. Such changes also could be induced by the presence of OH− and Y− and lead to the temporary establishment of a hydrogen bonded network including water molecules from E278 to the active site. Two protons might be pulled in from the D-pathway via E278 (step 9). This step is blocked in the E278Q mutants leading to the observed accumulation of the P-state. Uptake of the two protons would lead to electrostatic repulsion of the two protons at A and B and thus proton pumping. It is possible that in wild type only the first of the two protons is pulled in from the D-pathway. It would protonate the tyrosinate and reopen the K-pathway. The result of step 9 is the formation of the oxoferryl state F. Transfer of the fourth electron to heme a, accompanied by generation of AH (step 10), subsequent electron transfer from heme a to CuB or heme a and proton transfer from AH to B−, proton and electron rearrangements in the binuclear site (step 11) would lead to the formation of a hydroxy-intermediate (H), with OH− ions both at the iron and CuB atoms. Protonation of one of the OH− ions via the D- or K-pathway would lead to the expulsion of a proton from BH in step 12 (pumping of the fourth proton). It is possible that the O-state is formed directly, or via protonation of the CuB bound OH− first and subsequent proton transfer to the iron bound OH−. Now the cycle is completed.
Four protons were pumped; four protons were consumed in water formation. A fundamental aspect for this mechanism of proton pumping is that each reduction of heme a causes an uptake of one proton, which is stored in the A position between heme a and the outer surface, in a kind of electroneutrality principle already for heme a. Evidence for an electrogenic uptake of a proton during heme a reduction already had been obtained earlier (35). The number of protons taken up per reduction was, however, below 0.5. The reason for the discrepancy is that under these conditions the uptake of a proton during the third electron transfer is considered, with the OH− at CuB already neutralized and a proton at the B position. Both may partially stabilize reduction of heme a, so that only uptake of a fractional proton is observed. Theoretical calculations (A. Kannt, C. R. D. Lancaster, and H. Michel, unpublished data) similar to those of ref. 19 provide clear evidence that the first heme a reduction is accompanied by uptake of close to one proton.
Why does proton uptake upon heme a reduction not occur from the outside? The proton transfer from the inside may be faster, caused by optimization of hydrogen bonded networks during evolution. A proton in the A or B position would then electrostatically repel potentially incoming protons thereby increasing the energy barrier. However, when proton transfer from the inside is slowed down, uptake of protons from the outside may occur. These protons might reach the binuclear site via E278. This backflow would explain why D124N mutants still possess a considerable residual turnover in contrast to E278Q mutants. An electrochemical proton gradient would stimulate the backflow via the A-site to E278, and the turnover. Exactly such a behavior has been described for the R. sphaeroides mutant COX (31) corresponding to D124N.
An alternative route from the PM-state to the O-state.
If the connection between E278 and the active site would temporarily function already during the PM-state, a proton could convert the CuB-OH− to water in step 7A of Fig. 3B. This would be a proton-pumping step, and a state F′ would be established. Considering the possibility that the absorption maxima of P-states at 607 nm are caused by the strong hydrogen bond between the oxoferryl oxygen atom and the CuB-OH− ligand, and a shift to 580 nm in its absence, this state would be an F-state termed F′ (38) with one electron less in the binuclear site than a true F-state. The sequence of reactions presented in Fig. 3B would lead to a P′R-state. Such a state also could be an intermediate between PR and F in Fig. 3A, step 9. Then the route leads to the F-state with pumping of one proton and the cycle might be continued with step 10 of Fig. 3A. The conversion of F′ to O would therefore lead to pumping of two protons. However, since formation of F′ from PM is connected to pumping of only one proton, the overall yield from PM to O would be again three protons. The way via F′ should be favored upon delay of the third electron at low pH.
It must be noted that the F′-state can be directly formed upon reaction of H2O2 with the O-state under formation of one water molecule. The P-like state observed upon addition of substoichiometric amounts of H2O2 at high pH is then a state with an OH− instead of water as CuB-ligand. The only difference to the PM state (Fig. 3A) would be the absence of a proton in the B-site.
Acknowledgments
I thank E. Bamberg, J. Behr, A. Harrenga, A. Kannt, and C. R. D. Lancaster for reading the manuscript and discussion and D. Vinzenz, A. Harrenga, and A. Kannt for preparing the figures. Financial support was provided by the Deutsche Forschungsgemeinschaft (SFB472), the Fonds der Chemischen Industrie, and the Max-Planck-Gesellschaft.
ABBREVIATION
- COX
cytochrome c oxidase
Note Added in Proof
(i) It is well possible that in wild-type COX a proton is taken up via the K-pathway already immediately after the first electron transfer to the binuclear site, leading to proton pumping before the second electron transfer. (ii) In the alternative route, via F′ in Fig. 3B, the second proton is pumped immediately after the second electron transfer to the binuclear site. Combining i and ii leads to a variant of the basic proton pump mechanism by electrostatic repulsion in which one proton is pumped after each reduction of the binuclear site. This is in sharp contrast to the postulates of ref. 7 but well compatible with Fig. 2 of ref. 7 when one takes into account that only one proton is pumped during the F→O transition (see above). The alternative pathway is attractive because it avoids having the negative charges of the OH− and the tyrosinate close together in the PR-state.
Footnotes
A Commentary on this article begins on page 12747.
To whom reprint requests should be addressed. e-mail: michel@mpibp-frankfurt.mpg.de.
References
- 1. Wikström M. Nature (London) 1977;266:271–273. doi: 10.1038/266271a0. [DOI] [PubMed] [Google Scholar]
- 2.Ferguson-Miller, Babcock G T. Chem Rev (Washington, D C) 1996;7:2889–2907. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 3.Michel H, Behr J, Harrenga A, Kannt A. Annu Rev Biophys Biomol Struct. 1998;27:329–356. doi: 10.1146/annurev.biophys.27.1.329. [DOI] [PubMed] [Google Scholar]
- 4.Babcock, Wikström Nature (London) 1992;356:301–309. doi: 10.1038/356301a0. [DOI] [PubMed] [Google Scholar]
- 5.Chance C, Saronio C, Leigh J S. J Biol Chem. 1975;250:9226–9237. [PubMed] [Google Scholar]
- 6.Varotsis C, Babcock G T. Biochemistry. 1990;29:7357–7362. doi: 10.1021/bi00484a001. [DOI] [PubMed] [Google Scholar]
- 7.Wikström M. Nature (London) 1989;338:776–778. doi: 10.1038/338776a0. [DOI] [PubMed] [Google Scholar]
- 8.Mitchell R, Rich P R. Biochim Biophys Acta. 1994;1186:19–26. doi: 10.1016/0005-2728(94)90130-9. [DOI] [PubMed] [Google Scholar]
- 9.Rich P R. Aust J Plant Physiol. 1995;22:479–486. [Google Scholar]
- 10.Wikström M, Bogachev A, Finel M, Morgan J E, Puustinen A, Raitio M, Verkhovskaya M, Verkhovsky M I. Biochim Biophys Acta. 1994;1187:106–111. doi: 10.1016/0005-2728(94)90093-0. [DOI] [PubMed] [Google Scholar]
- 11.Morgan J E, Verkhovsky M I R, Wikström M. J Bioenerg Biomembr. 1994;26:599–608. doi: 10.1007/BF00831534. [DOI] [PubMed] [Google Scholar]
- 12.Proshlyakov D A, Ogura T, Shinzawa-Itoh K, Yoshikawa S, Kitagawa T. Biochemistry. 1996;35:8580–8586. doi: 10.1021/bi952096t. [DOI] [PubMed] [Google Scholar]
- 13.Kitagawa T, Ogura T. In: Progress in Inorganic Chemistry. Karlin K D, editor. Vol. 45. New York: Wiley; 1997. pp. 431–479. [Google Scholar]
- 14.Hellwig P, Behr J, Ostermeier C, Richter O-M H, Pfitzner U, Odenwald A, Ludwig B, Michel H, Mäntele W. Biochemistry. 1998;37:7390–7399. doi: 10.1021/bi9725576. [DOI] [PubMed] [Google Scholar]
- 15.Behr J, Hellwig P, Mäntele W, Michel H. Biochemistry. 1998;37:7400–7406. doi: 10.1021/bi9731697. [DOI] [PubMed] [Google Scholar]
- 16.Puustinen A, Bailey J A, Dyer R B, Mecklenburg S L, Wikström M, Woodruff W H. Biochemistry. 1997;36:13195–13200. doi: 10.1021/bi971091o. [DOI] [PubMed] [Google Scholar]
- 17.Riistama S, Hummer G, Puustinen A, Dyer R B, Woodruff W H, Wikström M. FEBS Lett. 1997;414:275–280. doi: 10.1016/s0014-5793(97)01003-x. [DOI] [PubMed] [Google Scholar]
- 18.Ostermeier C, Harrenga A, Ermler U, Michel H. Proc Natl Acad Sci USA. 1997;94:10547–10553. doi: 10.1073/pnas.94.20.10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kannt A, Lancaster C R D, Michel H. Biophys J. 1998;74:708–721. doi: 10.1016/S0006-3495(98)73996-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iwata S, Ostermeier C, Ludwig B, Michel H. Nature (London) 1995;376:660–669. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 21.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. Science. 1995;269:1069–1074. doi: 10.1126/science.7652554. [DOI] [PubMed] [Google Scholar]
- 22.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. Science. 1996;272:1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- 23.Yoshikawa S, Shinzawa-Itoh K, Nakashima R, Yaono R, Yamashita E, Inoue N, Yao M, Fei M J, Libei C P, Mizushima T, et al. Science. 1998;280:1723–1729. doi: 10.1126/science.280.5370.1723. [DOI] [PubMed] [Google Scholar]
- 24.Merrit E A, Murphy M E P. Acta Crystallogr D. 1994;50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]
- 25.Thomas J W, Puustinen A, Alben J O, Gennis R B, Wikström M. Biochemistry. 1993;32:10923–10928. doi: 10.1021/bi00091a048. [DOI] [PubMed] [Google Scholar]
- 26.Verkhovskaya M L, Garcia-Horsman A, Puustinen A, Rigaud J-L, Morgan J E, Verkhovsky M I, Wikström Proc Natl Acad Sci USA. 1997;94:10128–10131. doi: 10.1073/pnas.94.19.10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Riistama S, Puustinen A, Garcia-Horsman A, Iwata S, Michel H, Wikström M. Biochim Biophys Acta. 1996;1275:1–4. doi: 10.1016/0005-2728(96)00040-0. [DOI] [PubMed] [Google Scholar]
- 28.Michon T, Chenu M, Kellershon N, Desmadril M, Gueguen J. Biochemistry. 1997;36:8504–8513. doi: 10.1021/bi963168z. [DOI] [PubMed] [Google Scholar]
- 29.Vygodina T V, Pecoraro C, Mitchell D, Gennis R, Konstantinov A A. Biochemistry. 1998;37:3053–3061. doi: 10.1021/bi971876u. [DOI] [PubMed] [Google Scholar]
- 30.Karpefors M, Ädelroth P, Aagaard A, Sigurdson H, Ek M S, Brzezinski P. Biochim Biophys Acta. 1998;1365:159–169. doi: 10.1016/s0005-2728(98)00058-9. [DOI] [PubMed] [Google Scholar]
- 31.Mills D A, Ferguson-Miller S. Biochim Biophys Acta. 1998;1365:46–52. doi: 10.1016/s0005-2728(98)00040-1. [DOI] [PubMed] [Google Scholar]
- 32.Sacks V, Marantz Y, Aagaard A, Checover S, Nachliel E, Gutman M. Biochim Biophys Acta. 1998;1365:232–240. [Google Scholar]
- 33.Lübben M, Gerwert K. FEBS Lett. 1996;397:303–307. doi: 10.1016/s0014-5793(96)01174-x. [DOI] [PubMed] [Google Scholar]
- 34.Hinkle P, Mitchell P. Bioenergetics. 1970;1:45–60. doi: 10.1007/BF01516088. [DOI] [PubMed] [Google Scholar]
- 35.Artzatbanov V Y, Konstantinov A A, Skulachev V P. FEBS Lett. 1978;87:180–185. doi: 10.1016/0014-5793(78)80327-5. [DOI] [PubMed] [Google Scholar]
- 36.Ellis W R, Wang H, Blair D F, Gray H B, Chan S I. Biochemistry. 1986;25:161–167. doi: 10.1021/bi00349a023. [DOI] [PubMed] [Google Scholar]
- 37.Wikström M. Chemica Scripta. 1988;28A:71–74. [Google Scholar]
- 38.Moody A J, Rich P R. Eur J Biochem. 1994;226:731–737. doi: 10.1111/j.1432-1033.1994.tb20102.x. [DOI] [PubMed] [Google Scholar]
- 39.Fabian M, Palmer G. Biochemistry. 1995;34:13802–13810. doi: 10.1021/bi00042a011. [DOI] [PubMed] [Google Scholar]
- 40.Zaslavsky D, Kaulen A D, Smirnova I A, Vydogina T, Konstantinov A A. FEBS Lett. 1993;336:389–393. doi: 10.1016/0014-5793(93)80843-j. [DOI] [PubMed] [Google Scholar]
- 41.Fann Y C, Ahmed I, Blackburn N J, Boswell J S, Verkhovskaya M L, Hoffman B M, Wikström M. Biochemistry. 1995;34:10245–10255. doi: 10.1021/bi00032a019. [DOI] [PubMed] [Google Scholar]
- 42.Gennis R B. Biochim Biophys Acta. 1998;1365:241–248. doi: 10.1016/s0005-2728(98)00095-4. [DOI] [PubMed] [Google Scholar]
- 43.Hoganson C W, Pressler M A, Proshlyakov D A, Babcock G T. Biochim Biophys Acta. 1998;1365:170–174. doi: 10.1016/s0005-2728(98)00057-7. [DOI] [PubMed] [Google Scholar]