

Abstract
We solved the crystal structure of Streptococcus agalactiae serine/threonine phosphatase (SaSTP) using a combination of single-wavelength anomalous dispersion phasing and molecular replacement. The overall structure resembles that of previously characterized members of the PPM/PP2C STP family. The asymmetric unit contains four monomers and we observed two novel conformations for the flap domain among them. In one of these conformations, the enzyme binds three metal ions, whereas in the other it binds only two. The three-metal ion structure also has the active site arginine in a novel conformation. The switch between the two- and three-metal ion structures appears to be binding of another monomer to the active site of STP, which promotes binding of the third metal ion. This interaction may mimic the binding of a product complex, especially since the motif binding to the active site contains a serine residue aligning remarkably well with the phosphate found in the human STP structure.
Keywords: dephosphorylation, serine/threonine phosphatase, signaling, Streptococcus agalactiae, structure
Protein phosphatases are primarily classified on the basis of the type of the amino acid they dephosphorylate, serine/threonine phosphatases (STPs) act specifically on phosphoserine and phosphothreonine residues. Evolution has developed two main families of metalloenzymes for this purpose, phosphoprotein phosphatase P (PPP) and phosphoprotein phosphatase M (PPM) [1]. Based on sequence similarity, Streptococcus agalactiae STP (SaSTP) studied here belongs to a PPM subfamily called PP2C, because members of this subfamily resemble human phosphoprotein phosphatase 2C [1].
Serine/threonine phosphorylation/dephosphorylation is intimately linked with signaling events inside the cell. Many STPs expand the scope of signaling by recruiting additional domains into their structures. This is most common in PPP family enzymes, where both regulatory and targeting domains occur; for example, in phosphoprotein phosphatase 5, the STP domain is fused to four tetracotripeptide repeat protein –protein interaction modules [2]. PPM/PP2C enzymes may also have additional domains, for example, Arabidopsis thaliana ABI1 in which the catalytic domain is fused to an EF-hand motif, and human STP (HsSTP), which has an additional 8 kDa α-helical domain at the C-terminus [3,4].
The sizes of the catalytic domains of both PPP and PPM families are well conserved and structural studies have revealed significant similarities between them [4–9]. Both utilize two β sheets to help the enzymes orient their active site residues in a conformation where they bind active-site metal ions. The active-site ligands are, however, different; in PPP enzymes, histidine, aspartate and asparagine side chains bind the metal ions, whereas in PPM enzymes, aspartates and a glycine backbone carbonyl coordinate the metal ions [4]. The identity of metal ions within the groups varies and studies have sometimes shown slightly controversial results [1]. The PPM/PP2C studied to date have been shown to contain either Mg2+ or Mn2+ [1,4]. In addition, in the crystal structure of Toxoplasma gondii STP (TxSTP; PDB code 2I44), the metals are modeled as Ca2+ ions (unpublished).
Detailed biochemical analysis has revealed differences between these enzymes. Only PPPs are inhibited by the classical STP inhibitor okadaic acid [1]. Although similar, the mechanisms of these enzymes are not identical, because PPM and PPP class enzymes bind their substrates differently. In the PPP family, the substrate phosphoryl group is bound directly to the two metal ions via its oxygen residues, whereas PPM/PP2C family enzymes bind the substrate indirectly, via hydrogen-bonding interactions between the phosphoryl group and water molecules liganded to the metal ions [4,10,11].
The biochemistry of PPM/PP2C has been studied extensively using the human enzyme as a model [4,10,11]. It relies on two divalent metal ions and an activated bridging water molecule with a pKa of 7.5 [11] to achieve catalysis, a common feature in hydrolytic metalloenzymes [12]. One residue that appears to take part in catalysis in HsSTP is His62, which may act as a general acid and protonate the phosphate as it leaves [11], but this residue is missing from the prokaryotic Mycobacterium tuberculosis STP (MtSTP) [9]; and from SaSTP. This implies that, in these enzymes, some other residue or a water molecule would act as the general acid. HsSTP Arg33, conserved among STPs, has been proposed to take part in binding the phosphorylated protein substrate. The function of other conserved residues near the active site remains unclear [11]. Interestingly, MtSTP has been shown to bind a third metal ion near the active site [9]. A serine residue that takes part in binding the third metal ion is not conserved and the function of the third metal ion in MtSTP is unknown.
Recently, serine/threonine phosphorylation/dephosphorylation has been shown to occur in many prokaryotes, where it modulates cellular activities analogously to events found in eukaryotes. In Bacillus subtilis a PPM/PP2C STP activates sporulation transcription factor [13,14]. M. tuberculosis contains many serine/threonine kinases (STKs), including serine/threonine protein kinase G, which mediates survival of the bacteria [15]. Yersinia pseudotuberculosis and Yersinia enterocolitica YpkA STKs induce the secretion of many Yop virulence effector proteins [16,17], the Streptococcus pneumoniae stkP− strain has reduced infectivity in mice [18], and the Pseudomonas aeruginosa ppkA STK is needed for virulence in mice [19]. S. agalactiae has an active STP/STK system, which affects both the virulence and morphology of the bacteria [20–22].
The above-mentioned studies have thus shown that serine/threonine signaling cascades are linked to the virulence of organisms, leading to interesting possibilities for rational drug design against these pathogens. Drugs targeting S. agalactiae signaling enzymes may cure many severe diseases, such as sepsis and meningitis, which threaten the lives of newborn babies and immunocompromised adults. To support drug design, we need detailed structural information about the signaling proteins (STKs and STPs), and their complexes with their downstream targets, which in S. agalactiae include the response regulator CovR, adenylosuccinate synthase and a family II inorganic pyrophosphatase (SaPPase) [20–22]. We have previously crystallized one of the substrate molecules (SaPPase) [23]. Here we report the crystal structure of SaSTP. The structure revealed a third metal ion, as in MtSTP. However, unlike MtSTP, its presence correlates with binding of another STP monomer over the active site. This interaction may resemble the dephosphorylated product complex.
Results
Overall structure
The structure of SaSTP was solved at 2.65 Å resolution. The model consists of residues (1–242) in all four monomers in the asymmetric unit. Additional residues at the N-terminus (residues −4 to 0), introduced during the cloning step [23], were partly visible and the last residues of the polypeptide (residues 243–245) were not visible in electron-density maps. SaSTP has an αββα sandwich structure, consisting of two antiparallel β sheets packed against each other. The β sheets (β1 and β2) are surrounded by α helices (Fig. 1A,B). Similar to other PPM/PP2C STPs, both β sheets consist of antiparallel strands: these are formed by residues 1–8, 120–125, 128–135, 186–190 and 233–240 in β1, and 18–24, 30–38, 98–107, 110–116 and 175–180 in β2. β2 is flanked by two long antiparallel α helices (43–61 and 67–91). β1 is flanked by an α-helical region comprised of three separate helices (192–195, 200–207 and 213–226). The otherwise compact structure is interrupted by a flap subdomain (132–174) (Fig. 1B). This flap has two short α helices (138–144 and 150–154). The four monomers found in the asymmetric unit are similar but not identical to each other, and the largest deviations between them occur in this area (see below). Two conformations of the flap occur in the crystal; monomers A and B share a similar conformation, as do monomers C and D. Notably, these two groups also differ in metal content (see below).
Fig. 1.
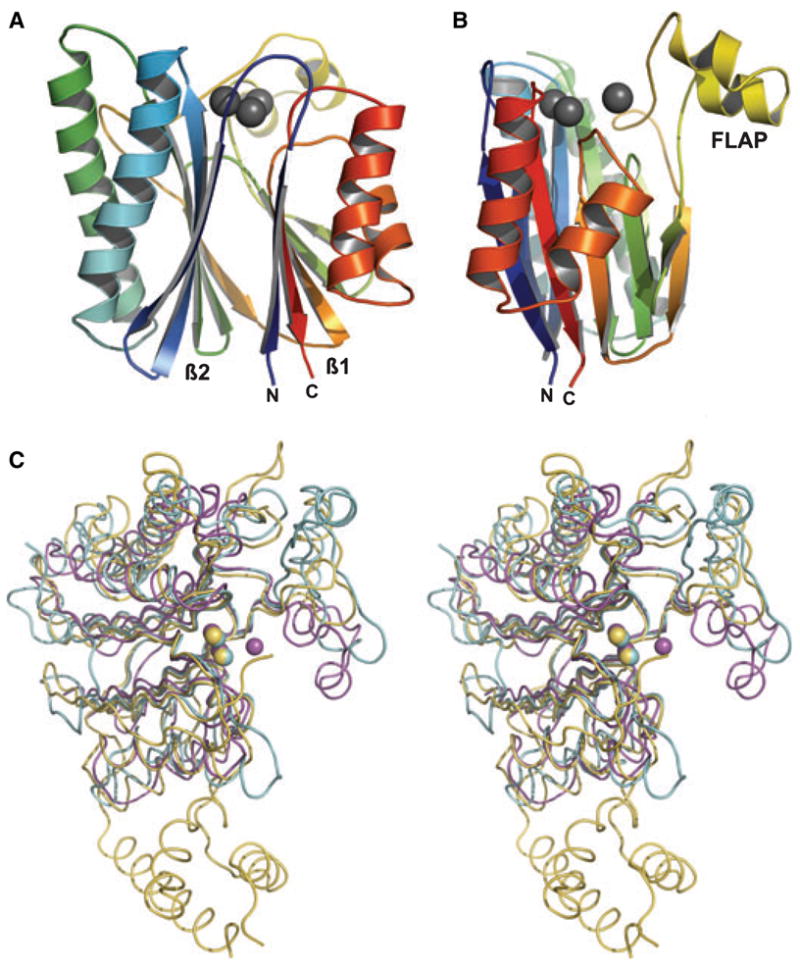
(A) Structure of the SaSTP (monomer C). The protein was drawn using a secondary structure representation in which the protein is colored from the N-terminus to the C-terminus using a spectrum of colors from blue to red. The three active site metal ions (M1, M2 and M3) are shown as gray spheres. The N- and C-termini, and the two β sheets are labeled. (B) The structure has been rotated through 90 ° around the y-axis. The flap subdomain is at right angles to the core structure. (C) Comparison of SaSTP (magenta), TxSTP (cyan) and HsSTP (yellow). Metal ions are shown as spheres and colored according to the corresponding protein. In SaSTP, the three metals are magnesium, the two metals in HsSTP are manganese and in TxSTP the two metals are calcium ions. This and the other figures were generated using PYMOL [39] and GIMP (http://www.gimp.org).
The rmsd/Cα values between different SaSTP monomers and known structures (PDB code 2I44) [4,9] show that the SaSTP is a member of the PPM/PP2C STP family (Table 1). The core structure is well preserved in these enzymes. The rmsd values range between 1.16 and 1.49 Å for the core β sheets. For the whole structure, the rmsd values are slightly higher, ranging from 1.78 to 2.31 Å. The evolutionarily less related TxSTP and HsSTP have remarkably different and larger flap domains than SaSTP and MtSTP, which is why the number of aligned residues is lower (Fig. 1C). In addition, both HsSTP and TxSTP have an extra strand in the β sheets, so that the arrangement is 5 plus 6 [4] (PDB code 2I44), and HsSTP also has a 75-residue C-terminal domain. Despite the low sequence identity (33%) between SaSTP and MtSTP, they are structurally closely related, both having a core of two five-stranded sheets put together in an identical manner.
Table 1.
rmsd values between different STP structures. Monomers A and D of SaSTP as well as the core β sheet of monomer A are compared with A monomers of HsSTP (PDB code 1A6Q), MtSTP (PDB code 1TXO), and TxSTP (PDB code 2I44). The rmsd calculations were carried out using SSM alignment program [38]. Values in parentheses show the number of aligned residues.
| MtSTP (Å) | HsSTP (Å) | TxSTP (Å) | |
|---|---|---|---|
| SaSTP (A) | 1.82 (223) | 2.31 (212) | 2.05 (208) |
| SaSTP (D) | 1.78 (223) | 2.24 (209) | 2.19 (207) |
| SaSTP (A) β sheet only | 1.21 (70) | 1.49 (70) | 1.16 (62) |
Active site
Overall, the active-site organization of SaSTP is very similar to that of the previously characterized PPM/PP2Cs. This was expected because the residues forming the active site are very well conserved and can be easily identified by sequence alignment. There are four monomers in the asymmetric unit in the SaSTP structure, and we found two catalytic metal ions (M1 and M2) in all the active sites (Fig. 2A). Based on the coordination and lack of an anomalous signal, we modeled the metal ions as Mg2+ ions (see Experimental procedures). Despite modest resolution of the structure, we observed all the coordinating water molecules of the M1 and M2 metal ions, including the water molecule bridging them. This water molecule/hydroxide ion is proposed to perform nucleophilic attack on the phosphorus atom of the substrate [4,9]. Similarly to MtSTP [9], there is no likely candidate residue for a general acid in the active site. This suggests that a water molecule may fill this role. Arg13, which has been proposed to participate in binding of the substrate, is in a similar position to the equivalent residue in HsSTP (Arg33), where it binds Pi found in the active site [4]. There are, however, important differences in the SaSTP monomers in this area (see below).
Fig. 2.
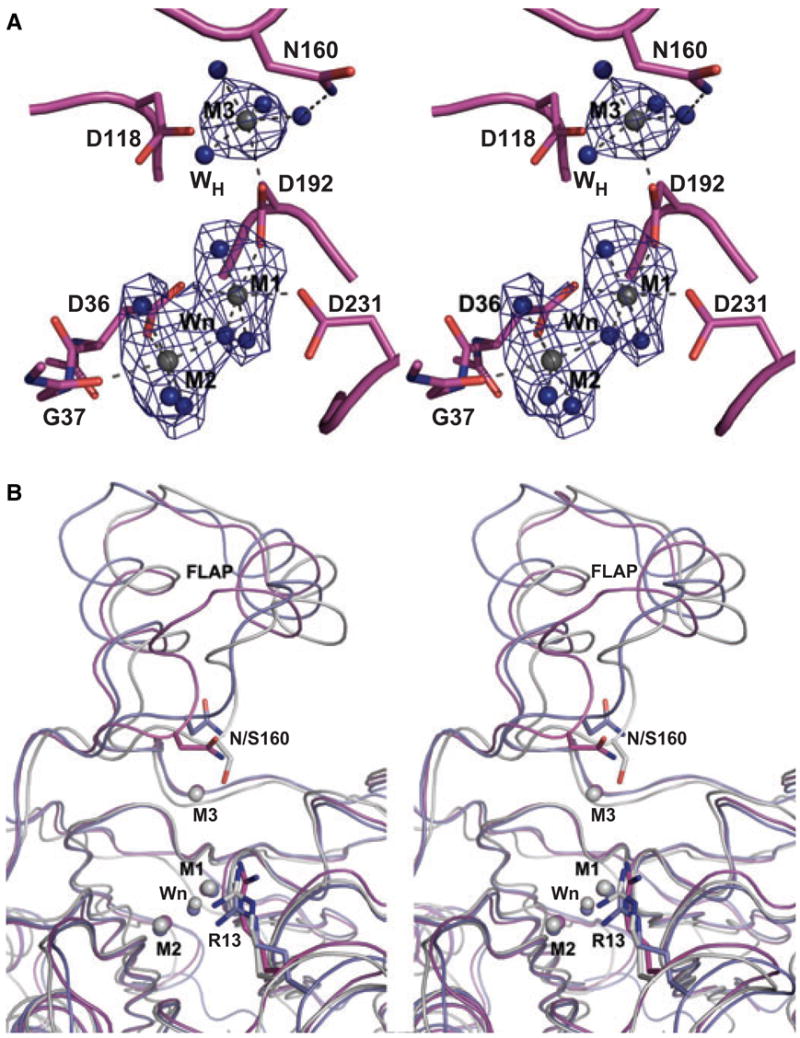
(A) Stereoimage of the active site of SaSTP monomer C. Residues are shown as combination of cartoon and stick models. Mg2+ ions are shown as gray spheres and water molecules as blue spheres. The density around the metal binding site is shown in blue mesh. The (Fo−Fc) omit map, contoured at 3σ was calculated after removing all the solvent atoms from the model. Before map calculation, the stripped model was refined for 20 cycles with REFMAC5. Coordinations for the metal ions and for Asn160 are indicated by dashed lines. (B) Superimposition of monomers A and C of SaSTP, and MtSTP monomer A [9]. Monomer C of SaSTP is magenta as in Fig. 2A, monomer A is blue and MtSTP is gray. Metal ions and nucleophilic water molecules are shown as spheres and colored according to the protein to which they are bound. The ‘additional’ metal, M3, is present in SaSTP monomer C and in MtSTP. In SaSTP all the metals are Mg2+, whereas in MtSTP they are Mn2+ ions. Arg13 and Asn160 (Ser in MtSTP) are shown as sticks.
In addition to the M1 and M2 metal ions, we observed an additional metal ion (M3) in two monomers of the asymmetric unit (monomers C and D; Fig. 2A). M3 was also modeled as a Mg2+ ion, based on the same principles as for M1 and M2 (see Experimental procedures). M3 is coordinated by Asp118, Asp192 and four water molecules (Fig. 2A). Asp192 thus bridges metals M1 and M3. The position of M3 between the flap subdomain and the active site is analogous to the third metal, Mn2+, found in MtSTP [9]. In order to bind the metal ion, the flap domain moves away from the core and the main chain conformation, especially that of the first helix, changes (Fig. 2B). In MtSTP, in addition to Asp118 and Asp191 (192 in SaSTP), M3 is coordinated by both the hydroxyl and the carbonyl groups of Ser160. The equivalent residue in SaSTP is Asn160, and it does not participate directly in metal coordination (Fig. 2A). Because most of the M3 ligands in SaSTP are water molecules, M3 in SaSTP may be less tightly bound than the M3 in MtSTP.
Crystal contacts at the active site
The differential occurrence of the metal ions correlates with the conformation of the flap subdomain (132–174) and the ‘binding’ of adjacent STP monomers over the active sites of monomers C and D, but not of A and B (Fig. 3A). For monomer C, which contains three metal ions, the monomer ‘binding’ over the active site is monomer A. Similarly, in monomer D the ‘binding’ molecule is monomer B. This crystal packing induces changes in the flap and near Arg13 (Fig. 3B), and correlates with binding of M3. Residues 147–157 of the flap, including a second short helix (150–156) in monomer A, bind to the active site of monomer C. The interactions in this contact include two salt bridges, Arg12(C)–Glu151(A) and His41(C)–Glu152(A), and three direct hydrogen bonds Ser14(C)–Glu152(A), Arg13(C)–Ser155(A), and Ile162(C)–Pro157(A) (Fig. 3B). These interactions may give clues as to how SaSTP interacts with its substrate (see below). Another crystal contact is formed by the flap subdomain of monomer C, but this contact region only contains a single hydrogen bond: Gln147(C) hydrogen bonds to the carbonyl group of His226(A). In monomers A and B, where His41 does not participate in a salt bridge, the area around His41 is poorly defined by the electron density.
Fig. 3.
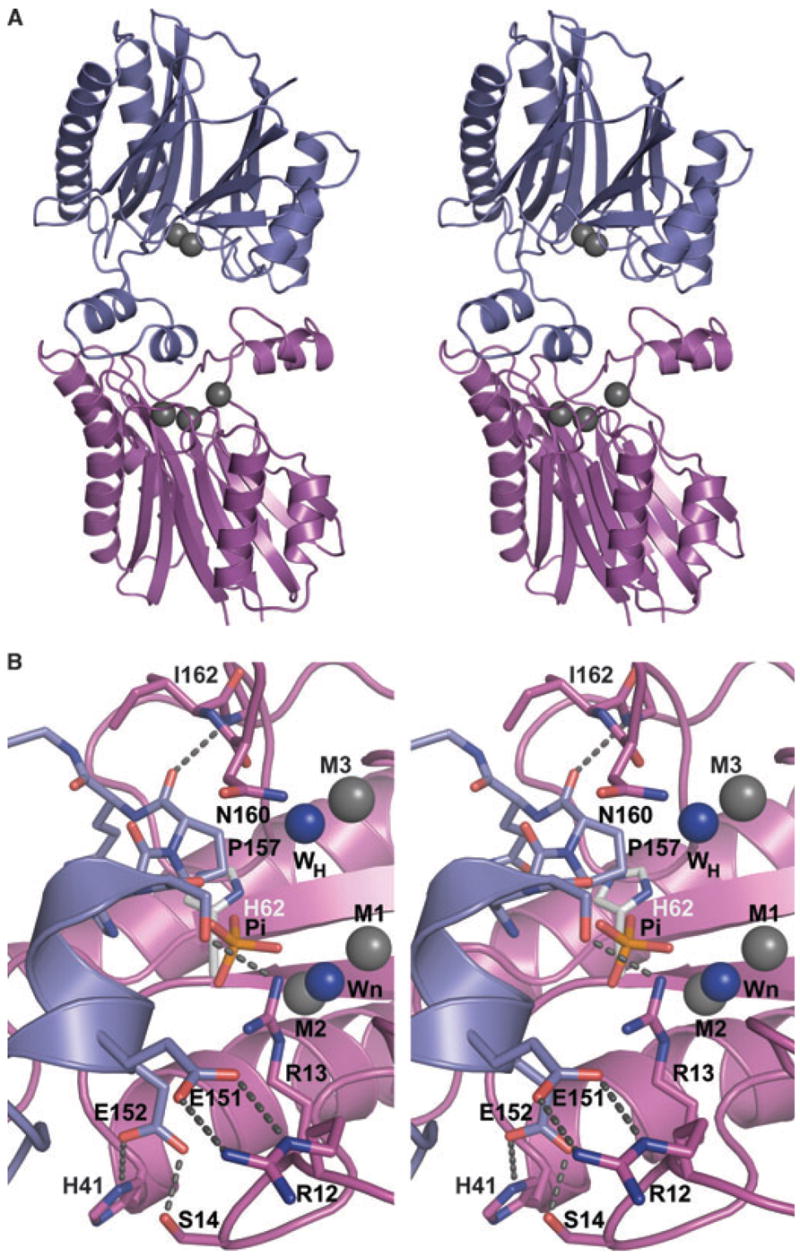
(A) Stereoimage showing the packing interactions between selected monomers (A and C) that mimics substrate binding. Monomer C (magenta) binds a peptide from the loop of monomer A (blue) to the active site. Metal ions are shown as gray spheres. Notably, monomer A binds two metals, whereas monomer C binds three. (B) Closeup view of crystal contact indicating the potential position of phosphate based on HsSTP. Coloring is as in (A) except that the nucleophilic (Wn) and putative general acid (WH) water molecules are shown as blue spheres, and residues participating in the interactions are shown as sticks. The direct interactions across the interface are indicated by dashed lines. The phosphate found in HsSTP was modeled into SaSTP by aligning the HsSTP and SaSTP monomers active sites. In the alignment metal ions M1 and M2, Wn, and carboxylate groups of Asp36, Asp192 and Asp231 were used. Alignment of 12 atoms resulted in an rmsd value of 0.3 Å. As shown, the modeled position of the phosphate from HsSTP is close to Ser155(A) and Arg13(C). Also, the general acid in HsSTP, His62, is shown as sticks with gray carbon atoms.
The Arg13 side chain shows two different conformations in the active site. In the monomer A and B structures, its conformation is similar to HsSTP, where it binds phosphate (Fig. 2B) [4]. In monomers C and D, Arg13 adopts a new conformation; the side chain is rotated so that the guanidine group points away from the active site. Interestingly, Arg13 now binds a serine residue (Ser155) in the monomer bound to the active site (Fig. 3B). This feature might be related to the mechanism of the enzyme (see Discussion).
Discussion
Overall structure
The structure of SaSTP is very similar to that of MtSTP, with an rmsd/Cα of 1.8 Å overall (Table 1). All the secondary structural elements are conserved between these enzymes, but there is variance, particularly in the conformation of the flap subdomain (see below). Interestingly, other PP2C-family STPs solved to date show variable numbers of strands in their core structure. HsSTP contains an additional β strand at the N-terminus extending the β1 sheet by one strand (Fig. 1). TxSTP also contains a similar, albeit much shorter, extra strand. With respect to SaSTP and MtSTP, there are other clear differences in the TxSTP structure at the first two N-terminal helices, of which there are three in TxSTP. The helices are also longer. The major difference within the PPM/PP2C STP structural family resides in the flap subdomain (Fig. 1C). The flap in TxSTP contains the antiparallel strands in addition to the two helices found in SaSTP and MtSTP. Furthermore, the flap is in a totally different conformation. In HsSTP, the flap consists of practically a single helix, because the second helical element involves only couple of residues.
The MtSTP crystal structure revealed for the first time a third metal ion in the active site of a PPM/PP2C STP and a flap domain conformation very different to that in HsSTP [9]. This raises the question of whether the difference in conformation of the flap domain is related to binding of the third metal ion or the substrate. The ligands for Mn3 in MtSTP were Asp118, Asp191 and Ser160, the last coming from the flap domain [9]. There are homologous residues to Asp118 and Asp191 in HsSTP (Asp146 and Asp239) and, of course, in SaSTP (Asp118 and Asp192). Our SaSTP crystal structure provides clear evidence that the flap subdomain is a mobile element. Furthermore, its conformation is linked with the binding of the third metal ion suggesting, like Pullen et al. [9], that rearrangement of the flap domain in HsSTP might lead to binding of a third metal ion. The implications of the M3 binding and flap subdomain conformations to the catalytic mechanism are discussed below.
The role of the third metal in catalysis
In MtSTP [9], Ser160 takes part in binding M3 metal by forming two of the coordinating interactions. In SaSTP, this residue has been replaced by Asn160 (Fig. 2B). This residue is in two different conformations, depending on the overall conformation of the flap subdomain. The conformations are correlated with the presence or absence of M3, but Asn160 does not bind directly to M3. Asn160 is, however, the closest flap domain residue to the M3 binding site. Because the M3 binding site is linked to the conformation of the flap, M3 might also exist in eukaryotic enzymes such as HsSTP. However, it was not observed in that structure [4], and an even larger movement of the flap domain would be required than the one observed in SaSTP. It would also require that the Asp146–Lys165 ion pair located in a position similar to the M3 site in SaSTP would be broken.
However, the most important reason to doubt the presence of an M3 site in eukaryotic STPs concerns the presence of a general acid. In HsSTP, His62 has been shown to be the general acid protonating the leaving group [11], but this residue is missing from prokaryotic STPs. However, our structure and a re-evaluation of the data presented by Pullen et al. [9] suggest that a water molecule may play this role in prokaryotic STPs. First, WH (Fig. 2A) is −4 Å away from the phosphate modeled by transferring the coordinates from the HsSTP structure. Because WH is coordinated to the M3 metal, its pKa value should be significantly less than 14, and so should be capable of protonating the phosphate leaving group. This is also consistent with the binding of M3 in SaSTP when a putative substrate-mimic loop binds into the active site (see below). Second, when the residues binding the metal ion in MtSTP are mutated, the Km value for metal binding was little changed. These mutations included the S160A mutation, making the MtSTP binding site similar to SaSTP. As our structure shows, this sequence does indeed bind metal ion at M3 – and the largest change they report in all their mutants at this site is less than a factor of four greater than wildtype (3.7–14.6 mM) [9]. Consequently, their measurements of MtSTP activity in the presence of 100 mM Mn2+ actually reflect enzyme activity with three metal ions present, and so the fact that mutations at the M3 site do not change kcat is unsurprising. There is probably no change in active site contents.
The WH in prokaryotic enzymes is located on the opposite side of the substrate phosphate in comparison with His62 in HsSTP (Fig. 3B), but at similar distances from the phosphate found in HsSTP structure. The nucleophilic water bridging metals M1 and M2 are at a reasonable distance (3.8 Å) from the phosphorous atom when compared with the HsSTP structure. The phosphate is bound indirectly to the metal ions via water molecules and the Wn–P–O angle (146°) is reasonably close to optimal when the roughness of the analysis is taken into account.
SaSTP has been shown to dephosphorylate three different substrates. These are a family II inorganic pyrophosphatase, a response regulator CovR, and a purine biosynthesis protein PurA [20–22]. Although the exact site of phosphorylation in these proteins is not known, it has been shown that SaPPase is phosphorylated at a serine residue [20]. Interestingly, we observed a crystal contact in SaSTP structure which involves the flap subdomain in the adjacent monomer. This contact correlates with binding of the M3 and, intriguingly, places a serine residue (Ser155) from a neighboring monomer close to the active site (Fig. 3B). An approximate location for the Pi can be obtained by superimposing the HsSTP structure [4], which contains a phosphate ion in the active site on SaSTP. When we did so, we found that Ser155 is located rather close to the phosphate with an Oγ-P distance of only 3.8 Å.
At the crystal contact of SaSTP, Ser155 also forms a hydrogen bond to the very same residue (Arg13), which is responsible for binding the phosphoserine residue in vivo [11]. This interaction suggests that the crystal structure might mimic a product complex. Although it is possible that the serendipitous interaction is only an artifact, it is apparently strong enough to appear in the crystal and adjust the enzyme in an induced-fit-like manner: the conformation of the flap domain changes, M3 metal is introduced and Arg13 turns so that it is binding the serine residue in the ‘substrate’. To our knowledge, this is the first time that the binding of a protein component at the active site has been described for a STP. Given that the interaction would resemble that of the actual complex, we were able to identify a sequence motif of the substrate molecule among the actual substrates of SaSTP. The motif, [ED]-hydrophil-X(1,2)-[ST]-X-P, allows similar interactions to those described here and is present in the S. agalactiae PPase, kinase and adenylosuccinate synthase, but not in S. agalactiae CovR/CsrR. The motif is located at the surface of the SaPPase (Rantanen, unpublished), and superposition of the prolines allows superposition of the serine Oγs – but not the rest of the putative motif. We are currently attempting to confirm the site of phosphorylation using biochemical methods.
Experimental procedures
Data collection and processing
Cloning, expression, purification, crystallization and data processing of the native protein have been described elsewhere [20,23]. The reported 2.65 Å native data set was processed using the program XDS [24] and the crystal was assigned to space group P21212 with four monomers per asymmetric unit (Table 1). We also produced selenomethionine-labeled protein, because molecular replacement with the best available molecular replacement probe (MtSTP [9] with 33% sequence identity) was not successful. The selenomethionine-labeled protein was produced using the same construct and purification protocol as for wild-type. Expression was, however, performed in M9 minimal medium (6.0 g Na2HPO4, 3.0 g KH2PO4, 1.0 g NH4Cl, 0.5 g NaCl per L), supplemented with 2 mM MgSO4, 0.2% glucose, 0.5 × 10−3% thiamine, 0.1 mM CaCl2, and 100 μg·mL−1 ampicillin. Just before induction, we added two mixtures of amino acids to shut down the biosynthetic pathways leading to methionine: lysine, threonine and phenylalanine at a concentration of 100 μg·L−1 and leucine, isoleucine and valine at a concentration of 50 μg·L−1. Simultaneously, we added selenomethionine at a concentration of 60 μg·L−1, and after 15 min, we started induction by adding 1 mM isopropyl thio-β-D-galactoside. Cells were transferred to 25 °C and harvested after 16 h. Purification and crystallization were performed as reported previously [20,23]. The protein yield was, however, considerably lower than for wild-type SaSTP (40 mg·L−1) [23]; we obtained only 5 mg of the protein per L of culture. MS analysis and a fluorescence scan showed that labeling the protein had succeeded (data not shown). Suppliers for reagents were as follows: ampicillin, thiamine and glucose, Sigma-Aldrich (St Louis, MO/YA-Kemia OY, Helsinki, Finland); Na2HPO4, KH2PO4 and CaCl2, JT Baker (Phillipsburg, NJ); NH4Cl, NaCl and MgSO4, Sigma-Aldrich; and amino acids, Merck/Calbiochem (Darmstad, Germany).
The labeled SaSTP crystal diffracted to 2.50 Å resolution. We collected single-wavelength anomalous dispersion (SAD) data at the selenium peak wavelength (0.97935 Å) and analyzed it using XDS [24] (Table 2). Data were collected at 100 K using radiation from the European Synchotron Radiation Facility tunable-wavelength beam line ID23-1. A total of 225° data were collected with 0.5° oscillation per frame. Data had an overall Rmerged(F) of 6.9% in space group P212121 (Table 1) [25]. Crystallization conditions were identical between native and selenomethionine-labeled crystals, but although the crystals were still primitive orthorhombic, the unit cell parameters and space group had changed with respect to the native data (Table 2).
Table 2.
Data collection statistics for native [23] and selenomethionine-labeled STP. Values in parentheses are for the highest resolution shell.
| Native | Selenomethionine | |
|---|---|---|
| Space group | P21212 | P212121 |
| Wavelength (Å) | 0.933 | 0.97935 |
| Unit-cell parameters (Å) | a = 91.8, b = 139.0, c = 86.7 | a = 49.1, b = 74.9, c = 137.3 |
| Resolution (Å) | 20–2.65 (2.7–2.65) | 20–2.5 (2.60–2.50) |
| Reflections measured | 169847 (7368) | 160419 (18194) |
| Unique reflections | 32767 (1731) | 33057 (3688) |
| Completeness (%) | 99.5 (97.5) | 97.8 (98.1) |
| Redundancy | 5.2 (4.3) | 4.9 (4.9) |
| I/σ (I) | 11.7 (3.7) | 14.2 (3.2) |
| s-norm/s-ano | 1.02 (1.04) | 1.26 (1.00) |
| Rmerged-F a (%) | 11.5 (43.7) | 6.9 (49.8) |
Rmerged−F = Σ |AI(h,P) − AI(h,Q)| = /0.5 Σ (AI(h,P)− AI(h,Q)), where if I ≥ 0 if I <0. P and Q are two subsets of data [25].
We used the programs SHELXC, SHELXD and SHELXE in the HKL2MAP program suite to solve the structure of SaSTP [26,27]. We found 12 sites, which suggested two monomers in the asymmetric unit as expected from the Matthews coefficient (Vm = 2.4 Å3·Da−1) [28]. Correlation coefficients from SHELXD were 50 and 28%, and the Patterson figure of merit was 19 (resolution cut-off 3.0 Å). Subsequent phasing with SHELXE resulted in a correlation coefficient of 54% and allowed us to build a model of the protein into the electron density map. The model was, however, rather incomplete; 20% of the residues were either missing or modeled in a wrong conformation (checked against the final coordinates) due to the poor quality of the maps, and refinement with REFMAC5 [29] stalled at R-factors of 26 and 30%. This may be due to real disorder in the crystal as the mean B-factor from the refinement was rather high (78.5 Å2) – as was the Wilson B-factor (77 Å2). It should be noted that the data did not show signs of twinning and other possible space groups were also tested for refinement. The structure that we obtained using SAD-phasing is reported only to clarify the structure solution process – the native data set was ultimately used to derive the structure that we present here (see below).
We used the best model we built to the experimental electron density as a model in molecular replacement to the 2.65 Å native data (Table 2) [23]. Molecular replacement was performed using the MOLREP [30] program of the CCP4 package [30,31] using the CCP4I graphical interface [32]. The Matthews coefficient [28] indicated four monomers in the asymmetric unit (Vm = 2.7 Å3·Da−1) and MOLREP was able to find all of them. The rotation searches initially hinted at the possible presence of the protein in two different conformations: two rotations had Rf/sigma values of 9.6, and the following two had lower values (7.8 and 6.7). Initial R-factor after the translation searches was 49.4% with a correlation coefficient of 0.394.
Model building and refinement
We refined the monomers using REFMAC [29] to an R-factor of 19.7% (for 5% test reflections Rfree = 27.1%). Model building between refinement cycles was done with COOT [33]. Because of the modest resolution and large asymmetric unit, we used strict NCS constraints to keep all four monomers similar at the beginning of the refinement. During refinement, it became apparent that there were differences between the molecules and finally NCS restraints with the default medium weight was applied between monomers A and B as well as between C and D. The stereochemistry of the final model is good as indicated by the Ramachandran plot calculated with PROCHECK [34]: 94.6% of the residues are in the most favored region, the rest of the residues in other allowed regions (Table 3).
Table 3.
Structure refinement statistics for SaSTP against the native P21212 and SAD data. SAD data collected at 0.97935 Å wavelength.
| Native | Selenomethionine | |
|---|---|---|
| Space group | P21212 | P212121 |
| Resolution range (Å) | 19.68–2.65 | 10–2.50 |
| NCS restraints | A to B & C to D | none |
| Number of reflections Rwork/Rfreea | 31351/1568 | 15989/799 |
| Atoms (total/water/metal) | 7780/298/10 | 3508/16/7 |
| Rwork (%)/Rfree (%) | 19.7/27.1 | 26.1/29.9 |
| rmsd bond length (Å) | 0.010 | 0.021 |
| rmsd bond angle (O) | 1.3 | 2.78 |
| Mean B-value (Å2) | 42.1 | 78.5 |
| Mean B-value (Å2), metal ions | 29.8 | 95.0 |
| Mean B-value (Å2), water molecules | 28.2 | 66.5 |
| Ramachandran plot | ||
| Residues in most favored region (%) | 94.6 | 71.9 |
| Residues in additionally and generously allowed regions | 5.4 | 25.3 |
R work = (Σ|Fobs − Fcalc|)/(Σ|Fobs|), where Fobs and Fcalc are observed and calculated structure factor amplitudes, respectively. Rfree is an R-factor for an unrefined subset of the data (5% of the data).
PPM class STPs are metalloenzymes and we found 2–3 metal ions bound near the active site in all monomers. We assigned these ions as Mg2+ for the following reasons. The crystallization solution contained 0.2 M magnesium acetate, making Mg2+ the most likely candidate. The coordination is octahedral, which is typical for Mg2+ [35–37] and the coordinating distances (1.83–2.47 Å) are close to those seen for Mg2+. We did not find any peaks in the anomalous maps at the metal binding sites (calculated at wavelengths of 0.979 and 0.933 Å) excluding the other metal ion, Mn2+, seen in PPM STPs. Furthermore, the B-factors of the metal ions are close to the B-factors of the coordinating residues (16–42 versus 27–40 Å2).
Acknowledgments
We would like to thank Professor Arto Annila for funding. We acknowledge the European Synchotron Radiation Facility for provision of synchrotron radiation and we would like to thank Didier Nurizzo for assistance in using beam line. This study was supported by grants from the Sigrid Juselius Foundation, and from the Academy of Finland (grant number 1105157) to AG, who is a member of the Biocentrum Helsinki research organization. It was also supported by the National Institutes of Health, Grant # RO1 AI056073 to CER and CHRMC Basic Science Steering Committee Award to LR.
Abbreviations
- MtSTP
Mycobacterium tuberculosis serine/threonine phosphatase
- PPM
phosphoprotein phosphatase M
- PPP
phosphoprotein phosphatase P
- SAD
single-wavelength anomalous dispersion
- SaSTP
Streptococcus agalactiae serine/threonine phosphatase
- STK
serine/threonine kinase
- STP
serine/threonine phosphatase
- TxSTP
Toxoplasma gondii STP
References
- 1.Barford D, Das AK, Egloff M-P. The structure and mechanism of protein phosphatases: insights into catalysis and regulation. Annu Rev Biophys Biomol Struct. 1998;27:133–164. doi: 10.1146/annurev.biophys.27.1.133. [DOI] [PubMed] [Google Scholar]
- 2.Chen MX, McPartlin AE, Brown L, Chen YH, Cohen PTW. A novel human protein serine/threonine phosphatase, which possesses four tetratricopeptide repeat motifs and localizes to the nucleus. EMBO J. 1994;13:4278–4290. doi: 10.1002/j.1460-2075.1994.tb06748.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leung J, Bouvier-Durand M, Morris PC, Guerrier D, Chefdor F, Giraudat J. Arabidopsis ABA response gene ABI1: features of a calcium-modulated protein phosphatase. Science. 1994;264:1448–1452. doi: 10.1126/science.7910981. [DOI] [PubMed] [Google Scholar]
- 4.Das AK, Helps NR, Cohen PT, Barford D. Crystal structure of the protein serine/threonine phosphatase 2C at 2.0 Å resolution. EMBO J. 1996;15:6798–6809. [PMC free article] [PubMed] [Google Scholar]
- 5.Goldberg J, Huang H, Kwon Y, Greengard P, Nairn AC, Kuriyan J. Three-dimensional structure of the catalytic subunit of protein serine/threonine phosphatase- 1. Nature. 1995;376:745–753. doi: 10.1038/376745a0. [DOI] [PubMed] [Google Scholar]
- 6.Griffith JP, Kim JL, Kim EE, Sintchak MD, Thomson JA, Fitzgibbon MJ, Fleming MA, Caron PR, Hsiao K, Navia MA. X-Ray structure of calcineurin inhibited by the immunophilin–immunosuppressant FKBP12–FK506 complex. Cell. 1995;82:507–522. doi: 10.1016/0092-8674(95)90439-5. [DOI] [PubMed] [Google Scholar]
- 7.Egloff M-P, Cohen PTW, Reinemer P, Barford D. Crystal structure of the catalytic subunit of human protein phosphatase 1 and its complex with tungstate. J Mol Biol. 1995;254:942–959. doi: 10.1006/jmbi.1995.0667. [DOI] [PubMed] [Google Scholar]
- 8.Egloff M-P, Johnson DF, Moorhead G, Cohen PTW, Cohen P, Barford D. Structural basis for the recognition of regulatory sub-units by the catalytic subunit of protein phosphatase 1. EMBO J. 1997;16:1876–1887. doi: 10.1093/emboj/16.8.1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pullen KE, Ng HL, Sung PY, Good MC, Smith SM, Alber T. An alternate conformation and a third metal in PstP/Ppp, the M. tuberculosis PP2C-family Ser/Thr protein phosphatase. Structure. 2004;12:1947–1954. doi: 10.1016/j.str.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 10.Jackson MD, Denu JM. Molecular reactions of protein phosphatases – insights from structure and chemistry. Chem Rev. 2001;101:2313–2340. doi: 10.1021/cr000247e. [DOI] [PubMed] [Google Scholar]
- 11.Jackson MD, Fjeld CC, Denu JM. Probing the function of conserved residues in the serine/threonine phosphatase PP2Calpha. Biochemistry. 2003;42:8513–8521. doi: 10.1021/bi034074+. [DOI] [PubMed] [Google Scholar]
- 12.Christianson DW, Cox JD. Catalysis by metal-activated hydroxide in zinc and manganese metalloenzymes. Annu Rev Biochem. 1999;68:33–57. doi: 10.1146/annurev.biochem.68.1.33. [DOI] [PubMed] [Google Scholar]
- 13.Yang X, Kang CM, Brody MS, Price CW. Opposing pairs of serine protein kinases and phosphatases transmit signals of environmental stress to activate a bacterial transcription factor. Genes Dev. 1996;10:2265– 2275. doi: 10.1101/gad.10.18.2265. [DOI] [PubMed] [Google Scholar]
- 14.Adler E, Donella-Deana A, Arigoni F, Pinna LA, Stragler P. Structural relationship between a bacterial developmental protein and eukaryotic PP2C phosphatases. Mol Microbiol. 1997;23:57–62. doi: 10.1046/j.1365-2958.1997.1801552.x. [DOI] [PubMed] [Google Scholar]
- 15.Walburger A, Koul A, Ferrari G, Nguyen L, Prescian-otto-Baschong C, Huygen K, Klebl B, Thompson C, Bacher G, Pieters J. Protein kinase G from pathogenic mycobacteria promotes survival within macrophages. Science. 2004;304:1800–1804. doi: 10.1126/science.1099384. [DOI] [PubMed] [Google Scholar]
- 16.Hakansson S, Galyov EE, Rosqvist R, Wolf-Watz H. The Yersinia YpkA Ser/Thr kinase is translocated and subsequently targeted to the inner surface of the HeLa cell plasma membrane. Mol Microbiol. 1996;20:593–603. doi: 10.1046/j.1365-2958.1996.5251051.x. [DOI] [PubMed] [Google Scholar]
- 17.Galyov EE, Hakansson S, Forsberg A, Wolf-Watz H. A secreted protein kinase of Yersinia pseudotuberculosis is an indispensable virulence determinant. Nature. 1993;361:730–732. doi: 10.1038/361730a0. [DOI] [PubMed] [Google Scholar]
- 18.Echenique J, Kadioglu A, Romao S, Andrew PW, Trombe MC. Protein serine/threonine kinase StkP positively controls virulence and competence in Streptococcus pneumoniae. Infect Immun. 2004;72:2434–2437. doi: 10.1128/IAI.72.4.2434-2437.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang J, Li C, Yang H, Mushegian A, Jin S. A novel serine/threonine protein kinase homologue of Pseudomonas aeruginosa is specifically inducible within the host infection site and is required for full virulence in neutropenic mice. J Bacteriol. 1998;180:6764–6768. doi: 10.1128/jb.180.24.6764-6768.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rajagopal L, Clancy A, Rubens CE. A eukaryotic-type serine/threonine kinase and phosphatase in Streptococcus agalactiae reversibly phosphorylate an inorganic pyrophosphatase and affect growth, cell segregation and virulence. J Biol Chem. 2003;278:14429–14441. doi: 10.1074/jbc.M212747200. [DOI] [PubMed] [Google Scholar]
- 21.Rajagopal L, Vo A, Silvestroni A, Rubens CE. Regulation of purine biosynthesis by a eukaryotic-type kinase in Streptococcus agalactiae. Mol Microbiol. 2005;56:1329–1346. doi: 10.1111/j.1365-2958.2005.04620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rajagopal L, Vo A, Silvestroni A, Rubens CE. Regulation of cytotoxin expression by converging eukaryotic- type and two-component signalling mechanisms in Streptococcus agalactiae. Mol Microbiol. 2006;62:941–957. doi: 10.1111/j.1365-2958.2006.05431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rantanen MK, Lehtiö L, Rajagopal L, Rubens CE, Goldman A. Crystallization and preliminary crystallographic analysis of two Streptococcus agalactiae proteins: the family II inorganic pyrophosphatase and the serine/threonine phosphatase. Acta Crystallogr F. 2006;62:891–894. doi: 10.1107/S174430910602954X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallogr. 1993;26:795–800. [Google Scholar]
- 25.Diederichs K, Karplus PA. Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat Struct Biol. 1997;4:269–275. doi: 10.1038/nsb0497-269. [DOI] [PubMed] [Google Scholar]
- 26.Sheldrick G, Schneider T. SHELXL: high-resolution refinement. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- 27.Pape T, Schneider TR. HKL2MAP: a graphical user interface for macromolecular phasing with SHELX-programs. J Appl Crystallogr. 2004;37:843–844. [Google Scholar]
- 28.Matthews BW. Solvent content of protein crystals. J Mol Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- 29.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 30.Vagin AA, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 31.CCP4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 32.Potterton E, Briggs P, Turkenburg M, Dodson EJ. A graphical user interface to the CCP4 program suite. Acta Crystallogr D. 2003;59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- 33.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 34.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283. [Google Scholar]
- 35.Harding MM. The geometry of metal–ligand interactions relevant to proteins. II. Angles at the metal atom, additional weak metal–donor interactions. Acta Crystallogr D. 2000;56:857–867. doi: 10.1107/s0907444900005849. [DOI] [PubMed] [Google Scholar]
- 36.Harding MM. The geometry of metal–ligand interactions relevant to proteins. Acta Crystallog D. 1999;55:1432–1443. doi: 10.1107/s0907444999007374. [DOI] [PubMed] [Google Scholar]
- 37.Harding MM. Geometry of metal–ligand interactions in proteins. Acta Crystallogr D. 2001;57:401–411. doi: 10.1107/s0907444900019168. [DOI] [PubMed] [Google Scholar]
- 38.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 39.Delano WL. The PyMOL Molecular Graphics System. Delano Scientific; Palo Alto, CA: 2002. [Google Scholar]