

Summary
The dynamic regulation of the structure, function and turnover of mitochondria is recognized as an immutable control node maintaining cellular integrity and homeostasis. The term ‘mitohormesis’ has recently been coined to describe the adaptive reprogramming of mitochondrial biology in response to low levels of metabolic substrate deprivation to augment subsequent mitochondrial and cellular tolerance to biological stress [1]. Disruption of these regulatory programs gives rise to cardiovascular and neurodegenerative diseases and augmentation or fine-tuning of these programs may ameliorate mitochondrial and global cellular stress-tolerance. This is in part via the regulation of reactive oxygen species, calcium homeostasis, and in response to caloric restriction, the capacity to augment DNA repair. The objective of this manuscript is to briefly review these regulatory programs and to postulate novel therapeutic approaches with the primary goal of modulating mitochondria to enhance tolerance to cardiac ischemic stress.
Introduction
Mitochondria orchestrate an extensive repertoire of cellular functions and have tissue specific programming to facilitate overall organ function [2]. In the heart, the roles of mitochondria include the generation of energy, the production and metabolism of reactive radical species and the regulation of apoptosis. These functions in turn are exquisitely controlled by regulatory programs governing mitochondrial copy number, functional content and activity, calcium metabolism, stress-tolerance and apoptotic pathways. The identification and characterization of these regulatory programs have promulgated the question of whether these can be modified to adapt mitochondrial function to improve cellular tolerance to biological stress.
In this review article we: 1) describe the innate regulatory pathways driving mitochondrial maintenance of cellular homeostasis, 2) discuss where these programs have been shown to be operational in enhancing stress tolerance in general and in the heart specifically, and 3) to identify current and potential mitochondrial targeted compounds that improve cardiac stress tolerance to prevent or treat ischemic heart disease.
Innate regulatory programs controlling mitochondrial plasticity
Mitochondrial biogenesis, the molecular control of mitochondrial turnover, content and number exquisitely coordinates diverse homeostatic demands via communication between the mitochondrial and nuclear genomes. The regulatory proteins and signaling pathways conducting this inter-genomic control has been reviewed recently [3,4]. The functional importance of mitochondrial biogenesis in maintaining cardiac homeostasis is evident in that the genetic ablation of master regulators of mitochondrial biogenesis, i.e. the peroxisome proliferator activated receptor γ coactivator 1 α and β (PGC1α/β) diminish cardiac adaptation to adrenergic stimulation and pressure-overload [5-7]. The disruption of downstream cognate transcription factors including the three major peroxisome proliferator activated receptor (PPAR) subtypes, also leads to cardiac contractile dysfunction and an enhanced susceptibly to oxidative damage [8-10]. Furthermore, the modest induction of transcription factor A of mitochondria (TFAM) evokes protection against ischemia induced heart failure [11]. However, excessive activation of this program becomes pathological as is shown by the generation of an over-abundance of mitochondria resulting in cardiomyopathy following sustained overexpression of murine heart PGC1α [12]. Moreover, chronic overexpression of PPARα in the heart blunts contractile recovery in response to ischemic stress [13]. An additional level of regulation of mitochondrial homeostasis is the molecular control of mitochondrial fusion and division (reviewed [14]) with unpublished data showing that enhancing mitochondrial fusion augments cardiomyocyte ischemia-tolerance [15]. Biological compounds that activate the mitochondrial biogenesis program will be discussed as putative modulators of ischemia-tolerance.
Caloric restriction orchestrated mitochondrial stress response
Hormesis is defined as the activation of cellular protective and reparative properties induced by mild physiologic stress. Caloric restriction is considered an hormetic process where chronic mild starvation enhances cellular DNA repair capacity and antioxidant defenses [16,17] and upregulates the mitochondrial biogenesis regulatory program [18]. Although the cardiac mitochondrial regulatory perturbations in response to caloric restriction have not been well characterized, short-term caloric restriction confers resistance to cardiac ischemia-reperfusion [19]. A candidate regulatory protein that may orchestrate this cardioprotective effect is the nutrient sensor protein sirt1. This NAD-dependent deacetylase enhances stress tolerance and lifespan extension during caloric restriction [20]. Accordingly sirt1 has been shown to attenuate constitutive and H2O2 mediated apoptosis in cardiomyocytes [21] and to protect the intact heart against paraquat mediated oxidative stress [22]. While this biology requires more extensive investigation, caloric-restriction mimetics may be candidates to blunt ischemia-reperfusion injury.
Ischemic preconditioning identifies mitochondrial targets to enhance ischemiareperfusion tolerance
A second stress-activated program operational in the heart and extensively investigated is the biological phenomenon termed ischemic preconditioning [23]. Here too, the regulation of mitochondria has been implicated in augmenting tolerance to ischemia-reperfusion. The preconditioning-induced mitochondrial perturbations identified to date include upregulation of antioxidant defense mechanisms including via the transient inhibition of mitochondrial respiration [24], via transient mitochondrial uncoupling [25,26] and by upregulation of antioxidant enzymes [27]. Furthermore, ischemic preconditioning reduces the susceptibility to mitochondrial permeability transition [15]. In the later part of this review we will discuss compounds that can mimic these mitochondrial effects as potential cardioprotective agents. The proposed mechanism enhancing mitochondrial ischemia tolerance in response to caloric restriction and preconditioning are schematized in figure 1.
Figure 1.
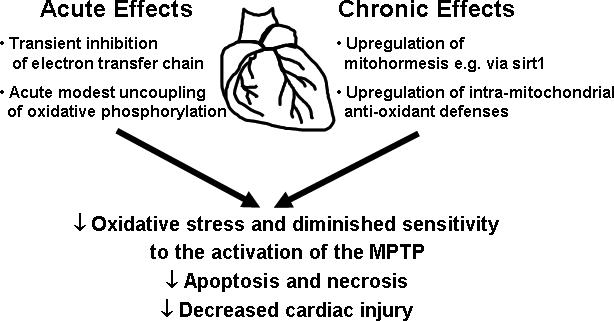
Schematic of modulations of mitochondria to augment cardiac ischemia-tolerance
Potential mitochondrial modulators to ameliorate ischemic injury
The mitochondrial adaptations to caloric restriction and ischemic preconditioning identify potential targets to explore as therapeutic modulators of ischemia-tolerance. In the remainder of this review we will review compounds with specific focus on potential caloric restriction mimetics, a class of compounds that may induce mitochondrial biogenesis, a novel anion known to transiently inhibit mitochondrial respiration under ischemia and a mitochondrial targeted antioxidant. Mitochondrial permeability inhibitors which have recently been reviewed are not discussed [23].
Resveratrol – a potential caloric restriction mimetic
The plant-derived polyphenol resveratrol (3,5,4’-trihydroxystilbene) is enriched in red wine and functions as a caloric restriction mimetic via upregulation of sirt1 and AMP-activated kinase (AMPK) [28-30]. Additionally it exhibits antioxidant properties [31] and upregulates the mitochondrial biogenesis program [28]. Signaling intermediates induced by this putative ‘mitohormetic’ compound have been identified [32,33]. Consistent with its known pleiotropic effects resveratrol administration confers protection against cardiac ischemia-reperfusion injury [31,34,35].
As a therapeutic agent, resveratrol has limitations in that it has a short initial half-life and limited in-vivo bioavailability [31]. To counter this, numerous investigators are exploring compounds to restrict its catabolism. Furthermore, to differentiate the cardioprotective properties from the pleiotropic effects of resveratrol, small molecules that may directly activate, for example, sirt1 are being actively pursued [36]. Once identified, the direct cardiac-tolerance effects of activation of sirt1 activators would need to be validated.
AMP-Kinase activating compounds and mitochondrial biogenesis
The activation of the fuel sensor AMPK by exercise or by the AMPK activators AICAR (5-aminoimidazole-4-carboxamide 1β-D-ribofuranoside) and β-guanidinopropionic acid enhances mitochondrial function and biogenesis in skeletal muscle [37,38]. With respect to AICAR, this is shown to be due to the direct upregulation of PGC-1α [39-41] and to the enhanced expression of mitochondrial proteins cytochrome c, UCP-3, and citrate synthase [42,43]. In the heart, ischemia increases AMPK activation [44], and loss of AMPK exacerbates ischemia-reperfusion injury [45]. However, whether the activation of AMPK augments mitochondrial biogenesis in the heart has not been established. Conversely, the genetic depletion of AMPKα2 isoform alters cardiac mitochondrial ultrastructure and disrupts complex I of the electron transfer chain [46]. Cumulatively these data suggest that the activation of AMPK may induce ameliorative effects on mitochondrial biology to enhance ischemia tolerance.
These cardioprotective effects of AMPK activation are likely not exclusively due to mitochondrial manipulations as activation of this fuel-sensing kinase evokes multiple additional metabolic effects (reviewed [47]). Furthermore, the use of AICAR as a cardiac therapeutic target is limited in that it promotes bradycardia and hypoglycemia [48]. Current therapeutic agents that indirectly activate AMPK include the biguanides and the thiazolidinediones [49]. Interestingly, both these anti-diabetic drugs have been shown to augment mitochondrial biogenesis [50,51] and in separate studies, to confer protection against cardiac ischemia [52,53]. As above, whether these effects are directly due to modulation of mitochondrial function in the heart is not established. Nevertheless, several drug discovery programs are pursuing more specific and potent AMPK activators [48] and with the development of these compounds, the capacity to directly link their activation with the modulation of mitochondria and cardiac tolerance can be explored.
Nitrite - a novel ischemia-inducible electron transfer chain inhibitor
Chronic uncoupling of oxidative phosphorylation or inhibition of the electron chain transfer would be incompatible with sustained ATP production essential for cardiac contraction. However, in the context of ischemia-reperfusion, acute uncoupled respiration and inhibition of complex I of the electron transfer chain (ETC) inhibit mitochondrial reactive oxygen species production, via reduced electron dissociation from the ETC (reviewed [54]) and via reduced electron flow through complex III resulting in a reduction in superoxide generation [55], respectively. A pharmacologic agent that could dynamically replicate either of these perturbations exclusively under ischemia-reperfusion conditions would introduce a novel therapeutic approach.
In this respect, the nitrite anion (NO2−) is an intriguing ‘candidate’ in that it is reduced to nitric oxide (NO), a known cardioprotective compound [56] and an inhibitor of mitochondrial electron transfer [57], primarily at low tissue pH and under hypoxic conditions. Under these conditions nitrite is either directly reduced to NO by disproportionation and/or by the enzymatic action of heme-containing proteins, including xanthine oxidoreductase, electron transfer proteins, deoxyhemoglobin, and dexoymyoglobin [58]. Indeed, administration of nitrite in preclinical studies confers protection against ischemia-reperfusion injury [59] in parallel with a reduction in post ischemic reactive oxygen species production and by reducing the activation of the MPTP [57]. In the latter study, it was shown that nitrite does not affect mitochondrial respiration under normoxic conditions, but rather inhibits complex I of the electron transfer chain in response to hypoxia and reoxygenation [57]. From a therapeutic perspective, the most exciting aspects of this study are that the administration of nitrite could precede the ischemic injury by as much as 24 hours and that this anion could be administered orally [57]. Pilot studies in human subjects are now being planned.
MitoQ - a mitochondrial-targeted antioxidant
Although the benefit of therapeutic antioxidants to limit oxidative damage during ischemic injury has not been realized, given the mitochondria's role as both source and target of ROS the use of the novel mitochondria-targeted antioxidants has gained interest (Reviewed [60]). Mitochondrial targeting is feasible by conjugating an antioxidant to a lipophilic cation. This exploits the high inner mitochondrial membrane potential enabling mitochondrial accumulation of this cation-conjugated target at 100−1000 times higher concentration than in cytoplasm [61,62]. MitoQ, based on the endogenous mitochondrial ubiquinone coenzyme Q is such a compoung. It has a redox chemistry closely regulated by the ETC possibly allowing MitoQ to simultaneously decrease oxidative damage and upregulate respiration.
In vivo animal studies have shown that MitoQ decreases ROS, apoptosis and ischemiareperfusion cardiac injury in conjunction with improved respiratory coupling and increased complex I and aconitase activities [62]. MitoQ has been proven to be well tolerated and toxic only at very high concentrations [63]. Trials are underway for neurological disorders, but the impact of this agent on human cardiac disease remains to be tested.
Conclusions
Mitochondrial plasticity, under both acute and chronic regulation, is increasingly recognized as integral to cellular and tissue tolerance of ischemic stress. Chronic modulation of mitochondrial content and function via the principle of mitohormesis underlies the protective effect of caloric restriction mimetics. Acutely, alteration of electron flow and inhibition of reactive species production remain attractive targets for cardioprotection. Importantly, better biologic understanding has led to therapeutics with increased specificity of action. This is illustrated by the action of nitrite to function as a putative dynamic regulator of electron flux, specifically under conditions of low oxygen and acidosis. This anion may potentially represent the first truly ischemia-activated agent. Exploiting mitochondrial biology has also enabled the development of mitochondrial-targeting agents such as antioxidants which may ultimately realize their long proposed benefits. The true impact of these developments on human cardiovascular health awaits further clinical study. Finally, the proposed compounds and their mechanisms of actions discussed in this review are shown schematically in figure 2.
Figure 2.
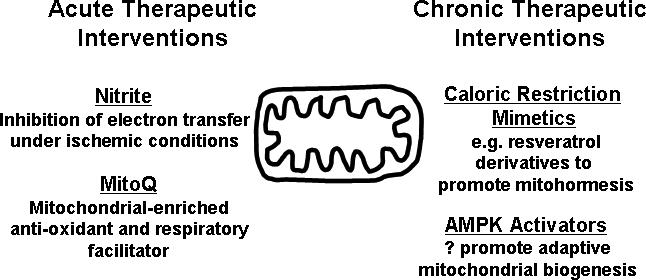
Mitochondrial directed therapeutic agents to enhance cardiac ischemia-tolerance
Acknowledgements
Michael Sack and Daniel Schwartz are funded by the Division of Intramural Research of the National Heart, Lung and Blood Institute of the NIH, Bethesda, MD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
• of Special Interest
•• of outstanding interest
- 1.Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extend Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007;6:280–293. doi: 10.1016/j.cmet.2007.08.011. [of outstanding interestThis original manuscript introduces the novel concept of mitohormesis and the role of caloric restriction in adaptation of the mitochondria.] [DOI] [PubMed] [Google Scholar]
- 2.Johnson DT, Harris RA, French S, Blair PV, You J, Bemis K, Wang M, Balaban RS. The Tissue Heterogeneity of the Mammalian Mitochondrial Proteome. Am J Physiol Cell Physiol. 2006 doi: 10.1152/ajpcell.00108.2006. [of outstanding interestThis original manuscript employs proteomics to demonstrate that mitochondrial proteins are distinctly regulated in different tissue types to reflect underlying functional preferences.] [DOI] [PubMed] [Google Scholar]
- 3.McLeod CJ, Pagel I, Sack MN. The mitochondrial biogenesis regulatory program in cardiac adaptation to ischemia-a putative target for therapeutic intervention. Trends Cardiovasc Med. 2005;15:118–123. doi: 10.1016/j.tcm.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 4.Finck BN, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) regulatory cascade in cardiac physiology and disease. Circulation. 2007;115:2540–2548. doi: 10.1161/CIRCULATIONAHA.107.670588. [DOI] [PubMed] [Google Scholar]
- 5.Arany Z, He H, Lin J, Hoyer K, Handschin C, Toka O, Ahmad F, Matsui T, Chin S, Wu PH, et al. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005;1:259–271. doi: 10.1016/j.cmet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 6.Lelliott CJ, Medina-Gomez G, Petrovic N, Kis A, Feldmann HM, Bjursell M, Parker N, Curtis K, Campbell M, Hu P, et al. Ablation of PGC-1beta results in defective mitochondrial activity, thermogenesis, hepatic function, and cardiac performance. PLoS Biol. 2006;4:e369. doi: 10.1371/journal.pbio.0040369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1alpha. Proc Natl Acad Sci U S A. 2006;103:10086–10091. doi: 10.1073/pnas.0603615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luptak I, Balschi JA, Xing Y, Leone TC, Kelly DP, Tian R. Decreased contractile and metabolic reserve in peroxisome proliferator-activated receptor-alpha-null hearts can be rescued by increasing glucose transport and utilization. Circulation. 2005;112:2339–2346. doi: 10.1161/CIRCULATIONAHA.105.534594. [DOI] [PubMed] [Google Scholar]
- 9.Cheng L, Ding G, Qin Q, Huang Y, Lewis W, He N, Evans RM, Schneider MD, Brako FA, Xiao Y, et al. Cardiomyocyte-restricted peroxisome proliferator-activated receptor-delta deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat Med. 2004;10:1245–1250. doi: 10.1038/nm1116. [DOI] [PubMed] [Google Scholar]
- 10.Ding G, Fu M, Qin Q, Lewis W, Kim HW, Fukai T, Bacanamwo M, Chen YE, Schneider MD, Mangelsdorf DJ, et al. Cardiac peroxisome proliferator-activated receptor gamma is essential in protecting cardiomyocytes from oxidative damage. Cardiovasc Res. 2007;76:269–279. doi: 10.1016/j.cardiores.2007.06.027. [DOI] [PubMed] [Google Scholar]
- 11.Ikeuchi M, Matsusaka H, Kang D, Matsushima S, Ide T, Kubota T, Fujiwara T, Hamasaki N, Takeshita A, Sunagawa K, et al. Overexpression of mitochondrial transcription factor a ameliorates mitochondrial deficiencies and cardiac failure after myocardial infarction. Circulation. 2005;112:683–690. doi: 10.1161/CIRCULATIONAHA.104.524835. [of Special InterestThis original manuscript demonstrates how modest induction of the mitochondrial biogenesis program can confer protection against ischemic injury.] [DOI] [PubMed] [Google Scholar]
- 12.Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–856. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sambandam N, Morabito D, Wagg C, Finck BN, Kelly DP, Lopaschuk GD. Chronic activation of PPARalpha is detrimental to cardiac recovery after ischemia. Am J Physiol Heart Circ Physiol. 2006;290:H87–95. doi: 10.1152/ajpheart.00285.2005. [DOI] [PubMed] [Google Scholar]
- 14.Detmer SA, Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol. 2007;8:870–879. doi: 10.1038/nrm2275. [DOI] [PubMed] [Google Scholar]
- 15.Hausenloy DJ, Scorrano L. Targeting cell death. Clin Pharmacol Ther. 2007;82:370–373. doi: 10.1038/sj.clpt.6100352. [DOI] [PubMed] [Google Scholar]
- 16.Sinclair DA. Toward a unified theory of caloric restriction and longevity regulation. Mech Ageing Dev. 2005;126:987–1002. doi: 10.1016/j.mad.2005.03.019. [DOI] [PubMed] [Google Scholar]
- 17.Lopez-Lluch G, Hunt N, Jones B, Zhu M, Jamieson H, Hilmer S, Cascajo MV, Allard J, Ingram DK, Navas P, et al. Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc Natl Acad Sci U S A. 2006;103:1768–1773. doi: 10.1073/pnas.0510452103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nisoli E, Tonello C, Cardile A, Cozzi V, Bracale R, Tedesco L, Falcone S, Valerio A, Cantoni O, Clementi E, et al. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science. 2005;310:314–317. doi: 10.1126/science.1117728. [DOI] [PubMed] [Google Scholar]
- 19.Shinmura K, Tamaki K, Bolli R. Short-term caloric restriction improves ischemic tolerance independent of opening of ATP-sensitive K+ channels in both young and aged hearts. J Mol Cell Cardiol. 2005;39:285–296. doi: 10.1016/j.yjmcc.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 20.Haigis MC, Guarente LP. Mammalian sirtuins--emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006;20:2913–2921. doi: 10.1101/gad.1467506. [DOI] [PubMed] [Google Scholar]
- 21.Alcendor RR, Kirshenbaum LA, Imai S, Vatner SF, Sadoshima J. Silent information regulator 2alpha, a longevity factor and class III histone deacetylase, is an essential endogenous apoptosis inhibitor in cardiac myocytes. Circ Res. 2004;95:971–980. doi: 10.1161/01.RES.0000147557.75257.ff. [DOI] [PubMed] [Google Scholar]
- 22.Alcendor RR, Gao S, Zhai P, Zablocki D, Holle E, Yu X, Tian B, Wagner T, Vatner SF, Sadoshima J. Sirt1 Regulates Aging and Resistance to Oxidative Stress in the Heart. Circ Res. 2007;100:1512–1521. doi: 10.1161/01.RES.0000267723.65696.4a. [of outstanding interestThis original manuscript demonstrates that the induction of a mediator of caloric restriction can protect the heart against oxidative stress.] [DOI] [PubMed] [Google Scholar]
- 23.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 24.Chen Q, Hoppel CL, Lesnefsky EJ. Blockade of Electron Transport before Cardiac Ischemia with the Reversible Inhibitor Amobarbital Protects Rat Heart Mitochondria. J Pharmacol Exp Ther. 2006;316:200–207. doi: 10.1124/jpet.105.091702. [of Special InterestThis original article demonstrates how the direct inhibition of electron transfer can confer protection against ischemic injury.] [DOI] [PubMed] [Google Scholar]
- 25.McLeod CJ, Aziz A, Hoyt RF, Jr., McCoy JP, Jr., Sack MN. Uncoupling proteins 2 and 3 function in concert to augment tolerance to cardiac ischemia. J Biol Chem. 2005;280:33470–33476. doi: 10.1074/jbc.M505258200. [DOI] [PubMed] [Google Scholar]
- 26.Nadtochiy SM, Tompkins AJ, Brookes PS. Different mechanisms of mitochondrial proton leak in ischaemia/reperfusion injury and preconditioning: implications for pathology and cardioprotection. Biochem J. 2006;395:611–618. doi: 10.1042/BJ20051927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamashita N, Nishida M, Hoshida S, Kuzuya T, Hori M, Taniguchi N, Kamada T, Tada M. Induction of manganese superoxide dismutase in rat cardiac myocytes increases tolerance to hypoxia 24 hours after preconditioning. J Clin Invest. 1994;94:2193–2199. doi: 10.1172/JCI117580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 29.Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, et al. Resveratrol improves health and survival of mice on a highcalorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dasgupta B, Milbrandt J. Resveratrol stimulates AMP kinase activity in neurons. Proc Natl Acad Sci U S A. 2007;104:7217–7222. doi: 10.1073/pnas.0610068104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov. 2006;5:493–506. doi: 10.1038/nrd2060. [DOI] [PubMed] [Google Scholar]
- 32.Das S, Alagappan VK, Bagchi D, Sharma HS, Maulik N, Das DK. Coordinated induction of iNOS-VEGF-KDR-eNOS after resveratrol consumption: a potential mechanism for resveratrol preconditioning of the heart. Vascul Pharmacol. 2005;42:281–289. doi: 10.1016/j.vph.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 33.Thirunavukkarasu M, Penumathsa SV, Koneru S, Juhasz B, Zhan L, Otani H, Bagchi D, Das DK, Maulik N. Resveratrol alleviates cardiac dysfunction in streptozotocin-induced diabetes: Role of nitric oxide, thioredoxin, and heme oxygenase. Free Radic Biol Med. 2007;43:720–729. doi: 10.1016/j.freeradbiomed.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Penumathsa SV, Thirunavukkarasu M, Koneru S, Juhasz B, Zhan L, Pant R, Menon VP, Otani H, Maulik N. Statin and resveratrol in combination induces cardioprotection against myocardial infarction in hypercholesterolemic rat. J Mol Cell Cardiol. 2007;42:508–516. doi: 10.1016/j.yjmcc.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goh SS, Woodman OL, Pepe S, Cao AH, Qin C, Ritchie RH. The red wine antioxidant resveratrol prevents cardiomyocyte injury following ischemia-reperfusion via multiple sites and mechanisms. Antioxid Redox Signal. 2007;9:101–113. doi: 10.1089/ars.2007.9.101. [DOI] [PubMed] [Google Scholar]
- 36.Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, Sinclair D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 2004;430:686–689. doi: 10.1038/nature02789. [DOI] [PubMed] [Google Scholar]
- 37.Reznick RM, Shulman GI. The role of AMP-activated protein kinase in mitochondrial biogenesis. J Physiol. 2006;574:33–39. doi: 10.1113/jphysiol.2006.109512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reznick RM, Zong H, Li J, Morino K, Moore IK, Yu HJ, Liu ZX, Dong J, Mustard KJ, Hawley SA, et al. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab. 2007;5:151–156. doi: 10.1016/j.cmet.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hood DA, Irrcher I, Ljubicic V, Joseph AM. Coordination of metabolic plasticity in skeletal muscle. J Exp Biol. 2006;209:2265–2275. doi: 10.1242/jeb.02182. [DOI] [PubMed] [Google Scholar]
- 40.Lee WJ, Kim M, Park HS, Kim HS, Jeon MJ, Oh KS, Koh EH, Won JC, Kim MS, Oh GT, et al. AMPK activation increases fatty acid oxidation in skeletal muscle by activating PPARalpha and PGC-1. Biochem Biophys Res Commun. 2006;340:291–295. doi: 10.1016/j.bbrc.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 41.Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou M, Lin BZ, Coughlin S, Vallega G, Pilch PF. UCP-3 expression in skeletal muscle: effects of exercise, hypoxia, and AMP-activated protein kinase. Am J Physiol Endocrinol Metab. 2000;279:E622–629. doi: 10.1152/ajpendo.2000.279.3.E622. [DOI] [PubMed] [Google Scholar]
- 43.Jorgensen SB, Treebak JT, Viollet B, Schjerling P, Vaulont S, Wojtaszewski JF, Richter EA. Role of AMPKalpha2 in basal, training-, and AICAR-induced GLUT4, hexokinase II, and mitochondrial protein expression in mouse muscle. Am J Physiol Endocrinol Metab. 2007;292:E331–339. doi: 10.1152/ajpendo.00243.2006. [DOI] [PubMed] [Google Scholar]
- 44.Baron SJ, Li J, Russell RR, 3rd, Neumann D, Miller EJ, Tuerk R, Wallimann T, Hurley RL, Witters LA, Young LH. Dual mechanisms regulating AMPK kinase action in the ischemic heart. Circ Res. 2005;96:337–345. doi: 10.1161/01.RES.0000155723.53868.d2. [DOI] [PubMed] [Google Scholar]
- 45.Russell RR, 3rd, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, Giordano FJ, Mu J, Birnbaum MJ, Young LH. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest. 2004;114:495–503. doi: 10.1172/JCI19297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Athea Y, Viollet B, Mateo P, Rousseau D, Novotova M, Garnier A, Vaulont S, Wilding JR, Grynberg A, Veksler V, et al. AMP-activated protein kinase alpha2 deficiency affects cardiac cardiolipin homeostasis and mitochondrial function. Diabetes. 2007;56:786–794. doi: 10.2337/db06-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 48.Young LH, Li J, Baron SJ, Russell RR. AMP-activated protein kinase: a key stress signaling pathway in the heart. Trends Cardiovasc Med. 2005;15:110–118. doi: 10.1016/j.tcm.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 49.Hardie DG. AMP-activated protein kinase as a drug target. Annu Rev Pharmacol Toxicol. 2007;47:185–210. doi: 10.1146/annurev.pharmtox.47.120505.105304. [DOI] [PubMed] [Google Scholar]
- 50.Suwa M, Egashira T, Nakano H, Sasaki H, Kumagai S. Metformin increases the PGC-1alpha protein and oxidative enzyme activities possibly via AMPK phosphorylation in skeletal muscle in vivo. J Appl Physiol. 2006;101:1685–1692. doi: 10.1152/japplphysiol.00255.2006. [DOI] [PubMed] [Google Scholar]
- 51.Wilson-Fritch L, Nicoloro S, Chouinard M, Lazar MA, Chui PC, Leszyk J, Straubhaar J, Czech MP, Corvera S. Mitochondrial remodeling in adipose tissue associated with obesity and treatment with rosiglitazone. J Clin Invest. 2004;114:1281–1289. doi: 10.1172/JCI21752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Legtenberg RJ, Houston RJ, Oeseburg B, Smits P. Metformin improves cardiac functional recovery after ischemia in rats. Horm Metab Res. 2002;34:182–185. doi: 10.1055/s-2002-26705. [DOI] [PubMed] [Google Scholar]
- 53.Yue Tl TL, Chen J, Bao W, Narayanan PK, Bril A, Jiang W, Lysko PG, Gu JL, Boyce R, Zimmerman DM, et al. In vivo myocardial protection from ischemia/reperfusion injury by the peroxisome proliferator-activated receptor-gamma agonist rosiglitazone. Circulation. 2001;104:2588–2594. doi: 10.1161/hc4601.099403. [DOI] [PubMed] [Google Scholar]
- 54.Sack MN. Mitochondrial depolarization and the role of uncoupling proteins in ischemia tolerance. Cardiovasc Res. 2006;72:210–219. doi: 10.1016/j.cardiores.2006.07.010. [of Special InterestThis review discusses the counterintuitive concept of how uncoupling of oxidative phosphorylation can confer resilience against ischemia-reperfusion injury in the heart.] [DOI] [PubMed] [Google Scholar]
- 55.Lesnefsky EJ, Chen Q, Moghaddas S, Hassan MO, Tandler B, Hoppel CL. Blockade of electron transport during ischemia protects cardiac mitochondria. J Biol Chem. 2004;279:47961–47967. doi: 10.1074/jbc.M409720200. [DOI] [PubMed] [Google Scholar]
- 56.Jones SP, Bolli R. The ubiquitous role of nitric oxide in cardioprotection. J Mol Cell Cardiol. 2006;40:16–23. doi: 10.1016/j.yjmcc.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 57.Shiva S, Sack MN, Greer JJ, Duranski M, Ringwood LA, Burwell L, Wang X, MacArthur PH, Shoja A, Raghavachari N, et al. Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J Exp Med. 2007;204:2089–2102. doi: 10.1084/jem.20070198. [of outstanding interestThis original manuscript illustrates the dynamic control of mitochondrial respiration by nitrite and its mitochondrial protective effects.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gladwin MT, Schechter AN, Kim-Shapiro DB, Patel RP, Hogg N, Shiva S, Cannon RO, 3rd, Kelm M, Wink DA, Espey MG, et al. The emerging biology of the nitrite anion. Nat Chem Biol. 2005;1:308–314. doi: 10.1038/nchembio1105-308. [DOI] [PubMed] [Google Scholar]
- 59.Duranski MR, Greer JJ, Dejam A, Jaganmohan S, Hogg N, Langston W, Patel RP, Yet SF, Wang X, Kevil CG, et al. Cytoprotective effects of nitrite during in vivo ischemiareperfusion of the heart and liver. J Clin Invest. 2005;115:1232–1240. doi: 10.1172/JCI22493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Victor VM, Rocha M. Targeting antioxidants to mitochondria: a potential new therapeutic strategy for cardiovascular diseases. Curr Pharm Des. 2007;13:845–863. doi: 10.2174/138161207780363077. [DOI] [PubMed] [Google Scholar]
- 61.Ross MF, Kelso GF, Blaikie FH, James AM, Cocheme HM, Filipovska A, Da Ros T, Hurd TR, Smith RA, Murphy MP. Lipophilic triphenylphosphonium cations as tools in mitochondrial bioenergetics and free radical biology. Biochemistry (Mosc) 2005;70:222–230. doi: 10.1007/s10541-005-0104-5. [of Special InterestThis original article introduces the concept that conjugation of compounds to lipophilic cations targets mitochondrial uptake.] [DOI] [PubMed] [Google Scholar]
- 62.Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, Sammut IA. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. Faseb J. 2005;19:1088–1095. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- 63.Smith RA, Porteous CM, Gane AM, Murphy MP. Delivery of bioactive molecules to mitochondria in vivo. Proc Natl Acad Sci U S A. 2003;100:5407–5412. doi: 10.1073/pnas.0931245100. [DOI] [PMC free article] [PubMed] [Google Scholar]