

Abstract
Lipid hydroperoxides (LOOHs) generated in cells and lipoproteins under oxidative pressure may induce waves of damaging chain lipid peroxidation near their sites of origin if O2 is readily available and antioxidant capacity is overwhelmed. However, recent studies have demonstrated that chain induction is not necessarily limited to a nascent LOOH’s immediate surroundings, but can extend to other cell membranes or lipoproteins by means of LOOH translocation through the aqueous phase. Mobilization and translocation can also extend the range of LOOHs as redox signaling molecules and in this sense they could act like the small, readily diffusible inorganic analogue, H2O2, which has been studied much more extensively in this regard. In this article, basic mechanisms of free radical- and singlet oxygen-mediated LOOH formation will first be reviewed, along with one-electron and two-electron LOOH reduction pathways and their biological consequences. The first studies to document spontaneous and protein-assisted LOOH transfer in model systems and cells will then be described. Finally, LOOH translocation will be discussed in the context of cytotoxicity vs. detoxification and also expanded effector action, i.e. redox signaling activity.
Keywords: Lipid peroxidation, Lipid Hydroperoxides, Hydrogen Peroxide, Biomembranes, Translocation, Lipid Transfer Proteins, Redox Signaling
Introduction: generation and possible fates of lipid hydroperoxides
Unsaturated lipids, including cholesterol (Ch), phospholipids (PLs), cholesteryl esters, and glycolipids in organized assemblies such as cell membranes and lipoproteins are prominent targets of oxidative modification [1,2]. This can occur non-enzymatically during a natural metabolic event such as mitochondrial electron transport or during exposure to exogenous chemical or physical agents, and typically develops under conditions of oxidative stress, i.e. when antioxidant defenses are overwhelmed [3]. Stress-induced lipid peroxidation could play a role in the development of atherosclerosis, neurodegeneration, cancer, and other disorders [1] . It may also be involved in the beneficial cytotoxic effects of oxidant-based chemotherapeutic and phototherapeutic drugs [1,4]. Like oxidative damage to proteins and nucleic acids, lipid peroxidation can be triggered by reactive oxygen species (ROS) such as hydroxyl radical (HO·), hydroperoxyl radical (HO2·), and singlet molecular oxygen (1O2), or by reactive nitrogen oxide species such as peroxynitrite (ONOO-) and nitrogen dioxide (·NO2) [1]. HO· is commonly generated by Fenton chemistry [5] and ONOO- by reaction of nitric oxide with superoxide [6]. Both HO· and ONOO- (as its conjugate acid ONOOH) are strong oxidants that can trigger free radical-mediated (chain) lipid peroxidation via abstraction of an allylic hydrogen from an unsaturated lipid, LH (Fig. 1). This might be a C-7 hydrogen in cholesterol or an sn-2 fatty acyl hydrogen in a phospholipid (PL). The resulting lipid radical (L·) with free electron delocalized over several carbons reacts rapidly with O2 to give a peroxyl radical (LOO·). The latter propagates peroxidation by abstracting hydrogen from a neighboring LH while being converted to a hydroperoxide (LOOH). 1O2 (1Δg O2) is typically generated by photodynamic action, i.e. sensitizer-mediated photoexcitation of ground state triplet O2 [7]. 1O2 can add directly to an unsaturated LH, converting it to LOOH with an allylic shift of the targeted double bound and retention of the LH hydrogen [4,7] (Fig. 1). By contrast, in an LOOH generated by HO· attack on a membrane LH, the oxygen atoms derive from ground-state O2 and the hydrogen from an adjacent LH.
Figure 1.
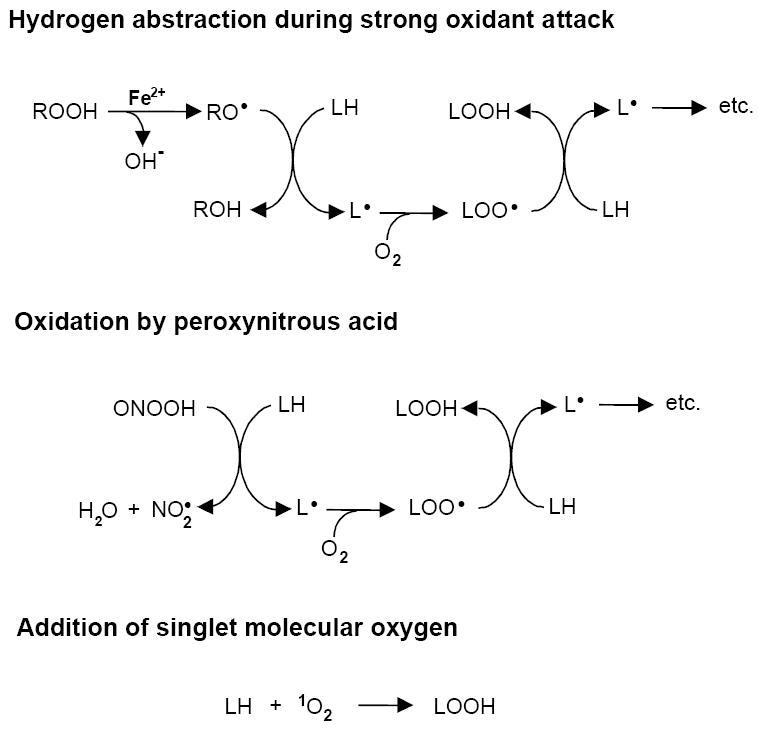
Three possible mechanisms of lipid peroxidation. The top scheme depicts initiation of free radical (chain) peroxidation by an oxyl radical (RO·) generated via Fenton-type reduction of a hydroperoxide (ROOH) by ferrous iron; when ROOH is H2O2, RO· is hydroxyl radical (HO·). The middle scheme depicts initiation by the non-radical oxidant peroxynitrous acid (ONOOH), which produces a radical, NO2·, which can also trigger chains via H-abstraction. The third scheme shows direct addition of 1O2 to LH (ene-addition) to give LOOH without free radical involvement.
Several different fates are possible for free radical- or 1O2-generated LOOHs. In the absence of reducing agents and/or catalytic metal ions, LOOHs will accumulate with stress duration (e.g. photodynamic dose) and in so doing perturb membrane structure/function due to their increased hydrophilicity. On the other hand, if reductants and redox-active iron are available, LOOHs can undergo Fenton-like one-electron reduction to highly reactive oxyl radical (LO·) intermediates. These might trigger peroxidation chains directly by abstracting hydrogens or might rapidly rearrange and autoxidize to epoxyallylic peroxyl radicals (OLOO·), which do this (Fig. 2). The latter mechanism is considered more likely, based on model studies with peroxidized fatty acids [8], although this has not yet been established for actual biomembrane systems. Chain peroxidation induced by one-electron reduction of a primary (or “priming”) LOOH generates new LOOHs which feed into the overall process (Fig. 2). These secondary reactions would expand and exacerbate the damaging effects of primary peroxidation alone, e.g. that produced by 1O2 attack or immediately following HO· attack [4,9]. It is important to note that interception of LOO· or OLOO· intermediates by chain-breaking antioxidants such as vitamin E also gives rise to LOOHs, but typically at a much lower rate than chain propagation reactions.
Figure 2.
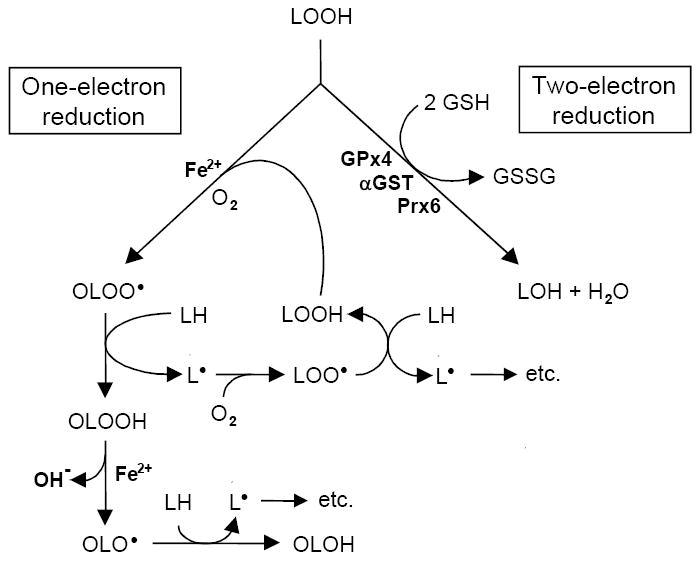
Scheme depicting two different routes of reductive LOOH turnover. Iron-catalyzed one-electron reduction accompanied by rapid O2 addition gives an epoxyallylic peroxyl radical (OLOO·) which triggers propagative free radical peroxidation as shown. For 1O2-generated LOOH, free radical chemistry would start here. Two-electron reduction, catalyzed by peroxidatic enzymes such as GPx4, α-GST, and Prx6 converts LOOH to redox-inactive LOH. One-electron turnover would exacerbate LOOH damage, whereas two-electron turnover would suppress it.
Newly formed LOOHs may also undergo enzyme-catalyzed two-electron reduction to relatively innocuous alcohol (LOH) products [10]. This is a detoxification process which acts in opposition to toxicity-enhancing one-electron reduction. In terms of cytoprotection, it is classified as a secondary (reparative) process to distinguish it from primary (preventative) reactions catalyzed by enzymes which intercept upstream ROS such as (superoxide dismutases) and H2O2 (catalase, glutathione peroxidases, peroxiredoxins). Since 1O2 has no known enzymatic scavengers, protection against its generated LOOHs must derive entirely from secondary reactions [4,10]. At least three different classes of intracellular enzyme have been implicated in reductive LOOH detoxification: GSH peroxidases, peroxiredoxins, and α-type GSH-S-transferase [11-13]. The selenium-dependent GSH peroxidases include GPx1 (the “classical” tetromeric isoform) and GPx4 (the monomeric isoform also known as PHGPx [11]. GPx1 and GPx4 both contain an active site selenocysteine that functions in the catalytic cycle, but differ significantly with regard to LOOH reactivity and specificity. GPx4 can act directly on phospholipid hydroperoxides (PLOOHs) in membranes, whereas GPx1 cannot unless sn-2 fatty acyl bonds are first hydrolyzed to liberate the peroxidized fatty acids [10,11]. Likewise, GPx4, but not GPx1, can catalyze the two-electron reduction of cholesterol hydroperoxides (ChOOHs) at the expense of GSH, either in Triton micelles, membrane bilayers, or lipoproteins [14]. Enzymatic transesterification may provide an additional pathway for eventual LOOH detoxification. In high density lipoprotein of plasma, for example, lecithin-cholesterol acyltransferase (LCAT) could transfer a peroxidized fatty acyl group from a PLOOH to cholesterol, giving a cholesteryl ester hydroperoxide which is sequestered from free radical turnover and subsequently inactivated by two-electron reduction [15]. Acyl group transfer from non-oxidized PL to a cholesterol hydroperoxide (e.g. 7-OOH) would likewise mediate peroxide detoxification by HDL. The degree of partitioning between the one- and two-electron reduction pathways could ultimately determine how a cell under oxidative stress responds to a LOOH challenge.
Compared with free radical precursors or products, LOOHs generated in membrane bilayers or lipoproteins should have relatively long lifetimes. Several years ago, Girotti et al. [16] predicted that this attribute, along with greater hydrophilicity compared with parent lipids, should allow LOOHs to depart their membranes of origin relatively rapidly and translocate to other membrane compartments (Fig. 3). A more localized variation of this phenomenon would be transbilayer (vectorial) LOOH translocation (Fig. 3), which can externalize certain cellular LOOHs for signaling purposes, one of the best studied examples being peroxidized phosphatidylserine (PSOOH) in cells destined for apoptosis [17-19]. Kagan et al. [19] have shown that upon oxidation, plasma membrane PS can act as a non-enzymatic “scramblase” that facilitates the externalization of both PSOOH and PS for recognition by macrophages. Intermembrane translocation of non-oxidized lipids such as cholesterol, phosphatidylcholine, and phosphatidylethanol has been studied extensively over the past thirty years [20-22]. As observed for the native lipids, one would expect LOOH transfer from one membrane to another to be favored by a strong negative concentration gradient and also by a relatively large acceptor compartment serving as a sink. In principle, LOOH transfer would be possible not only between membrane compartments in cells, but also between plasma lipoproteins, cells and lipoproteins, and plasma membranes of different cells (Fig. 3). For cholesterol and phospholipids, it is well established that intermembrane transfer at relatively low total lipid concentrations occurs via an aqueous transit pool rather than by collisions between donor and acceptor membranes [20]. Lipid departure from the donor membrane was typically found to be slow and rate limiting. Another key finding was that transfer can be accelerated by various intracellular lipid transfer proteins acting either as promoters of desorption from donor membranes or as delivery shuttles to acceptors [20, 23,24]. Transfer proteins for phosphatidylcholine, phosphatidylinositol, and cholesterol have been studied extensively [24,25]. One well-known example, sterol carrier protein-2 (SCP-2), also known as non-specific lipid transfer protein, facilitates the movement of cholesterol, fatty acids, and certain phospholipids [23-26]. In considering the occurrence of LOOH transfer in biological systems with its potential prooxidant vs. antioxidant and redox signaling implications, we predicted that many of the characteristics described for LH translocation would also apply for LOOHs [16].
Figure 3.
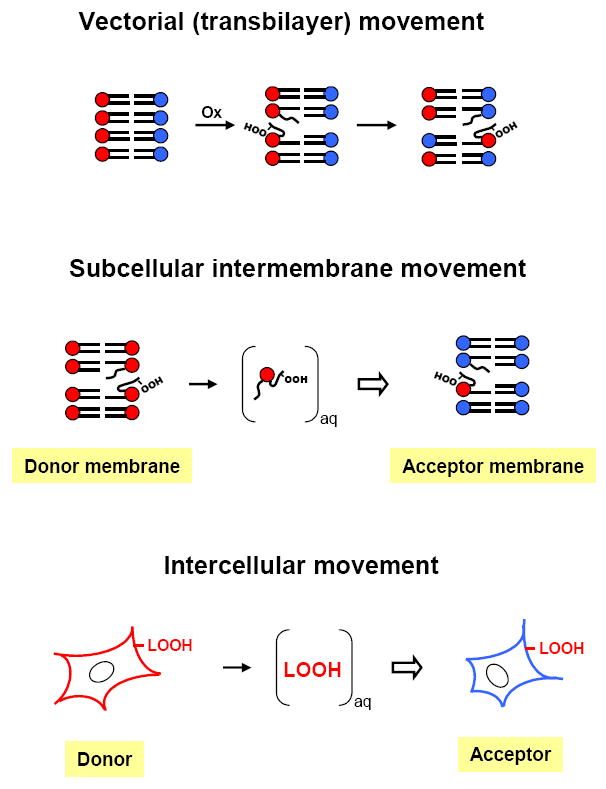
Three different modes of LOOH translocation: (a) vectorial in any given membrane (LOOH moves from one leaflet to the other); (b) intermembrane (LOOH desorbs slowly from a donor membrane, moves through the aqueous phase and is rapidly taken up by an acceptor membrane); (c) intercellular (LOOH desorbs from the plasma membrane of a donor cell and moves via the aqueous transit pool to the plasma membrane of an acceptor cell).
Spontaneous translocation of cholesterol-derived hydroperoxides
The first studies to demonstrate the existence of spontaneous intermembrane LOOH transfer were carried out using photodynamically peroxidized erythrocyte ghosts as donors, small unilamellar liposomal vesicles (SUVs) as acceptors, and cholesterol (Ch) and cholesterol-derived hydroperoxides (ChOOHs) as analytes [16]. Prior to photoperoxidation, ghost Ch was 14C-labeled to allow simultaneous monitoring of [14C]Ch and [14C]ChOOH by high-performance thin layer chromatography with phosphorimaging detection (HPTLC-PI). Ch and ChOOHs were readily separated by the HPTLC used, but not individual ChOOHs from one another [16]. SUV acceptors were typically in 5-10-fold lipid molar excess over ghost donors to assure that most of the transferred analyte was in this compartment at equilibrium. Transfer incubation was carried out under redox-silent conditions to prevent turnover of existing LOOHs or formation of new ones. At various incubation times, donors and acceptors were separated by centrifugation; lipids were extracted from the latter and analyzed by HPTLC-PI. Using this model system, Vila et al. [16,27] showed that the apparent first-order rate constant (k) for overall ChOOH transfer at 37 °C was at least 60-times greater than that for parent cholesterol (Ch). When departure from ghosts was tracked, the same k value was obtained for each analyte, confirming that no other process besides translocation was taking place. Transfer rates were found to be independent of SUV concentration over a wide range [27]. This indicated that for ChOOH transfer, like classical Ch [20], desorption from the donor membrane is rate-limiting and that diffusion through the aqueous phase is involved rather than membrane collision (Fig. 3). With SUV donors, transfer rate varied inversely with vesicle diameter, suggesting more facile departure with decreased lipid packing density. The ChOOH used in these experiments was a mixture of at least five different positional isomers: 3β-hydroxy-5α-cholest-6-ene-5-hydroperoxide (5α-OOH); 3β-hydroxycholest-4-ene-6α-hydroperoxide (6α-OOH); 3β-hydroxycholest-4-ene-6β-hydroperoxide (6β-OOH); 3β-hydroxycholest-5-ene-7α-hydroperoxide (7α-OOH); and 3β-hydroxycholest-5-ene-7β-hydroperoxide (7β-OOH). 5α-OOH, 6α-OOH, and 6β-OOH are generated exclusively by singlet oxygen (1O2) attack on Ch [28,29], whereas 7α-OOH and 7β-OOH derive from free radical attack or possibly 5α-OOH rearrangement [30]. The content order of these different isomers in photoperoxidized Ch is typically: 5α-OOH≫6α/β-OOH>7α/7β-OOH. In subsequent experiments, the transfer kinetics of individual isomers were compared [27], using reverse-phase high-performance liquid chromatography with a C18 column and mercury cathode electrochemical detection [HPLC-EC(Hg)] for their separation and analysis [31]. It was found that the apparent first-order rate constants for ghost-to-SUV transfer of these isomers decreased in the following order: 7α/7β-OOH ≫ 5α-OOH ≫ 6α-OOH>6β-OOH. (7α/7β-OOH represents an undefined mixture of these epimers, which could not be separated by the HPLC used.) Importantly, the rank order of these rate constants was the exact opposite of that observed for the reverse-phase HPLC retention times of these species [27]. Thus, 7α/7β-OOH with the shortest retention time (greatest hydrophilicity) had the highest rate constant, whereas 6β-OOH with the longest retention time (lowest hydrophilicity) had the lowest. A likely explanation (similar to that used for explaining why kCh is much greater than kPL (20)), is that the ChOOH desorption rate increases with increasing hydrophilicity, resulting in proportionately more peroxide in the aqueous transit pool [16,27]. The activation energy for ghost-to-SUV transfer of ChOOHs was found to be approximately the same as that of Ch (82 ± 3 kJ/mol) [27]. This indicates that even though the rate constant for ChOOH desorption greatly exceeds that for Ch, the net energy required for departure from the membrane surface is approximately the same. Intuitively, one might expect that the activation energy for ChOOH transfer to be lower than that for Ch because the latter is less hydrophilic. However, the A-or B-ring hydroperoxyl group of each ChOOH isomer is not too distant from the A-ring hydroxyl group. Thus, although the ChOOHs are more polar than Ch, they retain most of its amphiphilic properties, with the same hydrophobic “tail” portion and relatively rigid hydrophilic “head” portion. On this basis, it might not be surprising that Ch and the ChOOHs have similar energy requirements for transfer activation and desorption, where activation refers to movement of the intercalated hydrophobic portion to the bilayer-water interface. Based on hydrophobic effect considerations, however, ChOOH release into the aqueous phase would bring the system to a lower free energy than Ch release, and this would be reflected in the observed greater off-rates for ChOOHs [27].
There is now solid evidence that ChOOHs not only move from one membrane to another, but also from membranes to lipoproteins and vice versa. Using freshly prepared low density lipoprotein (LDL) with barely detectable LOOH, Vila et al. [32] showed that the LDL could progressively acquire ChOOHs from photoperoxidized erythrocyte membranes during incubation with the latter. It was found that the transfer rate constants decreased in the same rank order as observed with membrane acceptors, i.e. 7α/7β-OOH ≫ 5α-OOH ≫ 6α-OOH>6β-OOH. Reverse transfer to non-oxidized membranes was also demonstrated, but, interestingly, this occurred much more rapidly than forward transfer (~3-times faster for the overall ChOOH population). It is relevant to note that Ch is also known to depart more rapidly from LDL than from intact red blood cells (k ~0.0.95 h-1 vs. 0.15 h-1 at 37 °C) [20]. The LDL particle is ~250-times smaller in diameter than the red cell, and its Ch and PL are located in an outer monolayer rather than bilayer. Consequently, LDL’s outer lipids would be more loosely packed than red cell membrane lipids and this could account for the more rapid Ch departure. Faster ChOOH release from LDL than from red cell ghosts [32] can be explained along similar lines. The studies described [32] were the first to demonstrate that non-esterified ChOOHs can spontaneously move from membranes to LDL and vice versa. However, earlier work of related interest demonstrated that cholesteyl ester hydroperoxides (CEOOHs) can translocate between lipoproteins. For example, cholesteryl linoleate peroxidized mainly in its fatty acyl moiety (CLnOOH) was shown to move readily from high density lipoprotein (HDL) to hepatoma cells in serum-free medium [33]. The initial rate of CLnOOH uptake by cells was found to be ~9-times greater than that of unoxidized lipid (CLn) and at least 40-times greater than that of HDL itself, suggesting some type of selective uptake mechanism for CLnOOH species [34,35]. Once internalized, CLnOOH was rapidly metabolized via hydrolysis and peroxide reduction, consistent with the hypothesis that transfer of circulating CEOOHs from HDL to liver plays a key role in their detoxification [33-35]. Another study showed that CLnOOH can translocate from LDL to HDL at a rate strongly enhanced by cholesteryl ester transfer protein, presumably acting as a shuttle as in the case of unoxidized CLn [36]. ChOOH and CEOOH translocation between lipoproteins or lipoproteins and cell membranes, as described [32-36], may play an important role in modulating the potential toxicity and redox signaling activity of these species in the circulation.
Spontaneous translocation of phospholipid-derived hydroperoxides
Other studies carried out using photoperoxidized erythrocyte ghosts as donors and SUVs as acceptors showed that phospholipid-derived hydroperoxides (PLOOHs) are also capable of spontaneous intermembrane movement [32]. Liposomal acquisition of four different resolvable PLOOH families was monitored by HPLC-EC(Hg), using an amino (LC-NH2) column. These included hydroperoxides of phosphatidylcholine, phosphatidylethanolamine, phosphatidylserine, and sphingomyelin (PCOOH, PEOOH, PSOOH, and SMOOH, respectively). All of these families exhibited apparent first-order transfer kinetics, the rate constants for PCOOH, PEOOH, and PSOOH being essentially the same and approximately 4-times greater than that for SMOOH [32]. As observed for ChOOHs vs. Ch [16,27], PLOOHs translocated much more rapidly than their respective parent PLs, the k value for PCOOH, for example, being at least 30-times greater than that of PC (monitored with ghosts that had been initially labeled with a [14C]PC) [32]. In related work, Vila et al. [32] showed that ghost membrane PLOOHs can also migrate spontaneously to LDL, reaching an equilibrium far in favor of the lipoprotein when it was present in 5-10-fold lipid molar excess over the ghosts. The transfer rate constants for PCOOH, PEOOH, PSOOH and SMOOH were essentially the same as observed with SUV acceptors, again indicating that transfer kinetics are relatively insensitive to the physical properties of the acceptor compartment. Importantly, the PLOOHs collectively moved much more slowly than the ChOOHs, in keeping with the known slow transfer of PLs compared with Ch [20]. As an illustration, the first-order rate constant for PCOOH transfer was found to be approximately 1/5 that of 5α-OOH and 1/10 that of 7α/β-OOH. In most mammalian cell membranes except the plasma membrane, PL content and degree of unsaturation (oxidizability) greatly exceeds that of Ch so that actual transfer rates of PLOOHs may not necessarily be lower than those of ChOOHs. On the other hand, ChOOHs are less susceptible to enzymatic reduction (e.g. by GPx4) than PLOOHs [10,14], resulting in longer ChOOH lifetimes for translocation. Thus, the actual extent of intermembrane ChOOH vs. PLOOH transfer in an oxidatively stressed cell depends on a number of complex factors, including relative content of oxidizable lipid in the donor membrane, oxidant accessibility, peroxide hydrophilicity, and peroxide susceptibility to one- or two-electron turnover in the donor and transit compartments.
Prooxidant effects of translocated LOOHs
Movement of LOOHs from one membrane compartment to another could greatly expand their range of signal transduction and toxic damage, a prospect that was recognized only recently. Heretofore, these species were largely believed to exert their effects while still situated on their membranes of origin, e.g. one-electron turnover of LOOHs produced by membrane-localized 1O2 [4]. Free radical-mediated prooxidant effects might accrue if the acceptor compartment is relatively rich in redox-active iron and deficient in chain-breaking antioxidants such as α-tocopherol or LOOH-detoxifying enzymes such as GSH-dependent GPx4 (Fig. 4). In cells, LOOHs might migrate from mitochondria, which are under relatively high oxidative pressure, to the nucleus, where genomic DNA could be damaged by LOOH-derived radicals or non-radical breakdown products [37]. The first experiments to demonstrate the damaging prooxidant effects of translocated LOOHs were carried out using photoperoxidized erythrocyte ghost donors and [14C]Ch-containing SUV acceptors that were exposed to ascorbate and a lipophilic iron complex to trigger one-electron LOOH reduction [16]. The [14C]Ch served as a resident probe for free radical-mediated chain peroxidation, as determined by HPTLC-PI analysis of characteristic [14C]Ch oxidation products (ChOx) [38]. The intensity of chain peroxidation in SUV acceptors increased progressively with transfer incubation time, most of it being due to ChOOHs rather than PLOOHs [37]. The most important triggering ChOOH was 5α-OOH, the major adduct of photogenerated 1O2. No significant ChOx were detected in SUVs treated with iron/ascorbate after transfer preincubation with non-photoperoxidized ghosts, indicating that the reaction was entirely dependent on transfer priming.
Figure 4.
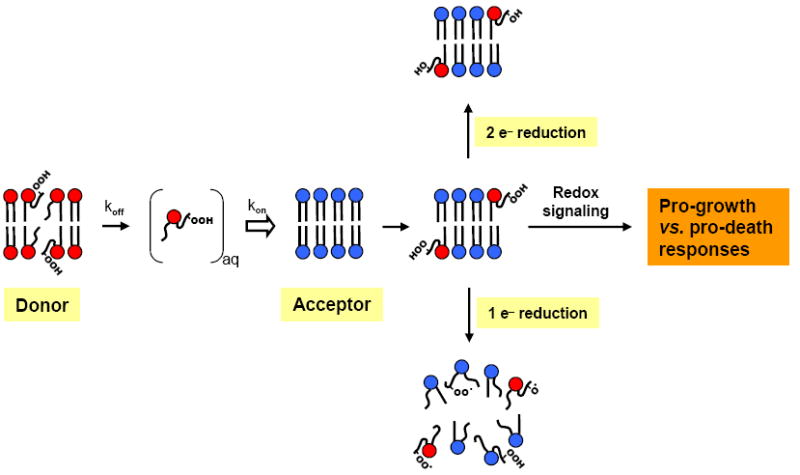
Possible fates or effects of intermembrane-translocated LOOHs. In the acceptor compartment, a transferred LOOH could undergo one-electron reduction, thus initiating damaging chain peroxidation. Alternatively, two-electron detoxification could occur if antioxidant capacity is sufficient, e.g. GSH/GPx4. For PLOOHs, resulting PLOHs could be hydrolyzed and reacylated to restore PL status. Two-electron reactions resulting in pro-death or pro-survival signaling cascades are also possible, e.g. −CySH oxidation to −CySOH in sensor proteins.
Preexisting LOOHs are known to increase the sensitivity of LDL to metal ion-catalyzed chain peroxidative damage, which, in vivo, could increase its atherogenic potential [39]. Low levels of LOOHs (mainly CEOOHs) are invariably detected in freshly isolated LDL, even when extreme care is used to prevent oxidation during preparation, so it is believed that they accumulate in vivo [40]. How this occurs has been the subject of much investigation and debate. It has been reported that LDL can be oxidatively modified by reactive oxygen or reactive nitrogen oxide species produced by leukocytes it encounters in the circulation [41,42]; in this case, preexisting LOOHs would derive mainly from LDL lipids. Other evidence suggests that cellular lipoxygenases, e.g. 15-LOX of monocytes or macrophages, play a crucial role [43]. This could reflect some level of physical contact between the LDL particles and cells [43], with LDL lipids having access to the LOX active site. Another possibility is translocation of LOX-generated LOOHs from cells to LDL. This would not necessarily require cell-to-LDL contact, but could occur via an aqueous transit pool as demonstrated for an oxidized red cell membrane/LDL model; see above [32]. This model is physiologically meaningful because erythrocytes and also activated phagocytic cells are under relatively high peroxidative pressure in the circulation and this could be relieved by LOOH transfer to LDL and other lipoproteins. Erythrocytes are also deficient in GPx4, which would allow LOOHs to accumulate and translocate. Vila et al. [32] showed that erythrocyte ghost-derived LOOHs greatly increased the sensitivity of LDL to Cu2+-induced free radical peroxidation, as assessed by conjugated diene, ChOX, and CEOOH formation . Although this was not reported, it is likely that transfer-acquired LOOHs also triggered the oxidative modification of the apoB-100 protein, a pro-atherogenic reaction associated with increased LDL recognition by the scavenger receptor of vascular macrophages [39]. In addition to propagating chain reactions in an acceptor particle, downstream LOOHs arising from LDL lipids might themselves translocate to other particles in relay-like fashion, thus extending damage far beyond the initial acceptor. Relatively rapid departure of ChOOHs and CEOOHs from oxidizing LDL would tend to favor this, and plasma lipid transfer proteins might participate [36].
Up to this point, we have focused on the prooxidant potential of LOOH translocation through long-range dissemination of oxidative damage and stress signaling. However, it is possible that this process could play an antioxidant role under certain circumstances. In oxidatively stressed cells, for example, directed LOOH translocation to subcellular sites where antioxidants such as GSH and GPx4 are relatively abundant might provide a means for efficient LOOH inactivation (Fig. 4). Whether this can take place in cells has not yet been documented. However, there is strong in vitro evidence that protein-mediated CEOOH transfer from LDL to HDL and from HDL to liver cells facilitates reductive CEOOH detoxification [33-35]. Such transfer in vivo could play an important anti-atherogenic role. Thus, LOOH translocation could have either a prooxidant (stress expanding) or antioxidant (stress containing) outcome, depending on conditions in the acceptor environment. At present, little is known about the actual determinants of either outcome in a biological system, e.g. an oxidatively stressed cell.
Protein-facilitated LOOH transfer
Natural (non-oxidized) lipids, particularly those in the phospholipid family, translocate relatively slowly on their own, but this can be accelerated by proteins with lipid transfer capabilities [23]. These proteins play a crucial role in lipid metabolism and in membrane and lipoprotein biogenesis/homeostasis. Intracellular transfer proteins with varying specificities for various phospholipids, fatty acids, and cholesterol have been identified [23-26]. A well studied example is sterol carrier protein-2 (SCP-2), also known as non-specific lipid transfer protein, which not only facilitates the intermembrane transfer of Ch and other sterols, but also various fatty acids, fattyl-CoAs, and phospholipids (26,44,45). SCP-2 is a small (13.2 kDa) translation product of a fusion gene encoded for 58 kDa SCP-x (45 kDa of which represents a peroxisomal 3-ketoacyl-CoA thiolase) and 15 kDa pro-SCP-2 [44]. Mature SCP-2 derives from post-translational proteolytic cleavage of pro-SCP-2, although some formation by cleavage at the SCP-x level appears possible. Most of the SCP-2 of mammalian cells resides in peroxisomes, but significant amounts can also be found in the cytosol, lysosomes, and mitochondria [44]. Numerous studies with model systems have shown that SCP-2 facilitates the transfer of various sterols and phospholipids from donor to acceptor membranes [44,45]. One possible mechanism involves interaction of the basic N-terminal domain with the negative donor membrane surface and binding of an available lipid, followed by detachment, migration to the acceptor membrane, and unloading [46]. However, there is also evidence that SCP-2 can bind at least some desorbed lipids in the aqueous compartment and thereby accelerate delivery in this fashion [44,47].
Relatively rapid LOOH transfer from erythrocyte ghost membranes to liposomal (SUV) membranes was recently shown to be further enhanced by either natural bovine liver SCP-2 or human recombinant SCP-2 [48]. This was the first reported evidence for such action by a cellular lipid transfer protein. For several ChOOH regio-isomers, the net rate constants for SCP-2-accelerated transfer (i.e. spontaneous-corrected) decreased in the same rank order as observed for spontaneous transfer, i.e. 7α/7β-OOH > 5α-OOH > 6α-OOH>6β-OOH. The net k values with SCP-2 present increased progressively and dramatically with increasing ChOOH hydrophilicity, suggesting better protein recognition of the more water-soluble species [48]. To illustrate this point for 7α/β-OOH (the most hydrophilic) vs. 6β-OOH (the least hydrophilic), the k7-OOH/k6β-OOH ratio was found to be ~8 for spontaneous transfer, but >60 for net SCP-2-enhanced transfer. SCP-2 also accelerated PLOOH transfer from ghosts to SUVs, the net k value for PCOOH, PEOOH, or PSOOH being 4-6-fold greater than that for spontaneous transfer, but only 1/5 that of 5α-OOH and 1/10 that of 7α/β-OOH [48]. Thus, the net rate constants among the different PLOOHs differed by no more than ~50%, whereas the corresponding ChOOH values differed by a wide margin, up to nearly 70-fold. This might be explained by hydrophilicity considerations, the PLOOHs being similar to one another in this regard, but the ChOOHs differing substantially. As previously observed with non-oxidized Ch or PLs [44], SCP-2-facilitated LOOH transfer was promoted by the following donor membrane properties: increasing PL unsaturation, and increasing net negative charge, e.g. from PS [48].
With regard to extracellular lipid transfer proteins, Christison et al. [36] have shown that the serum cholesteryl ester transfer protein strongly stimulates the rate of CEOOH movement from LDL to HDL, suggesting an important role for this protein in anti-atherosclerotic defense. Whether other serum lipid transfer proteins act similarly on other LOOHs remains to be established.
LOOH translocation in cellular and subcellular systems, and consequences thereof
Transfer-dependent cytotoxicity of ChOOHs has been demonstrated in vitro on COH-BR1 cells, a subline of breast epithelial tumor cells [27]. These cells express little, if any, GPx4 [49], making them unusually sensitive to LOOH pressure. Vila et al. [27] found that the time-dependent degree of cell killing induced by three different SUV-donated ChOOHs decreased in parallel with their rates of spontaneous transfer uptake, i.e. 7α-OOH >5α-OOH >6β-OOH, thus indicating that transfer-limited cytotoxicity is possible. Although prooxidant and potentially lethal ChOOH translocation has important implications for all types of oxidative challenge, it has special meaning in 1O2-mediated photodynamic reactions. Special attention should be given to 5α-OOH in this regard. In a cell under 1O2 attack, e.g. a skin fibroblast exposed to UVA radiation (320-400 nm), 5α-OOH would be generated much more rapidly than any other ChOOH, but detoxified relatively slowly by the GSH/GPx4 system [10]. Thus, one would expect 5α-OOH to have a longer lifetime and accumulate to higher steady state levels than other ChOOHs, and possibly also PLOOHs, which are much better GPx4 substrates than ChOOHs [10,14]. Under these conditions, intermembrane transfer of 5α-OOH would be favored and this could greatly expand its range of activity as a toxic oxidant and/or signaling molecule. It is important to point out that 5α-OOH can undergo allylic rearrangement to 7α-OOH [29]. This becomes more favorable as the extent of membrane peroxidation increases, but is usually insignificant when <5 mol % of the lipid is oxidized, as in a moderately stressed cell [10]. 5α-OOH to 7α-OOH rearrangement is also repressed in relatively polar environments [29], as would be the case for desorbed 5α-OOH during transit in the aqueous compartment.
Translocation between subcellular compartments plays a vital role in the metabolism of various natural lipids (20,21]. For example, Ch movement from endoplasmic reticulum to mitochondria in adrenocortical cells is crucial for steroid hormone biosynthesis and SCP-2 has been implicated in this [44]. Under oxidative stress conditions, SCP-2-mediated transfer/exchange of ChOOHs and other LOOHs might exacerbate oxidative damage in an acceptor organelle, e.g. mitochondrion. Vila et al. [48] first tested this hypothesis using a model system consisting of [14C]7α-OOH-bearing SUVs and isolated mouse liver mitochondria. Two parameters were examined: (a) HPTLC-PI-assessed kinetics of spontaneous and SCP-2-enhanced uptake of [14C]7α-OOH by the mitochondria; and (b) effect of peroxide acquisition on mitochondrial membrane potential (ΔΨ), as detected with the fluorescent probe rhodamine 123 (Rh123). Control SUVs containing the two-electron reduction product, [14C]7α-OH, instead of [14C]7α-OOH were studied alongside. It was found that 7α-OOH translocated to mitochondria with apparent first-order kinetics which could be strongly enhanced by SCP-2 [48]. SCP-2 also accelerated 7α-OH uptake, the rate at the same analyte and protein concentration being about one-third that of 7α-OOH. Cytotoxic relevance was demonstrated by showing that transfer-acquired 7α-OOH caused a loss of ΔΨ that was significantly increased by SCP-2 in time- and dose-dependent fashion [48]. Identical incubation with 7α-OH-containing SUVs produced a relatively insignificant drop in ΔΨ, consistent with this oxysterol being redox-inactive. Importantly, when the mitochondria were treated with a lipophilic iron complex in low concentration prior to exposure to 7α-OOH-SUVs plus SCP-2, the ΔΨ loss was significantly enhanced, along with a more rapid conversion of incoming 7α-OOH to 7α-OH, which was likely due to iron-catalyzed one-electron reduction. These findings are consistent with a mechanism in which one-electron turnover of translocated 7α-OOH induced damaging chain peroxidation in mitochondrial membranes, thus impairing the maintenance of membrane potential and other key functions. It is apparent from these findings that by trafficking LOOHs, SCP-2 (and possibly other lipid transfer proteins) might exacerbate cell injury under oxidative stress conditions unless the LOOHs are delivered to sites at which efficient detoxification occurs (see Fig. 4).
This notion is supported by more recent studies in which an SCP-2-overexpressing cell line was challenged with an exogenous LOOH. A transfectant clone (SC2A) of rat hepatoma cells stably expressing ~10-times more immunodetectable SCP-2 than a vector control (VC) was found to be substantially more sensitive to thiazolyl blue (MTT)-assessed killing induced by liposomal 7α-OOH than VC [50]. 7α-OOH is a major intermediate generated by free radical peroxidation of plasma membrane Ch and this system could model its transfer uptake from activated leukocytes at tumor inflammatory sites. SCP2 hypersensitivity was demonstrated in both increasing [7α-OOH]-fixed time and fixed [7α-OOH]-increasing time format. SC2A and VC cells were less sensitive, but equally so, to the non-lipid hydroperoxide, tert-butyl-OOH, implying that the 7α-OOH effects were SCP-2-specific [50]. The initial rate of 7α-OOH uptake (measured as [14C]7α-OOH radioactivity) was found to be ~40% greater for SC2A cells than VC, an effect attributed to faster internalization rather than mere association with the cell surface. Overexpressing cells accumulated [14C]7α-OOH radioactivity more rapidly in mitochondria than in other subcellular compartments, suggesting possible preferential SCP-2-mediated delivery to mitochondria. Faster internalization and mitochondrial targeting of [14C]7α-OOH was accompanied by more extensive damaging turnover of the peroxide, as demonstrated by more rapid (a) HPTLC-PI-detected accumulation of [14C]7α-OH and other one-electron reduction products, (b) buildup of cellular ROS, as determined with 2’,7’-dichlorofluorescin diacetate (DCFH-DA), (c) mitochondria-localized chain lipid peroxidatin, as visualized with C11-BODIPY, (d) loss of mitochondrial ΔΨ, as probed with JC-1 fluorophore, and (e) initiation of Hoechst-detectable apoptosis [50]. Thus, SCP-2-overexpressing cells clearly showed greater sensitivity to an incoming LOOH, most of the toxic, pro-apoptotic activity occurring in mitochondria. This was the first study to demonstrate that subcellular peroxidative stress damage can be selectively targeted and exacerbated by a lipid transfer protein [50].
Recent experiments have shown that isolated recombinant SCP-2 strongly inhibits the two-electron reduction of peroxidized linoleic acid (LAOOH) by the GSH/GPx1 system (Girotti et al., unpublished data). Lysozyme, which exhibits no lipid binding or transfer properties, had no effect on LAOOH reduction. Thus, association with SCP-2 appears to have protected LAOOH from reductive inactivation, presumably because of hindered access of GSH and/or GPx1. Whether SCP-2-bound LAOOH or other LOOHs would also be resistant to radical-producing one-electron reduction remains to be established, but if so, these findings collectively would argue that a lipid transfer protein not only accelerates LOOH translocation, but prolongs LOOH lifetime in transit.
Role of LOOH translocation in signal transduction: comparison with H2O2 signaling
Cellular hydroperoxides in general, including ubiquitous H2O2 generated by primary oxidative reactions and LOOHs generated secondarily have long been known for their damaging prooxidant effects associated with pathologies such as atherogenesis and neurodegeneration. It is becoming increasingly clear, however, that these species can also function as important regulators of signal transduction pathways in prokaryotic and eukaryotic cells [51,52]. Such signaling could result in increased proliferation on the one hand or programmed cell death (apoptosis) on the other, depending on cell type, peroxide pressure, and a number of other variables [51]. As already mentioned, transbilayer externalization of PSOOH in cells undergoing oxidant-induced apoptosis also plays an important signaling role, viz. in macrophage recruitment [17-19]. Hydrogen peroxide is the most widely studied peroxide known to be involved in redox signaling, and much more is known about H2O2 signaling than that of any other peroxides, including LOOHs. When applied in low concentration ranges (0.1-10 μM) to cultured mammalian cells, H2O2 can elicit pro-growth/survival responses often accompanied by transcriptional activation of antioxidant genes [50]. This came as somewhat of a surprise when first discovered [53], given H2O2’s more widely known cytotoxic effects. At higher concentrations, e.g. 10-100 μM, H2O2 can induce stress signaling that results in growth arrest and apoptotic cell death with minimal initial disruption of cell metabolism or membrane integrity. Above these levels, i.e. >0.1 mM, H2O2 can produce an oxidative stress that overcomes or negates more restrained signaling activity, resulting in membrane disruption and necrotic death. This is typically associated with metal ion-catalyzed reduction of H2O2 to HO·, which triggers gross non-specific damage such as free radical-mediated lipid peroxidation. Actual transition points for these various responses vary with cell type as determined by factors such as content and distribution of chemical and enzymatic antioxidants [51,52]. H2O2 signaling operates via the intermediacy of “sensor” molecules, which are either activated directly by reaction with H2O2 or indirectly by some mediator (e.g. a peroxidase) which reacts with H2O2 [51]. The most extensively studied H2O2 sensor to date is the prokaryotic transcriptional activator OxyR. OxyR controls intracellular peroxide tone by regulating the expression of several antioxidant genes (including alkyl hydroperoxide reductase, glutathione reductase, and glutaredoxin) as well as its own expression [54,55]. Activation of tetrameric OxyR by H2O2 is triggered by oxidation of a specific cysteine residue in each subunit (CySH 199) to a sulfenic acid derivative (CySOH), followed by reaction with CySH 208 to give a disulfide linkage [56]. This produces a conformational change that results in transcriptional activation, reversal of which is effected by glutaredoxin-1-catalyzed reduction of the disulfide. The key determinant of the highly selective oxidation at CySH 199 is its low pKa compared with ordinary CySH residues, meaning that it exists predominantly in the thiolate form, which is more susceptible to oxidation than the thiol form [56]. Site-specific oxidation of CySH to CySOH has emerged as a recurring mechanism in CySH-based H2O2 signaling [51,52]. A more recently described signaling mechanism not based on CySH oxidation pertains to the bacterial PerR transcriptional repressor [57,58]. PerR coordinates iron in a His-rich regulatory domain, making these His residues highly sensitive to oxidation by H2O2, another example of a site-specific effect on a sensor molecule [51]. The reaction in this case is presumably mediated by Fenton-generated hydroxyl radical, resulting in a conformational change in PerR that leads to derepression of genes encoded for catalase and other antioxidant enzymes [58].
Eukaryotic cells also exhibit various modes of H2O2-mediated redox signaling, some of which have been partially characterized [51,52]. The level of complexity for eukaryotes is typically much greater than that observed for prokaryotes. One of the most widely studied mammalian cell examples involves the two cysteinyl residues in the -CySH-X-X-CySH- active site of thioredoxin (Trx) or classical glutaredoxin (Grx) [59-61]. The CySH nearer to the N-terminus is surface-exposed and has an abnormally low pKa, making it readily oxidizable to CySOH by H2O2. Once formed, CySOH reacts rapidly with the proximal CySH to give an intramolecular disulfide bond. Conversion of fully oxidized Trx back to the fully reduced form is catalyzed by thioredoxin reductase, whereas Grx is regenerated by the GSH/glutathione reductase system [59,60]. A well known example of how Trx’s redox cycle plays a regulatory role pertains to ASK1, the primary activating kinase in the JNK and p38 stress-activated protein kinase pathways that typically signal for pro-apoptotic gene expression [62]. Fully reduced Trx binds near the N-terminus of ASK1, thereby preventing its autophosphorylation-activation and marking it for ubiquitination and degradation [60,63,64]. On the other hand, -S-S-linked Trx formed by oxidative stress/cytokine-induced increases in [H2O2] dissociates from ASK1, allowing it to become activated for induction of a stress-signaling cascade. A similar mechanism applies to Grx, although reduced Grx inactivates ASK1 by binding near its C-terminus rather than N-terminus [60,65]. Thus, Trx and Grx can act as sensor proteins, their active site CySH residues serving as “redox switches” for regulating ASK1-mediated gene expression under normal vs. oxidative/nitrosative stress conditions. Protein tyrosine phosphatases (PTPs) are another important example of redox-regulated sensor proteins in mammalian cells. PTPs play a key role in the regulation of tyrosine-dependent kinases, including stress-activated protein kinases and growth factor receptors, which are active in their phosphorylated tyrosine form and inactive in dephosphorylated form [51]. As in the case of Trx and Grx, PTPs typically contain a low pKa CySH in their active sites which can be either directly or indirectly oxidized to CySOH by H2O2. There is recent evidence that certain peroxidized peptides and proteins can also accomplish this [66]. The CySOH reacts readily with a neighboring CySH or backbone amide nitrogen to fully, yet reversibly, inactivate the PTP, thus resulting in greater activity of target phosphoproteins [51,52]. In the case of JNK, for example, this mechanism may be a key determinant of proapoptotic signaling under stress conditions [52]. It is of interest to note that selenocysteine (Sec) analogues of Trx have recently been described (SelL proteins), which contain a -Sec-X-X-Sec-active site motif instead of -CySH-X-X-CySH- [67]. SelL proteins are typically found in aquatic organisms and are redox-active like Trx, complete oxidation converting them to the diselenide form [67]. Whether these proteins also serve as redox sensors in signaling pathways has not yet been established.
Hydrogen peroxide is similar to H2O in various physical properties and diffuses readily in aqueous environments [68]. Like LOOHs, H2O2 is a non-radical ROS and on this basis may persist longer in cells than , for example. As supporting evidence, the half-lives of H2O2 and in activated lymphocytes have been reported to be ~1 ms and ~1 μs, respectively [69]. Relative longevity could be a key factor in H2O2’s distal signaling activity, e.g. ability to delocalize from a site of generation (near an activated NADPH oxidase complex on a plasma membrane, for example) and move to other sites where it might activate resident sensor proteins. Translocation signaling such as this might not only occur within a given cell, but also between cells (e.g. activated macrophages and tumor cells), in which it takes on the appearance of a “bystander effect” [70,71]. Of course, the signaling lifetime of H2O2 will depend on the distribution of and access to detoxifying enzymes such as catalase, glutathione peroxidases, and peroxiredoxins, i.e. how much of the peroxide is inactivated in transit before it reaches a sensor site (cf. Fig. 4). Encounters with redox-active metal ions, e.g. appropriately chelated ferrous iron, would also reduce H2O2’s signaling range, but in this case secondary mobile species could arise via Fenton chemistry, e.g. LOOHs from HO· attack on membrane lipids (cf. Fig. 4). Although H2O2 is known to traverse biological membranes, it does not do this by moving through the bilayer per se, at least at any significant rate due to its high polarity [67]. Instead, as indicated by recent evidence [67], H2O2 moves preferentially through transmembrane protein channels previously known to facilitate H2O passage, the so-called aquaporins. Aquaporins are typically found in plasma membranes of animal cells, but at least one homologue, AQP8, is known to exist in the mitochondrial inner membrane [72]. In addition to handling H2O, AQP8 is postulated to be an export portal for H2O2 generated in mitochondria [68], a major source of this species in aerobic cells. Although much is known about H2O2-mediated stress signaling as a general phenomenon, key aspects such as the determinants of its origin, lifetime, and subcellular movement/distribution are still not clearly established for any given cell type under stress.
Even less is known about LOOHs in these respects. However, the recent recognition that an LOOH’s action may not be confined to where it originates, but may be “broadcasted” by spontaneous or protein-mediated translocation [16,27,32,48] has opened up many possibilities for disseminated redox signaling that could resemble those described for H2O2. Relatively short range LOOH translocation is exemplified by the externalization of plasma membrane PSOOH in oxidatively stressed cells fated for apoptosis [17-19]. Externalized PSOOH signals for recognition by macrophages expressing scavenger receptors such as CD36, which are required for engulfment of the apoptotic cells [73]. Since fatty acid-, phospholipid-, and cholesterol-derived hydroperoxides are much more massive than H2O2, their spontaneous diffusion coefficients will be lower. Intermembrane movement would be slower still if an LOOH is carried by a transfer protein. However, the signaling advantages in this case would be (a) longer lifetime due to sequestration (see above) and (b) site-specific targeting to elicit a response, neither of which applies for free diffusion of LOOHs or H2O2. It is important to note that H2O2 can be rapidly decomposed by various intracellular enzymes that it encounters, including catalase, selenoperoxidases, and peroxiredoxins, and the localization of these enzymes relative to where H2O2 is generated and diffuses outward has a critical bearing on it signaling potency. Similar reasoning applies to mobilized free LOOHs and their inactivating enzymes, although in this case there could be a broad range of intrinsic LOOH reactivities for any given enzyme and this could be a strong determinant of signaling lifetime. In the case of GPx4, for example, PLOOHs collectively are much better substrates than ChOOHs, and in the latter group, 5α-OOH (the only known tertiary peroxide among the common natural LOOHs) is the least reactive. Thus, as suggested previously, for cells in which GPx4 is the main LOOH detoxifying agent, 1O2-generated 5α-OOH would be expected to have a long signaling lifetime compared with other ChOOHs and also PLOOHs.
The two membranes of mitochondria comprise an ideal system for studying the effects of relatively short-range LOOH translocation between membranes. Cardiolipin (CL), an acidic phospholipid with four unsaturated fatty acyl chains, is located exclusively in the mitochondrial inner membrane (IM) of non-stressed cells, where, in the outer leaflet, it plays an important role in tethering cytochrome c (cyt c) and other respiratory chain proteins. CL is highly sensitive to peroxidation (conversion to CLOOH) and this weakens its interaction with cyt c, allowing the latter to be released into the intermembrane space as an early event in the intrinsic pathway of oxidative apoptosis [74-76]. Recent studies have revealed that in stressed mitochondria with accumulating H2O2, cyt c can act as a CL peroxidase, thereby catalyzing its own release [77,78]. However, CL’s role in apoptosis is more complex than this, since it is also known to provide specificity for binding of truncated Bid (tBid) at the outer membrane (OM). tBid in turn recruits Bax, which oligomerizes into an OM-spanning megapore through which cyt c can pass into the cytosol for activation of the apoptosome [79]. Other proapoptotic proteins can traverse this pore as well. How CL, which originates in the IM, can materialize in the OM for tBid/Bax targeting is a question of major importance. CL exists at IM-OM contact sites, but whether this is sufficient for apoptotic requirements has been controversial [80]. One attractive possibility is that CL peroxidation not only mobilizes cyt c, but also CL itself in the form of relatively hydrophilic CLOOHs. Various CLOOH molecular species (possibly along with other CL-oxides) could translocate from the IM to OM in spontaneous or protein-facilitated fashion, thus promoting tBid/Bax recruitment. Recent evidence obtained with IM-OM models (Girotti et al., unpublished) has indicated that this short-range LOOH translocation mechanism is entirely feasible for mitochondrion-initiated apoptosis.
It is important to point out that proteins are also susceptible to peroxidation. Davies et al. [81] have demonstrated that protein hydroperoxides are generated in oxidatively stressed mammalian cells, and point out that this is consistent with the abundance of protein in cells and the strong susceptibility of amino acids like histidine, tyrosine and tryptophan to peroxidation, e.g. by 1O2 attack. Like LOOHs and H2O2, peroxides of amino acid residues in proteins may be subject to selenoperoxidase- or peroxiredoxin-catalyzed reduction to redox-inactive alcohol derivatives [82]. Protein-associated peroxides would not be free to translocate or be transported like LOOHs, since their translational mobility is restricted by the proteins to which they are attached. On the other hand, small peroxidized proteins as a whole may diffuse at significant rates from one site to another in cells or in plasma, and this could disseminate their biological effects. However, this translocation would necessarily be much slower than that of smaller LOOHs. In the case of a peroxidized membrane protein, most of the peroxide-induced damage or signaling activity would be limited by the protein’s ability to diffuse within the confines of the bilayer, but this is yet to be documented. Clearly, much remains to be learned about the transfer-associated physiological effects of these different peroxide classes in oxidatively stressed cells or plasma, and this promises to be an area of intensive investigation in the coming years.
Alternative possibilities for the “broadcasting” of LOOH effects
Other mechanisms for disseminating LOOH effects besides direct translocation have been proposed. Redmond et al. [37,83,84], using a human cell line and deuteroporphyrin, a 1O2-generating, plasma membrane-localizing sensitizer, recently reported that photoactivation of the sensitzer resulted in oxidative damage not only to the plasma membrane, but to nuclear DNA as well. Some spillover of the applied sensitizer into the nucleus was ruled out as a possible explanation for the DNA damage, detected as single strand breaks and alkali-sensitive sites by Comet assay. Similarly, direct attack of 1O2 on DNA was ruled out because of 1O2’s very short lifetime and limited diffusion range [37]. Consequently, it was postulated that the DNA damage was due to secondary oxidizing species that arise downstream from the primary 1O2 targets on the plasma membrane. The finding that free radical-mediated lipid peroxidation was induced in the irradiated cells and that this could be inhibited by radical-scavenging antioxidants such as trolox and α-tocopherol suggested that the DNA effects were mediated by relatively long-lived radicals (LOO·, LO·) arising from one-electron turnover of nascent 1O2-generated LOOHs on the plasma membrane (37,84]. The observed inhibitory effects of hydrophilic trolox acting mainly in the cytosol were consistent with such a scenario, although it is difficult to imagine that lipid-derived radicals (in contrast to primary LOOHs) would survive long enough to reach the nucleus. Contributions by electrophilic end-products of free radical peroxidation, particularly malonaldehyde and 4-hydroxy-2-nonenal, were also postulated (37,841).
Another proposed mechanism by which membrane LOOHs might induce distal DNA damage involves the peroxide-mediated activation of phospholipase, cyclooxygenase (COX) and lipoxygenase (LOX) enzymes [85]. It is well known, for example, that phospholipase A2, is more active in hydrolyzing membrane phospholipids in peroxidized than non-oxidized membranes [10]. This results in liberation of arachidonate and other fatty acids in both non-oxidized and peroxidized form. By increasing the “peroxide tone”, liberated fatty acid hydroperoxides can activate LOXs and COXs which act on arachidonate to generate new peroxides as intermediates in prostanoid and leukotriene formation [85]. In a mechanism of this type, small amounts of primary or “priming” LOOHs could lead to increasingly greater amounts of successor LOOHs via an enzymatic amplification process wherein the enzymes involved (LOXs, COXs) are positively modulated by an increasing peroxide tone. By simple analogy, this would operate somewhat like a self-perpetuating relay system in which LOOH pressure expands inwardly from the plasma membrane, eventually damaging secondary targets such as nuclear DNA. This relay-type model [85] is distinct from that involving direct transfer of a primary LOOH (see preceding sections). The applicability of these different mechanisms and their relative importance in any given system under oxidative pressure are key subjects of continuing investigation.
Conclusions and future directions
It is now clear that the ubiquitous inorganic hydroperoxide, H2O2, can function not only as a redox-active cytotoxic agent, but also as an activator or deactivator of sensor proteins that regulate signal transduction pathways [51,52]. Hydrogen peroxide generated in mitochondria or at the plasma membrane by NOX enzymes can diffuse to new sites, limited by encounters with peroxidases, and this is an important facet of its cytotoxic as well as signaling activity. Similarly, downstream peroxides like LOOHs can dissociate from oxidizing cell membranes or lipoproteins and diffuse, be transported (or possibly relayed) to new sites, where toxic reactions, detoxification, or signaling actions can ensue. As in the case of H2O2, high LOOH pressure in cells under relatively intense oxidative stress is expected to result in lethal damage, whereas low LOOH pressure may elicit the opposite, i.e. a pro-growth response. LOOHs are much less hydrophilic than H2O2 and can be sequestered and shuttled by lipid transfer proteins. Factors such as these will have an important bearing on LOOH signaling activity, for example, compared with that of H2O2. Stress-induced “bystander effects” within and between cells have been attributed to ROS movements, e.g. or H2O2 translocation [68-70], and it is appears likely that LOOH translocation can contribute to these effects. The ability of LOOHs to translocate was recognized only recently and much less is known about this process than that of H2O2. Clearly, more work needs to be done in an effort to better understand the mechanism, cellular occurrence, and biological ramifications of this phenomenon.
Acknowledgments
Studies in the author’s laboratory were supported by USPHS grants CA72630 and HL085677. Contributions to these studies by Andrew Vila, Tamas Kriska, Robert Kernstock, Vlad Levchenko, and Witek Korytowski are greatly appreciated.
Abbreviations
- ROS
reactive oxygen species
- Ch
cholesterol
- CE
cholesteryl ester
- CEOOH
cholesteryl ester hydroperoxide
- ChOOH
cholesterol hydroperoxide
- CL
cardiolipin
- CLOOH
cardiolipin hydroperoxide
- LH
unsaturated lipid
- LOOH
lipid hydroperoxide
- PL
phospholipid
- PLOOH
phospholipid hydroperoxide
- PCOOH
phosphatidylcholine hydroperoxide
- PEOOH
phosphatidylethanolamine hydroperoxide
- PSOOH
phosphatidylserine hydroperoxide
- SMOOH
sphingomyelin hydroperoxide
- LDL
low density lipoprotein
- SCP-2
sterol carrier protein-2 (non-specific lipid transfer protein)
- SUV(s)
small unilamellar vesicle(s)
- GPx4
glutathione peroxidase isotype 4
- HPLC-EC(Hg)
high-performance liquid chromatography with mercury cathode electrochemical detection
- HPTLC-PI
high-performance thin layer chromatography with phosphorimaging detection
- 5α-OOH
3β-hydroxy-5α-cholest-6-ene-5-hydroperoxide
- 6α-OOH
3β-hydroxycholest-4-ene-6α-hydroperoxide
- 6β-OOH
3β-hydroxycholest-4-ene-6β-hydroperoxide
- 7α-OOH
3β-hydroxycholest-5-ene-7α-hydroperoxide
- 7β-OOH
3β-hydroxycholest-5-ene-7β-hydroperoxide
- 7α/7β-OOH
undefined mixture of 7α-OOH and 7β-OOH
- 7α-OH
cholest-5-ene-3β,7α-diol
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Halliwell B, Gutteridge JMC. Free radicals in biology and medicine. Oxford, UK: Clarendon Press; 1999. [Google Scholar]
- 2.Porter NA, Caldwell SE, Mills KA. Mechanisms of free radical oxidation of unsaturated lipids. Lipids. 1995;30:277–290. doi: 10.1007/BF02536034. [DOI] [PubMed] [Google Scholar]
- 3.Sies H. Biochemistry of oxidative stress. Angew Chem Int Ed Engl. 1986;25:1058–1071. [Google Scholar]
- 4.Girotti AW. Photosensitized oxidation of membrane lipids: reaction pathways, cytotoxic effects, and cytoprotective mechanisms. J Photochem Photobiol B. 2001;63:103–113. doi: 10.1016/s1011-1344(01)00207-x. [DOI] [PubMed] [Google Scholar]
- 5.Bors W, Saran M, Tait D. Oxygen radicals in chemistry and biology. W. deGruyter; New York: 1984. [Google Scholar]
- 6.Pryor WA, Squadrito G. The chemistry of peroxynitrite: a product from the reacton of nitric oxide with superoxide. Am J Physiol. 1995;268:L669–L722. doi: 10.1152/ajplung.1995.268.5.L699. [DOI] [PubMed] [Google Scholar]
- 7.Foote CS. Mechanisms of photosensitized oxidation. Science. 1968;162:963–970. doi: 10.1126/science.162.3857.963. [DOI] [PubMed] [Google Scholar]
- 8.Wilcox AL, Marnett LJ. Polyunsaturated fatty acid alkoxyl radicals exist as carbon-centered epoxyallylic radicals: a key step in hydroperoxide-amplified lipid peroxidation. Chem Res Toxicol. 1993;6:413–416. doi: 10.1021/tx00034a003. [DOI] [PubMed] [Google Scholar]
- 9.Porter NA, Caldwell SE, Mills KA. Mechanisms of free radical oxidation of unsaturated lipids. Lipids. 1995;30:277–290. doi: 10.1007/BF02536034. [DOI] [PubMed] [Google Scholar]
- 10.Girotti AW. Lipid hydroperoxide generation, turnover, and effector action in biological systems. J Lipid Res. 1998;39:1529–1542. [PubMed] [Google Scholar]
- 11.Ursini F, Mairoino M, Brigelius-Flohe R, Aumann KD, Roveri A, Schomburg D, Flohe L. Diversity of glutathione peroxidases. Methods Enzymol. 1995;252:38–53. doi: 10.1016/0076-6879(95)52007-4. [DOI] [PubMed] [Google Scholar]
- 12.Rhee SG, Chae HZ, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med. 2005;38:1543–1552. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 13.Yang Y, Cheng J-Z, Singhal SS, Saini M, Pandya U, Awasthi S, Awasthi YC. Role of ghutathione S-transferases in protection against lipid peroxidation. J Biol Chem. 2001;276:19220–19230. doi: 10.1074/jbc.M100551200. [DOI] [PubMed] [Google Scholar]
- 14.Thomas JP, Maiorino M, Ursini F, Girotti AW. Protective action of phospholipid hydroperoxide glutathione peroxidase against membrane-damaging lipid peroxidation. J Biol Chem. 1990;265:454–461. [PubMed] [Google Scholar]
- 15.Nagata Y, Yamamoto Y, Niki E. Reaction of phosphatidylcholine hydroperoxide in human plasma: the role of peroxidase and lecithin-cholesterol acyltransferase. Arch Biochem Biophys. 1996;329:24–30. doi: 10.1006/abbi.1996.0187. [DOI] [PubMed] [Google Scholar]
- 16.Vila A, Korytowski W, Girotti AW. Dissemination of peroxidative stress via intermembrane transfer of lipid peroxides: model studies with cholesterol hydroperoxides. Arch Biochem Biophys. 2000;380:208–218. doi: 10.1006/abbi.2000.1928. [DOI] [PubMed] [Google Scholar]
- 17.Kagan VE, Gleiss B, Tyurina YY, Tyurin VA, Elenstrom-Magnusson C, Liu S-X, Serinkan FB, Arroyo A, Chandra J, Ohhrnius S, Fadeel B. A role for oxidative stress in apoptosis: oxidation and externalization of phosphatidylserine is required for macrophage clearance of cells undergoing Fas-mediated apoptosis. J Immunol. 2002;169:487–499. doi: 10.4049/jimmunol.169.1.487. [DOI] [PubMed] [Google Scholar]
- 18.Tyurina YY, Kawai K, Tyurin VA, Liu S-X, Kagan VE, Fabisiak JP. The plasma membrane is the site of selective phosphatidylserine oxidation during apoptosis: role of cytochrome c. Antiox Redox Signal. 2004;6:209–225. doi: 10.1089/152308604322899288. [DOI] [PubMed] [Google Scholar]
- 19.Tyurina YY, Tyurin VA, Zhao Q, Djukic M, Quinn PJ, Pitt BR, Kagan VE. Oxidation of phosphatdiylserine: a mechanism for plasma membrane phospholipid scrambling during apoptosis? Biochem Biophys Res Commun. 2004;324:1059–1064. doi: 10.1016/j.bbrc.2004.09.102. [DOI] [PubMed] [Google Scholar]
- 20.Phillips MC, Johnson WJ, Rothblat GH. Mechanisms and conswquences of cellular cholesterol exchange and transfer. Biochim Biophys Acta. 1987;906:223–276. doi: 10.1016/0304-4157(87)90013-x. [DOI] [PubMed] [Google Scholar]
- 21.Dawidowicz EA. Lipid exchange: transmembrane movement, spontaneous movement, and protein-mediated transfer of lipids and cholesterol. In: Klausner RD, Kempf C, van Renswoude J, editors. Current Topics in Membranes and Transport. Vol. 29. New York: Academic Press; 1987. pp. 175–202. [Google Scholar]
- 22.Brown RE. Spontaneous lipid transfer between organized lipid assemblies. Biochim Biophys Acta. 1992;1113:375–389. doi: 10.1016/0304-4157(92)90007-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zilversmit DB. Lipid transfer proteins. J Lipid Res. 1984;25:1563–1569. [PubMed] [Google Scholar]
- 24.Wirtz KWA. Phospholipid transfer proteins. Annu Rev Biochem. 1991;60:73–99. doi: 10.1146/annurev.bi.60.070191.000445. [DOI] [PubMed] [Google Scholar]
- 25.Wirtz KWA. Phospholipid transfer proteins revisited. Biochem J. 324:353–360. doi: 10.1042/bj3240353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scallen TJ, Pastuszyn A, Noland BJ, Chanderbhan R, Kharroubi A, Vanhouny GV. Sterol carrier and lipid transfer proteins. Chem Phys Lipids. 1985;38:239–261. doi: 10.1016/0009-3084(85)90019-2. [DOI] [PubMed] [Google Scholar]
- 27.Vila A, Korytowski W, Girotti AW. Spontaneous intermembrane transfer of various cholesterol-derived hydroperoxide species: kinetic studies with model membranes and cells. Biochemistry. 2001;40:14715–14726. doi: 10.1021/bi011408r. [DOI] [PubMed] [Google Scholar]
- 28.Schenck GO, Gollnick K, Neumuller OA. Zur photosensibilisieren autoxydation der steroide. Darstellung von steroid-hydroperoxyden mittels phototoxischer photosensibilisatoren. Justus Liebigs Ann Chem. 1957;603:46–59. [Google Scholar]
- 29.Kulig MJ, Smith LL. Sterol metabolism XXV. Cholesterol oxidation by singlet molecular oxygen. J Org Chem. 1973;38:3639–3642. doi: 10.1021/jo00960a050. [DOI] [PubMed] [Google Scholar]
- 30.Smith LL, Teng JI, Kulig MJ, Hill FL. Sterol metabolism XXIII. Cholesterol oxidation by radical-induced processes. J Org Chem. 1973;38:1763–1765. doi: 10.1021/jo00949a041. [DOI] [PubMed] [Google Scholar]
- 31.Korytowski W, Geiger PG, Girotti AW. High performance liquid chromatography with mercury cathode electrochemical detection; application tolipid hydroperoxide analysis. J Chromatogr B. 1995;670:189–197. doi: 10.1016/0378-4347(95)00182-4. [DOI] [PubMed] [Google Scholar]
- 32.Vila A, Korytowski W, Girotti AW. Spontaneous transfer of phospholipid and cholesterol hydroperoxides between cell membranes and low density lipoprotein: assessment of reaction kinetics and prooxidant effects. Biochemistry. 2002;41:13705–13716. doi: 10.1021/bi026467z. [DOI] [PubMed] [Google Scholar]
- 33.Sattler W, Stocker R. Greater selective uptake by HepG2 cells of high density lipoprotein cholesteryl ester hydroperoxides than of unoxidized cholesteryl esters. Biochem J. 1993;294:771–778. doi: 10.1042/bj2940771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fluiter K, Vietsch H, Biessen EAL, Kostner GM, van Berkel TJC. Increased selective uptake in vivo and in vitro of oxidixed cholesteryl esters from high density lipoprotein by rat liver parenchymal cells. Biochem J. 1996;319:471–476. doi: 10.1042/bj3190471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fluiter K, Sattler W, De Beer MC, Connell PM, van der Westhuyzen DR, van Berkel TJC. Scavenger receptor BI mediates the selective uptake of oxidized cholesteryl esters by rat liver. J Biol Chem. 1999;274:8893–8899. doi: 10.1074/jbc.274.13.8893. [DOI] [PubMed] [Google Scholar]
- 36.Christison JK, Rye KA, Stocker R. Exchange of oxidized cholesteryl linoleate between LDL and HDL mediated by cholesteryl ester transfer protein. J Lipid Res. 1995;36:2017–2026. [PubMed] [Google Scholar]
- 37.Ouedraogo GD, Redmond RW. Secondary reactive oxygen species extend the range of photosensitization effects incells: DNA damage produced via initial membrane photosensitization. Photochem Photobiol. 2003;77:192–203. doi: 10.1562/0031-8655(2003)077<0192:sroset>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 38.Korytowski W, Wrona M, Girotti AW. Radiolabeled cholesterol as a reporter for assessing one-electron turnover of lipid hydroperoxides. Anal Biochem. 1999;270:123–132. doi: 10.1006/abio.1999.4070. [DOI] [PubMed] [Google Scholar]
- 39.Steinberg D, Parthasarathy S, Carew TE, Khoo JC, Witztum JL. Beyond cholesterol: modifications of low density lipoprotein that increase its atherogenicity. N Engl J Med. 1989;320:915–924. doi: 10.1056/NEJM198904063201407. [DOI] [PubMed] [Google Scholar]
- 40.Thomas JP, Kalyanaraman B, Girotti AW. Involvement of preexisting lipid hydroperoxides in Cu(II)-stimulated oxidation of low density lipoprotein. Arch Biochem Biophys. 1994;315:244–254. doi: 10.1006/abbi.1994.1496. [DOI] [PubMed] [Google Scholar]
- 41.Cathcart MK, McNally AK, Morel DW, Chisholm GM. Supeoxide anion participates in human monocyte-mediated oxidation of low density lipoprotein and conversion of low density lipoprotein to a cytotoxin. J Immunol. 1989;142:1963–1969. [PubMed] [Google Scholar]
- 42.Schmitt D, Shen Z, Shang R, Colles SM, Wu W, Salomon RG, Chen Y, Chisholm GM, Hazen SL. Leukocytes utilize myoloperoxidase-generated nitrating intermediates as physiological catalysts for the generation of biologically active oxidized lipids and sterols in serum. Biochemistry. 1999;38:16904–16915. doi: 10.1021/bi991623w. [DOI] [PubMed] [Google Scholar]
- 43.Benz DJ, Mol M, Ezaki M, Mori-Ito N, Zelan I, Miyanohara A, Friedmann T, Parthasarathy S, Steinverg D, Witztum JL. Enhanced levels of lipoperoxides in low density lipoprotein incubated with murine fibroblasts expressing high levels of human 15-lipoxygenase. J Biol Chem. 1995;270:5191–5197. doi: 10.1074/jbc.270.10.5191. [DOI] [PubMed] [Google Scholar]
- 44.Gallegos AM, Atshaves BP, Storey SM, Starodub O, Petrescu AD, Huang H, McIntosh AL, Martin GG, Chao H, Kier AB, Schroeder F. Gene structure, intracellular localization, and functional roles of sterol carrier protein-2. Prog Lipid Res. 2001;40:498–563. doi: 10.1016/s0163-7827(01)00015-7. [DOI] [PubMed] [Google Scholar]
- 45.Stolowich NJ, Petrescu AD, Huang H, Martin GG, Scott AI, Schroeder F. Sterol carrier protein-2: structure reveals function. Cell Mol Life Sci. 2002;59:193–212. doi: 10.1007/s00018-002-8416-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Woodford JK, Colles SM, Myers-Payne SC, Billheimer JT, Schroeder F. Sterol carrier portein-2 stimulates intermembrane sterol transfer by direct membrane interaction. Chem Phys Lipids. 1995;76:73–84. doi: 10.1016/0009-3084(95)02436-m. [DOI] [PubMed] [Google Scholar]
- 47.Colles SM, Woodford JK, Moncecchi D, Myers-Payne SC, McLean LR, Billheimer JT, Schroeder F. Cholesterol interaction with recombinant human sterol carrier protein-2. Lipids. 1995;30:795–803. doi: 10.1007/BF02533954. [DOI] [PubMed] [Google Scholar]
- 48.Vila A, Levchenko VV, Korytowski W, Girotti AW. Sterol carrier protein-2-facilitated intermembrane transfer of cholesterol- and phospholipid-derived hydroperoxides. Biochemistry. 2004;43:12592–12605. doi: 10.1021/bi0491200. [DOI] [PubMed] [Google Scholar]
- 49.Hurst R, Korytowski W, Kriska T, Esworthy RS, Chu F-F, Girotti AW. Hyperresistance to cholesterol hydroperoxide-induced peroxidative injury and aopototic death in a tumor cell line that overexpresses glutathione peroxidase isotype-4. Free Radic Biol Med. 2001;31:1051–1065. doi: 10.1016/s0891-5849(01)00685-2. [DOI] [PubMed] [Google Scholar]
- 50.Kriska T, Levchenko VV, Korytowski W, Atshaves BP, Schroeder F, Girotti AW. Intracellular dissemination of peroxidative stress: internalization, transport, and lethal targeting of a cholesterol hydroperoxide species by sterol carrier protein-2-overexpressing hepatoma cells. J Biol Chem. 2006;281:23643–23651. doi: 10.1074/jbc.M600744200. [DOI] [PubMed] [Google Scholar]
- 51.Stone JR, Yang S. Hydrogen peroxide: a signaling messenger. Antiox Redox Signal. 2006;8:243–270. doi: 10.1089/ars.2006.8.243. [DOI] [PubMed] [Google Scholar]
- 52.Veal EA, Day AM, Morgan BA. Hydrogen peroxide sensing and signaling. Molecular Cell. 2007;26:1–14. doi: 10.1016/j.molcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 53.Burdon RH, Alliangana D, Gill V. Hydrogen peroxide and the proliferatin of BHK-21 cells. Free Radical Res. 1995;23:471–486. doi: 10.3109/10715769509065268. [DOI] [PubMed] [Google Scholar]
- 54.Storz G, Tartaglia LA, Ames BN. Transcriptional regulator of oxidative stress-inducible genes: direct activation by oxidation. Science. 1990;248:189–194. doi: 10.1126/science.2183352. [DOI] [PubMed] [Google Scholar]
- 55.Aslund F, Zheng M, Beckwith J, Storz G. Regulation of the OxyR transcription factor by hydrogen peroxide and the cellular thiol-disulfide status. Proc Natl Acad Sci USA. 1999;96:6161–6165. doi: 10.1073/pnas.96.11.6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee C, Lee SM, Mukhopadhyay P, Kim SJ, Lee SC, Ahn WS, Yu MH, Storz G, Ryu SE. Redox regulation of OxyR requires specific disulfide bond formation involving a rapid kinetic reaction path. Nature Struct Mol Biol. 2004;11:1179–1185. doi: 10.1038/nsmb856. [DOI] [PubMed] [Google Scholar]
- 57.Herbig AF, Helmann JD. Roles of metal ions and hydrogen peroxide in modulating the interaction of the Bacillus subtilis PerR peroxide regulon repressor with operator DNA. Mol Microbiol. 2001;41:849–859. doi: 10.1046/j.1365-2958.2001.02543.x. [DOI] [PubMed] [Google Scholar]
- 58.Lee JW, Helmann JD. The PerR transcription factor senses hydrogen peroxide by metal-catalyzed histidine oxidation. Nature. 2006;440:363–367. doi: 10.1038/nature04537. [DOI] [PubMed] [Google Scholar]
- 59.Fernandes AP, Holmgren A. Glutaredoxins: glutathione-dependent redox enzymes with functions far beyond a simple thioredoxin backup system. Antiox Redox Signal. 2004;6:63–74. doi: 10.1089/152308604771978354. [DOI] [PubMed] [Google Scholar]
- 60.Lillig CH, Holmgren A. Thioredoxin and related molecules: from biology to health and disease. Antiox Redox Signal. 2007;9:25–47. doi: 10.1089/ars.2007.9.25. [DOI] [PubMed] [Google Scholar]
- 61.Kaimul AM, Nakamura H, Masutani H, Yodoi J. Thioredoxin and thioredoxin-binding protein-2 in cancer and metabolic syndrome. Free Radic Biol Med. 2007;43:861–868. doi: 10.1016/j.freeradbiomed.2007.05.032. [DOI] [PubMed] [Google Scholar]
- 62.Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192:1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- 63.Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Yl, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK-1) EMBO J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu Y, Min W. Thioredoxin promotes ASK-1 ubiquitination and degradation to inhibit ASK-1-mediated apoptosis in a redox activity-independent manner. Circ Res. 2002;90:1259–1266. doi: 10.1161/01.res.0000022160.64355.62. [DOI] [PubMed] [Google Scholar]
- 65.Song JJ, Rhee JG, Suntharalingam M, Walsh SA, Spitz DR, Lee YJ. Role of glutaredoxin in metabolic oxidative stress: glutaredoxin as a sensor of oxidative stress mediated by H2O2. J Biol Chem. 2002;277:46566–46575. doi: 10.1074/jbc.M206826200. [DOI] [PubMed] [Google Scholar]
- 66.Gracanin M, Davies MJ. Inhibition of protein tyrosine phosphatases by amino acid, peptide, and protein hydroperoxides: Potential modulation of cell signaling by protein oxidation products. Free Radic Biol Med. 2007;42:1543–1551. doi: 10.1016/j.freeradbiomed.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 67.Shchedrina VA, Novoselov SV, Malinouski MY, Gladyshev VN. Identification and characterization of a selenoprotein family containing a diselenide bond in a redox motif. Proc Natl Acad Sci USA. 2007;104:13919–13924. doi: 10.1073/pnas.0703448104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bienert GP, Schjoerring JK, Jahn TP. Membrane transport of hydrogen peroxide. Biochim Biophys Acta. 2006;1758:994–1003. doi: 10.1016/j.bbamem.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 69.Reth M. Hydrogen peroxide as a second messenger in lymphocyte activaton. Immunology. 2002;3:1129–1134. doi: 10.1038/ni1202-1129. [DOI] [PubMed] [Google Scholar]
- 70.Pelle E, Mammone T, Maes D, Frenkel K. Keratinocytes act as a source of reactive oxygen species by transferring hydrogen peroxide to melanocytes. J Invest Dermatol. 2005;124:793–797. doi: 10.1111/j.0022-202X.2005.23661.x. [DOI] [PubMed] [Google Scholar]
- 71.Waghray M, Cui Z, Horowitz JC, Subramanian IM, Martinez FJ, Toews GB, Thannickal VJ. Hydrogen peroxide is a diffusible paracrine signal for the induction of epithelial cell death by activated myofibroblasts. FASEB J. 2005;19:854–856. doi: 10.1096/fj.04-2882fje. [DOI] [PubMed] [Google Scholar]
- 72.Calamita G, Ferri D, Gena P, Liquori GE, Cavalier A, Thomas D, Svelto M. The inner mitochondrial membrane has aquaporin-8 water channels and is highly permeable to water. J Biol Chem. 2005;280:17149–17153. doi: 10.1074/jbc.C400595200. [DOI] [PubMed] [Google Scholar]
- 73.Greenberg ME, Sun M, Zhang R, Febbraio M, Silverstein R, Hazen SL. Oxidized phosphatidylserine-DC36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J Exp Med. 2006;203:2613–2625. doi: 10.1084/jem.20060370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nomura K, Imai H, Koumura T, Kobayashi T, Nakagawa Y. Mitochondrial phospholipid hydroperoxide glutathione peroxidase inhibits the release of cytochrome c from mitochondria by suppressing the peroxidation of cardiolipin in hypoglycemia-induced apoptosis. Biochem J. 2000;351:183–193. doi: 10.1042/0264-6021:3510183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Petrosillo G, Ruggiero FM, Paradies G. Role of reactive oxygen species and cardiolipin in the release of cytochrome c from mitochondria. FASEB J. 2003;17:2202–2208. doi: 10.1096/fj.03-0012com. [DOI] [PubMed] [Google Scholar]
- 76.Iverson SL, Orrenius S. The cardiolipin-cytochrome c interaction and the mitochondrial regulation of apoptosis. Arch Biochem Biophys. 2004;423:37–46. doi: 10.1016/j.abb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 77.Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, Vlasova II, Zhao Q, Zou M, Di P, Svistunenko DA, Kurnikov IV, Borisenko GG. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nature Chem Biol. 2005;1:223–232. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- 78.Bayir H, Tyurin VA, Tyurina YY, Viner R, Ritov V, Amoscato AA, Zhao Q, Zhang XJ, Janesko-Feldman KL, Alexander H, Basova LV, Clark RSB, Kochanek PM, Kagan VE. Selective early cardiolipin peroxidation after traumatic brain injury: an oxidative lipodomics analysis. Ann Neurol. 2007;62:154–169. doi: 10.1002/ana.21168. [DOI] [PubMed] [Google Scholar]
- 79.Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- 80.Mannella CA. The relevance of mitochondrial membrane topology to mitochondrial function. Biochim Biophys Acta. 2005;1762:140–147. doi: 10.1016/j.bbadis.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 81.Davies MJ. Singlet oxygen-mediated damage to proteins and its consequences. Biochem Biophys Res Commun. 2003;305:761–770. doi: 10.1016/s0006-291x(03)00817-9. [DOI] [PubMed] [Google Scholar]
- 82.Morgan PE, Dean RT, Davies MJ. Protective mechanisms against peptide and protein peroxides generated by singlet oxygen. Free Radic Biol Med. 2004;36:484–496. doi: 10.1016/j.freeradbiomed.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 83.Ouedraogo GD, Redmond R. Photosensitized membrane oxidation leads to remote DNA damage by secondary reactive oxygen species. Free Radic Biol Med. 2002;33:S421. [Google Scholar]
- 84.Redmond R, Kochevar IE. Spatially resolved cellular responses to singlet oxygen. Photochem Photobiol. 2006;82:1178–1186. doi: 10.1562/2006-04-14-IR-874. [DOI] [PubMed] [Google Scholar]
- 85.Booth L, Redmond R. Can lipid peroxidation of plasma membranes induce DNA strand breaks? Free Radic Biol Med. 2002;33:S390. [Google Scholar]