

Abstract
Histone deacetylase inhibitors (HDACIs) are novel anticancer agents with potent cytotoxicity against a wide range of malignancies. We have previously demonstrated that either Calphostin C (CC) (a protein kinase C (PKC) inhibitor) or Parthenolide (an NF-κB inhibitor) abrogates HDACI-induced transcriptional activation of NF-κB and p21, which is associated with profound potentiation of HDACI-mediated induction of apoptosis. Valproic acid (VA), a commonly used antiepileptic agent, has recently been shown to be an HDACI. This study was aimed to evaluate the anticancer property of VA in thoracic cancer cells and the development of clinically relevant strategies to enhance VA-mediated induction of apoptosis using kinase inhibitors Staurosporine (STP) or its analogue UCN-01. Treating cultured thoracic cancer cells with VA (0.62–10.0 mM) resulted in significant cell line- and dose-dependent growth inhibition (IC50 values: 4.1–6.0 mM) and cell cycle arrest at G1/S checkpoint with profound accumulation of cells at G0/G1 phase but little induction of apoptosis. Valproic acid, being an HDACI, caused significant dose-dependent accumulation of hyperacetylated histones, following 24 h of treatment. Valproic acid-mediated 5–20-fold upregulation of transcriptional activity of NF-κB was substantially (50–90%) suppressed by cotreatment with CC, STP or UCN-01. Whereas minimal death (<20%) was observed in cells treated with either VA (1.0 or 5.0 mM) alone or kinase inhibitors alone, 60–90% of cells underwent apoptosis following exposure to combinations of VA+kinase inhibitors. Kinase inhibitor-mediated suppression of NF-κB transcriptional activity played an important role in sensitising cancer cells to VA as direct inhibition of NF-κB by Parthenolide drastically synergised with VA to induce apoptosis (VA+Parthenolide: 60–90% compared to <20% following single-drug treatments). In conclusion, VA, a well-known antiepileptic drug, has mild growth-inhibitory activity on cultured cancer cells. The weak VA-mediated induction of apoptosis of thoracic cancer cells can be profoundly enhanced either by Parthenolide, a pharmacologic inhibitor of NF-κB, or by UCN-01 a kinase inhibitor that has already undergone phase I clinical development. Combinations of VA with either a PKC inhibitor or an NF-κB inhibitor are promising novel molecularly targeted therapeutics for thoracic cancers.
Keywords: histone deacetylase inhibitor, valproic acid, UCN-01, Staurosporine, protein kinase C, Parthenolide, NF-κB, apoptosis
Chromatin remodelling via histone acetylation by histone acetyltransferases and histone deacetylases (HDACs) is considered as a key element in the dynamic regulation of many genes that play crucial roles in cell growth and differentiation (Kramer et al, 2003; Rosato and Grant, 2004). Chromatin structure, in its compact hypoacetylated state, may affect gene expression by blocking the access of transcription factors to their cognate DNA sequences to initiate gene transcription. On the other hand, chromatins with hyperacetylated core histones, due to acetylation-mediated neutralisation of positive charges on lysine residues and disruption of nucleosome structures, assume a relaxed conformation with unfolding of associated DNA that facilitates transcriptional activity (Grunstein, 1997; Richon et al, 1998; Struhl, 1998; Wolffe and Kurumizaka, 1998; Sambucetti et al, 1999). Histone deacetylases have been shown to be involved in oncogenic transformation by mediating the function of transcription factors in certain forms of haematologic malignancies and possibly in the pathogenesis of solid tumours (Rosato and Grant, 2004). Therefore, identification and clinical development of pharmacologic inhibitors of HDACs for anticancer therapy have attracted a great deal of interest.
Histone deacetylase inhibitors (HDACIs) are structurally diverse chemical compounds that share common biological properties of inducing core histone hyperacetylation leading to gene expression and of mediating potent antitumour effects. Some of HDACIs are either naturally occurring compound like sodium butyrate, a fatty acid metabolite found in high concentration in the lumen of large intestine, or pharmacologic compound such as valproic acid (VA), a commonly prescribed antiepileptic drug whose pharmacokinetics and toxicity profiles are well documented (Brodie and Dichter, 1996; Blaheta and Cinatl, 2002), whereas others are complex chemicals isolated from culture broths of microorganisms (such as Depsipeptide, Apicidin or Trichostin A (TSA)) or synthetic derivatives (for instance MS-275, CI-994). Histone deacetylase inhibitors are subdivided into four fundamental groups: short-chain fatty acids (sodium butyrate, phenylbutyrate, valproic acid), synthetic benzanide derivatives (MS-275, CI-994), cyclic tetrapeptides (Depsipeptide, Trapoxin, Apicidin) and hydroxamic acids (TSA, suberoylanilide hydroxamic acid (SAHA), LAQ8240) (Rosato and Grant, 2004). Histone deacetylase inhibitors induce differentiation, cell cycle arrest and/or apoptosis of cancer cells in culture and in vivo animal models (Jaboin et al, 2002; Rosato and Grant, 2004; Schrump and Nguyen, 2005). Multiple HDACIs (sodium butyrate, SAHA, Depsipeptide, MS275) have been shown to have anticancer properties in phase I and II clinical trials (Schrump et al, 2002; Kelly et al, 2003; Piekarz and Bates, 2004; Rosato and Grant, 2004).
The antitumour activity of HDACIs has been attributed to both their ability to inhibit deacetylases (leading to accumulation of hyperacetylated histones and alteration of gene transcription) and their ability to downregulate phenotypic expression of oncoproteins as well as activation of the intrinsic apoptosis-inducing cascade (Ruefli et al, 2001; Yu et al, 2002; Marks and Jiang, 2005). Histone deacetylase inhibitor-mediated cell cycle arrests or induction of apoptosis are dependent on treatment conditions such as drug concentrations and duration of drug exposure, as well as the intrinsic sensitivity of malignant cells to this class of anticancer agents (Yu et al, 2002; Maxhimer et al, 2005). Activation of NF-κB transcriptional activity and upregulation of p21 gene expression are frequently observed following HDACI treatments in cancer cells (Burgess et al, 2001; Dai et al, 2003; Mayo et al, 2003). Histone deacetylase inhibitor-mediated activation of NF-κB activity and/or upregulation of p21 gene expression have actually been shown to impede drug-induced apoptosis (Burgess et al, 2001; Mayo et al, 2003; Nguyen et al, 2003, 2004; Rosato et al, 2004; Rundall et al, 2004; Maxhimer et al, 2005). In fact, inhibition of HDACI-induced NF-κB activation by pharmacologic inhibitors of IκB kinase inhibitor (IKK) or by overexpression of kinase-resistant IκB resulted in substantial induction of apoptosis of HDACI-treated cells (Rundall et al, 2004; Maxhimer et al, 2005). Similarly, depletion of p21 by antisense oligonucleotides sensitises cultured cancer cells to HDACIs (Burgess et al, 2001). The molecular pathways that regulate HDACI-mediated NF-κB activation seem to involve PI3K/Akt (Mayo et al, 2003) and/or protein kinase C (PKC) signallings (Kim et al, 2003; Maxhimer et al, 2005). Mayo et al (2003) have demonstrated that the ability of HDACIs to increase NF-κB transcriptional activity was not associated with events that stimulated nuclear translocation, but rather related to modulation of transactivation potential of the RelA/p65 subunit of NF-κB via a PI3K/Akt-dependent mechanism. Maxhimer et al (2005) have shown that the PKC inhibitor Calphostin C (CC) completely abrogated TSA-mediated NF-κB activation in cultured thoracic cancer cells and this was associated with profound induction of apoptosis following treatment with CC+TSA drug combination. Moreover, Grant and his co-workers (Rosato et al, 2002; Rahmani et al, 2003a, 2003b) as well as our group (Nguyen et al, 2003, 2004) have published multiple reports that described the synergistic interactions between HDACIs and different kinase inhibitors (for instance, Flavopiridol or LY294002) to induce massive apoptosis in leukaemic or thoracic cancer cells in vitro. These findings support further preclinical and clinical development of targeted therapeutic strategies that combine inhibition of either NF-κB or PI3K or PKC signallings with HDACIs by using clinically relevant pharmacologies.
Staurosporine (STP) and its analogue UCN-01 were initially developed as PKC inhibitors for cancer therapy (Seynaeve et al, 1994). More recent studies have identified these compounds as kinase inhibitors with a broader range of molecular targets in vivo, including chk1/2 and PDK1, a kinase that act directly upstream of Akt and MAP/extracellular signal-regulated kinase (ERK) kinase (MEK) (Sato et al, 2002). Exposure of cultured cancer cells to UCN-01 (250–1000 nM) resulted in complete abrogation of phosphorylation at Ser473 and Thr308 residues of Akt, both of which are essential for full activation of Akt kinase activity (Amornphimoltham et al, 2004; Kondapaka et al, 2004). Clinical trials of UCN-01 in patients with refractory neoplasms, as monotherapy or in combination with cytotoxic chemotherapeutics, have been reported (Sausville et al, 2001). Continuous 72-h infusion of UCN-01 resulted in ‘free’ (after ultracentrifugation) plasma drug concentrations of 200–600 nM and tissue drug concentration (via measurement of salivary UCN-01 concentrations as surrogates) of 110 nM (Sausville et al, 2001). In view of the facts that STP and its clinically relevant analogue UCN-01 inhibit multiple kinases known to influence the cytotoxic effect of HDACIs in cancer cells, we hypothesised that STP or UCN-01 would synergise with HDACIs to promote profound induction of apoptosis. To this end, we sought to investigate the cytotoxicity of the UCN-01+VA combination in cultured thoracic cancer cells in vitro. These agents have been used in human for different indications, the former as a phase I anticancer agent and the latter as an established antiepileptic drug that was recently demonstrated to have anticancer activity in vitro and in vivo animal model (Gottlicher et al, 2001). This combination has the potential to be developed into a clinically applicable novel targeted molecular therapeutics.
MATERIALS AND METHODS
Cells and reagents
The oesophageal cancer (EsC) cells TE2, TE12; the non-small-cell lung cancer (NSCLC) cells H322, H460 and the malignant pleural mesothelioma (MPM) cells H513, H211 were maintained in RPMI-1640 culture medium supplemented with fetal calf serum (10% vol vol−1), streptomycin (100 μg ml−1), penicillin (100 U ml−1) and glutamine (2 mM). Trichostatin A (Sigma, St Louis, MO, USA), Calphostin C, STP, (Calbiochem, La Jolla, CA, USA) and the NF-κB inhibitor Parthenolide (Alexis, San Diego, CA, USA) were dissolved in dimethyl sulphoxide and stored at −20°C. Valproic acid (Alexis) was dissolved in distilled water to make 500 mM stock and stored in 4°C. UCN-01 was provided by the Developmental Therapeutic Programme of the National Cancer Institute, NIH, Bethesda, MD, USA.
Determination of apoptosis
Cancer cells seeded at 3 × 105 cells per well in six-well plates were treated either with VA alone (1.0. or 5.0 mM) or in combination with CC (1.0–2.0 μM), UCN-01 (250–500 nM), STP (200 nM) or Parthenolide (20 μM). The second drug was added into the VA-treated cells 12 h after the onset of VA treatment as 10 × stock solutions with VA remained in the cultured medium. Treated cells were harvested at 48 h after the onset of VA treatment, fixed in 1% paraformaldehyde and 70% ethanol, and assayed for apoptosis by using the terminal deoxynucleotidyltransferase-mediated dUTP nick-end labelling (TUNEL)-based ApoBrdU assay (BD Pharmingen, Torrance, CA, USA), as per the protocol provided by the manufacturer.
NF-κB transcriptional activity
The transcriptional activity of NF-κB was evaluated by transient transfection of NF-κB-luciferase reporter plasmid (Invitrogen, Carlsbad, CA, USA) into cells before drug treatments. Luciferase activity of cell lysates is directly correlated with the transcriptional activity of NF-κB. Cells were plated onto 24-well plates at a density of 8 × 104 cells well−1. For transcription experiments, cells were transfected with 200 ng of NF-κB-Luc plasmid per well with Fugene (Promega, Madison, WI, USA). At 24 h after transfection, cells were treated with different experimental conditions as outlined in the respective figure legends. The transcriptional activity of NF-κB was quantified by measuring the luciferase activity from cell lysates with Luciferase Assay System (Promega, Madison, WI, USA) and the Lumat LB 9507 lucinometer (EG&G Berthold, Gaithersburg, MD, USA). The NF-κB activity was normalised with the total cellular protein in cell lysates determined by BCA protein assay (Pierce Biotechnology, Rockford, IL, USA) and expressed as fold of the NF-κB activity of untreated control cells.
Western blotting
Cultured NSCLC and EsC cells were treated with different experimental conditions as described in the figure legends. Whole-cell extracts were prepared in cell lysis buffer (Cell Signaling Technologies, Beverly, MA, USA) supplemented with 1 mM PMSF (Sigma, St Louis, MO, USA). In total, 50 μg of cell extracts were separated on a gradient 4–20% SDS–PAGE and transferred onto nitrocellulose membranes. The membranes were blocked in 5% Blotto before immunoblotting with antibodies targeting apoptotic proteins Bax, Bak, Bcl2, BclXL (Cell Signaling Technology, Beverly, MA, USA at 1 : 1000 dilution) and cIAP1 (R&D, Minneapolis, MN, USA, 1 : 2000 dilution). Nitrocellolose membranes were also immunoblotted with antibodies targeting phospho-Adducin (Upstate, Waltham, MA, USA, 1 : 1000 dilution with 3% Blotto), Akt and phospho-Akt Ser473 (Cell Signaling, 1 : 500 dilution with 5% BSA), p42/p44 and phospho-p42/p44 (Cell Signaling, 1 : 1000 dilution with 5% BSA) and acetylated-Histones 3 and 4 (Upstate, 1 : 1000 dilution with 3% Blotto). The primary antibodies were detected by HRP-conjugated anti-mouse and anti-rabbit secondary antibodies and detected with West Dura chemiluminescence (Pierce Biotechnology, Rockford, IL, USA).
Data analysis
Supra-additive apoptosis is defined as the apoptosis induced by the drug combinations that is, by statistical analysis, significantly greater than the algebraic sum of apoptosis induced by individual drug treatment. Data are expressed as means±standard error of the means (s.e.m.) of at least three independent experiments that yielded similar results. Statistical analysis was performed with the GraphPad InStat software (GraphPad, San Diego, CA, USA). Statistical analysis was performed using analysis of variance (ANOVA) and pairwise comparison using Bonferroni test or by two-tailed Student's t-test, with P<0.05 indicating statistical significance.
RESULTS
Staurosporine or UCN-01 suppresses TSA-mediated NF-κB activation and enhances TSA-induced apoptosis
As we have previously demonstrated that pharmacologic inhibition of PKC activity by using CC resulted in abrogation of TSA-mediated NF-κB activation and enhancement of apoptosis in cells treated with the TSA+CC combination (Maxhimer et al, 2005), we sought to determine if STP or UCN-01, via being PKC inhibitors as indicated by their ability to markedly reduce the expression of phosphorylated α/γ adducin (p-adducin) (Figure 1A), could duplicate the effect of CC in suppressing NF-κB activation and enhancing the cytotoxic effect of TSA in cultured thoracic cancer cells. Staurosporine (200 nM) or UCN-01 (500 nM) significantly inhibited NF-κB transcriptional activation in H322 and TE12 cells that were treated with TSA, quite similar to the effect of the PKC inhibitor CC (Figure 1A). Whereas less than 20% of cultured thoracic cancer cells exposed to either CC, STP or UCN-01 alone or TSA alone were apoptotic, 55% to more than 90% of cells treated with the respective drug combinations had undergone apoptosis (Figure 1B). However, the supra-additive induction of apoptosis by UCN-01 or STP in combination with TSA may also be attributable to their function as negative modulators of Akt and MEK/ERK1/2 activation via inhibition of PDK1 (Sato et al, 2002), in addition to being PKC inhibitors. To further investigate the effect of STP or UCN-01 on MAPK ERK1/2 or Akt activation in cultured thoracic cancer cells, dose–response and time-course studies were performed on cells treated with STP (100–400 nM) or UCN-01 (250–1000 nM) and harvested at 1 or 24 h after the onset of drug exposure (Figure 2). UCN-01 treatment resulted in a significant dose-dependent reduction of pAkt as early as 1 h after drug exposure. At 24 h, higher phosphorylated Akt levels were observed in UCN-01-treated cells than in untreated controls. Staurosporine, on the other hand, mediated a dose-dependent activation of Akt throughout the whole time-course experiment. Dose-dependent suppression of ERK1/2 activation, however, was observed in both STP- and UCN-01-treated cells at both time points. As expected, PKC activity in cultured thoracic cancer cells, as indicated by p-adducin levels, was completely abrogated by either STP or UCN-01, with STP mediating a very rapid onset of PKC inhibition and UCN-01 mediating a slower onset of PKC inhibition.
Figure 1.
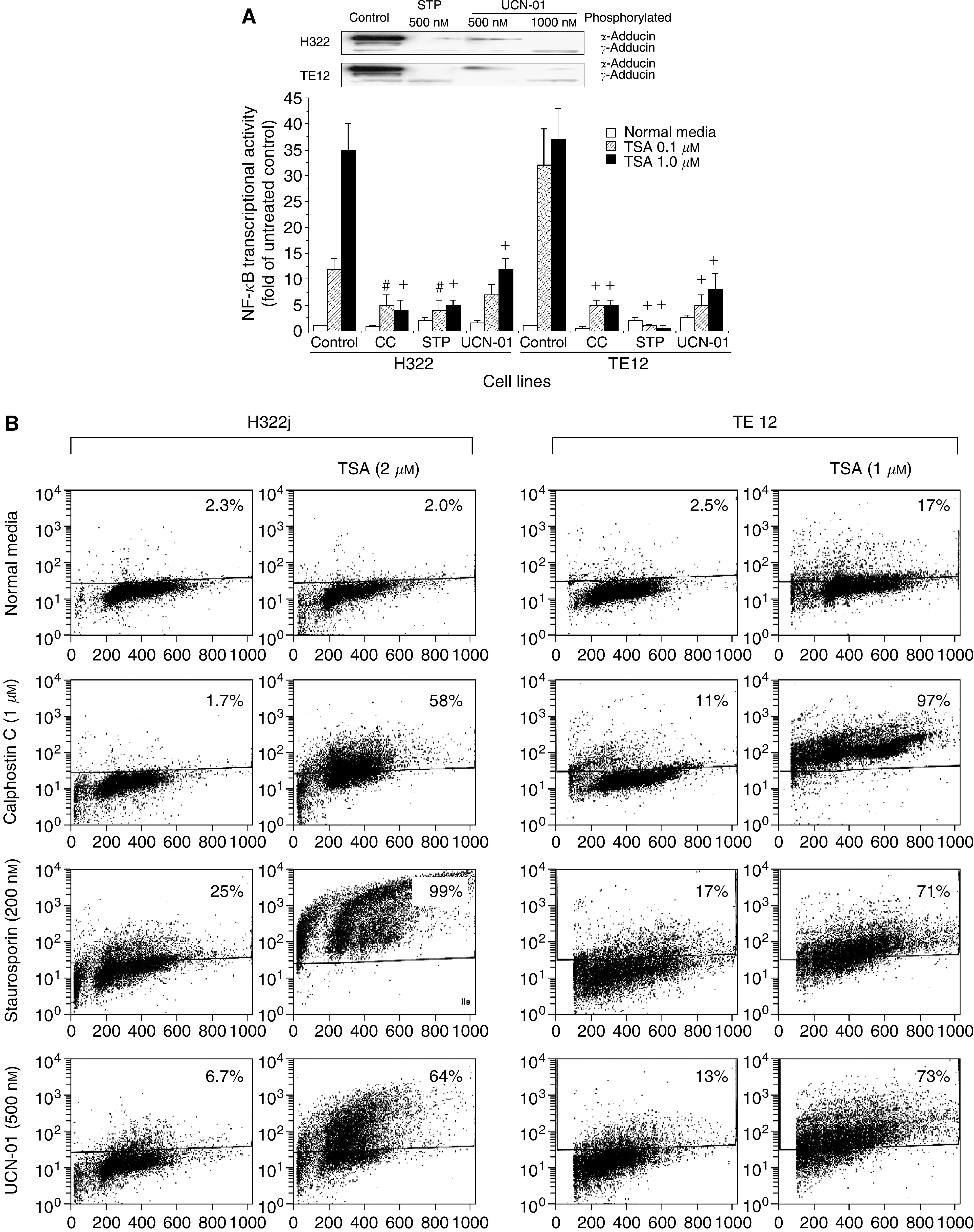
(A) Suppression of TSA-mediated upregulation of NF-κB transcriptional activity by Calphostin C, Staurosporine and its analogue UCN-01 in NSCLC cell H322 and EsC cell TE12. Cells were transiently transfected with NF-κB-Luc plasmid as described in Materials and Methods and then subsequently treated with TSA (0.1 and 1.0 μM) with or without concurrent Calphostin C (CC – 2 μM), Staurosporine (STP – 200 nM) or UCN-01 (500 nM). Staurosporine and UCN-01 exerted a potent PKC-inhibitory activity as evidenced by the complete depletion of p-adducin following 12 h of exposure to the respective drugs. Luciferase activity assayed 24 h after drug treatments were normalised for proteins of cell lysates and expressed as fold of activity of untreated control cells. Data are expressed as mean±s.e.m. of three independent experiments (#P<0.05–0.01 and +P<0.001 vs controls by ANOVA and pair-wise comparison by Bonferroni test). (B) Profound induction of apoptosis in H322 and TE12 cells treated with TSA+CC, TSA+STP and TSA+UCN-01 combinations. Cells were exposed to TSA (1 or 2 μM) for 12 h, to CC (1 μM) or STP (200 nM) or UCN-01 (500 nM) continuously for 36 h or TSA followed by other drugs. Cells were then harvested 48 h after the onset of treatment to assay for apoptosis. Representative data of three independent experiments that yielded similar results are shown here.
Figure 2.

Time-course (1 and 24 h) and dose-dependent STP (50, 100, 200 nM)- or UCN-01 (250, 500, 1000 nM)-mediated modulation of the PKC activity and of phosphorylated Akt, ERK1/2 levels in H460 and TE12 cells. β-Actin of cell lysates was blotted to indicate equal loadings of proteins. Profound depletion of pERK1/2, p-adducin and pAkt (in early time points) following UCN-01 exposure. Staurosporine effect on these molecular targets was similar to the effect of UCN-01, except that it stimulated (instead of inhibited) Akt phosphorylation.
The growth-inhibitory effect of VA in cultured thoracic cancer cells
Subsequent experiments were devoted to evaluating the antitumour effect of VA in cultured thoracic cancer cells. Valproic acid has been shown to have HDACI activity and to mediate differentiation and/or apoptosis in different carcinoma cell lines in vitro and in vivo (Gottlicher et al, 2001; Phiel et al, 2001; Kramer et al, 2003; Gurvich et al, 2004). Valproic acid also exhibited HDACI activity in cultured thoracic cancer cells. Treating representative NSCLC cells H460, EsC cells TE12 and MPM cells H513 with VA resulted in a dose-dependent accumulation of hyperacetylated H3 and H4 histones (Figure 3A). Valproic acid mediated a mild reduction of cell proliferation, the magnitude of which was cell line-dependent with the VA IC50 values ranging from 4.5 to 8.0 mM (Figure 3A). The effect of VA on the progression of cancer cells through the cell cycle was determined by PI staining and flow cytometry. Treating cultured thoracic cancer cells with VA (1.0 or 5.0 mM) for 48 h resulted in accumulation of cells in G0/G1 phase and a concomitant reduction of cell in S and G2/M phases, indicating that VA mediated cell cycle arrest at the G1/S checkpoint (Figure 3B). Similar to previous reports (Gottlicher et al, 2001; Kramer et al, 2003), VA was a weak inducer of apoptosis in thoracic cancer cell lines as less than 25% of cells were apoptotic following 48 h of continuous exposure to high concentration of VA at 5.0 mM (Figure 3C). By itself, VA exerted a very mild growth-inhibitory effect, yet we postulated that the cytotoxic effect of VA, being an HDACI, could be enhanced by the PKC inhibitor CC or the broader spectrum kinase inhibitor STP or UCN-01. These kinase inhibitors negatively regulate PKC and ERK1/2 activation, both of which have been shown to potentiate the cytotoxic effect of HDACIs (Rahmani et al, 2003b; Maxhimer et al, 2005).
Figure 3.

(A) The HDAC-inhibitory activity of VA in H460, TE12 and H513 cells. Cells were treated with VA (0.5, 1.0 and 5.0 mM) for 12 h and hyperacetylated H3 and H4 histone proteins were determined by Western blot analysis. Mild growth-inhibitory effect of VA on cultured thoracic cancer cells. Cells, seeded in 96-well microtiter plates, were continuously exposed to VA (0.62–10.0 mM) for 96 h. Cell viability was measured by MTT (4,5-dimethylthiazo-2-yl)-2,5-diphenyl tetrazolium bromide) and cell viability was expressed as percentages of untreated control cells. Data are expressed as mean±s.e.m. of three independent experiments. (B) Cell cycle arrest at G1/S checkpoint with accumulation of cells at G0/G1, little induction of apoptosis only at high concentrations of VA (5 mM) for 48 h. (C) Mild but statistically significant induction of apoptosis of cultured thoracic cancer cells was only observed following exposure to high concentration of VA (5.0 mM; # or +P<0.05–0.01 vs control cells or VA (1mM)-treated cells, respectively, by ANOVA and pairwise comparison by Bonferroni test). Cells were continuously treated with VA at either 1.0 or 5.0 mM for 48 h and harvested for quantitation of apoptosis by the TUNEL-based ApoBrdU assay and flow cytometry. Data are expressed as mean±s.e.m. of three independent experiments.
Profound enhancement of apoptosis induction by combining VA with kinase inhibitors
We first determined if VA, as an HDACI, would induce activation of NF-κB transcriptional activity that could be suppressed by kinase inhibitor CC, STP or UCN-01 as was previously demonstrated with TSA. Treating NSCLC cells H460, EsC cells TE12 or the MPM cells H211 with VA (1.0 or 5.0 mM) for 24 h resulted in a cell line- and dose-dependent 5–20-fold increase of NF-κB transcriptional activity (Figure 4). Such robust upregulation of NF-κB activity was significantly suppressed by CC (2 μM), STP (200 nM) or UCN-01 (500 nM) when assayed 24 h after exposure to the respective drug combinations (Figure 4). Staurosporine was most effective among the kinase inhibitors at inhibiting VA-mediated NF-κB activation. UCN-01, on the other hand, in best experimental conditions mediated 50% (two-fold) to 70% (three-fold) reduction of VA-mediated NF-κB activation. Moreover, there was significant reduction in the levels of the antiapoptotic proteins cIAP1, BclXL and Bcl2 without discernible alterations in the levels of proapoptotic proteins Bax, Bak in H460 or TE12 cells treated with the VA+UCN-01 combinations (Figure 5). The lack of alteration of Bax and Bak, taken together with reduction of the levels of antiapoptotic proteins in cancer cells exposed to the VA+UCN-01 combination, would favour a more proapoptotic milieu within treated cells. Moreover, the effect of UCN-01 on ERK1/2, Akt and PKC activity in VA-treated cells was evaluated by Western blotting for levels of pERK1/2, pAkt and p-adducin (Figure 6). Valproic acid mediated a dose-dependent upregulation of PKC kinase activity as indicated by increased p-adducin levels in H460 and H513 cells, whereas there was no discernable alteration of the already high basal p-adducin level in TE12 cells and these were completely abrogated by UCN-01. Similarly, UCN-01 significantly inhibited ERK1/2 activation in VA-treated H460 and TE12 cells but not H513 cells. On the other hand, UCN-01 mediated a clear depletion of pAkt in H513, H460 and TE12 cells. The VA-induced accumulation of hyperacetylated histone H4 in H460, H513 and TE12 cells was not altered by UCN-01 (data not shown), implying that the synergistic interaction between STP or UCN-01 and VA to mediate suppression of NF-κB transcriptional activity and induction of apoptosis was independent of the HDAC-inhibitory activity of VA (Figure 6).
Figure 4.

Suppression of VA-mediated activation of NF-κB by Calphostin C (CC), Staurosporine (STP), UCN-01 or Parthenolide in H460, TE12 and H211 cells. Cells were transiently transfected with NF-κB-luciferase reporter plasmid and concurrently treated with VA (1 or 5 mM) in combination with CC (2 μM), STP (200 nM), UCN-01 (500 nM) or Parthenolide (20 μM). Cells were harvested 24 h after the onset of drug treatment and assayed for luciferase activity. Data are presented as fold increase of luciferase activity, normalised for cellular proteins, from baseline activity of untreated controls (means±s.e.m. of three independent experiments; +P<0.05–0.01 and #P<0.001 vs controls by ANOVA and pairwise comparison by Bonferroni test).
Figure 5.

Reduction of Bcl2, BclXL, cIAP1 levels without alteration of the expression of Bak or Bax in TE12 or H460 cells treated with VA (1.0 or 5.0 mM) and UCN-01 (500 nM) concurrent combinations. Representative data of two independent experiments with similar results are shown here.
Figure 6.

Suppression of pERK1/2, pAkt and p-adducin levels in VA (1.0 or 5.0 mM)-treated H460, TE12 and H513 cells by UCN-01 (500 nM). Representative data of two independent experiments with similar results are shown here.
Suppression of VA-mediated NF-κB activation by the PKC inhibitor CC or by the kinase inhibitors STP and UCN-01 was correlated with significant synergistic induction of apoptosis in NSCLC cells H460, EsC cells TE12 or MPM cells H211 at both the clinically relevant VA concentration of 1.0 mM and particularly at higher VA concentration of 5.0 mM (Figure 7A). We further investigated the ability of UCN-01 to mediate synergistic cytotoxic effect in combination with VA in H322, H460, TE2 and H513 (Figure 7B). Whereas <25% of cultured thoracic cancer cells treated with either VA (1.0 or 5.0 mM) or UCN-01 (500 nM) underwent apoptosis, 60–85% of these cells were TUNEL-positive 48 h after the onset of exposure to combinations of VA+UCN-01. The magnitude of the interaction between UCN-01 (at 500 nM) and VA (1.0 or 5.0 mM) to mediate additive (H460 and H513) or supra-additive (H322, TE2, TE12, H211) apoptotic cell death varied between cell lines at VA at 1.0 mM but uniformly profound at higher concentration of VA of 5.0 mM in all cell lines (Figure 7B). At the indicated concentrations, STP (200 nM) was a more rapid and potent inhibitor of PKC than UCN-01 (500 nM) (Figure 2). As PKC was previously demonstrated to be a positive regulator of HDACI-mediated NF-κB activation (Maxhimer et al, 2005), it was not surprising to observe that STP was significantly more potent than UCN-01 in abrogating VA-mediated activation of NF-κB (Figure 4). This was positively correlated with the more pronounced induction of apoptosis by the STP+VA (1.0 mM) combination (Figure 8). We next investigated the effect of direct inhibition of NF-κB by using the pharmacologic inhibitor Parthenolide that interferes with NF-κB activation by targeting the IκB kinase complex via inhibition of NIK- and MEKK1-induced activation of IKKα and IKKβ (Murphy et al, 1988; Bork et al, 1997; Hehner et al, 1999; Curry et al, 2004). Parthenolide at 30 μM completely abrogated VA-induced NF-κB transcriptional activation in the representative cultured thoracic cancer cells (Figure 4). This was associated with substantial increase of apoptosis of cells treated with VA+Parthenolide, particularly at the low, clinically relevant concentration of VA of 1.0 mM (Figures 7A and 9). These findings were in complete agreement with observations previously reported by our group (Maxhimer et al, 2005) as well as by others (Mayo et al, 2003, Rundall et al, 2004) and confirmed the notion that activation of NF-κB in HDACI-treated cells impeded the ability of HDACI to effectively mediate cell death.
Figure 7.
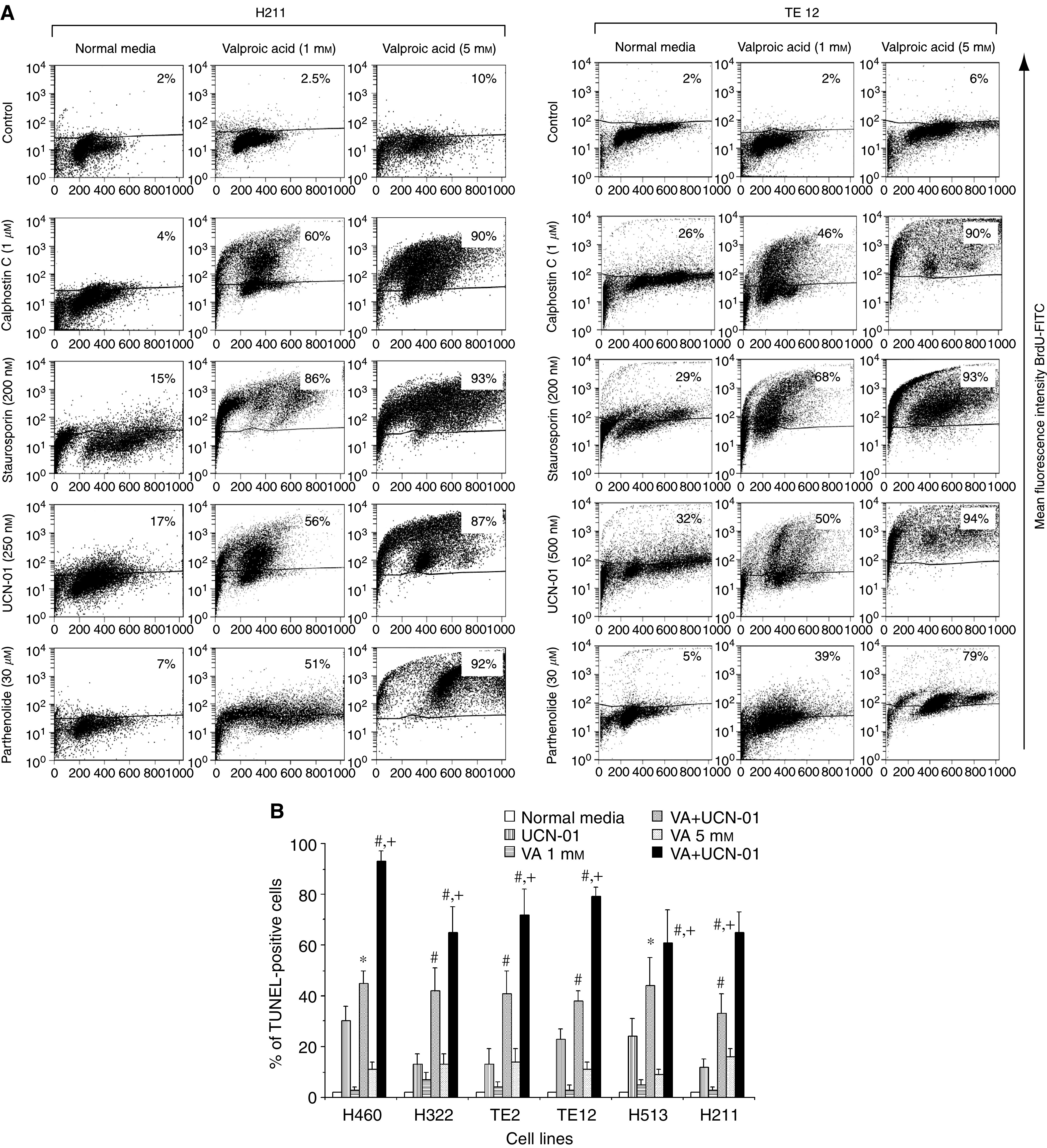
(A) Profound enhancement of apoptosis by the combinations of VA (1.0 or 5.0 mM) with CC, STP, UCN-01 or Parthenolide in the representative cultured thoracic cancer cells TE12 or H211. Apoptosis was quantified by TUNEL-based ApoBrdU assay and flow cytometry. (B) Summary of the VA dose-dependent additive or supra-additive induction of apoptosis in the panel of six cultured thoracic cancer cells treated with VA and UCN-01 (500 nM) combinations. Additive enhancement of apoptosis was observed in H460 and H513 cells treated with VA (1.0 mM+UCN-01; *P>0.05 combination effects vs sum of individual drug effects) and supra-additive enhancement of apoptosis was observed in other cell lines and combinations, especially at the clinically relevant concentration of VA of 1.0 mM (# P<0.05–0.001 combination effects vs sum of individual drug effects). The magnitude of apoptosis induced by VA+UCN-01 was clearly dependent on VA concentrations (+P<0.05–0.001 VA(1.0)+UCN-01 vs VA(5 mM)+UCN-01). Data are expressed as mean±s.e.m. of three independent experiments.
Figure 8.

Staurosporine (200 nM) is more potent than UCN-01 (500 nM) in mediating supra-additive enhancement of apoptosis in combination with low concentration of VA of 1.0 mM (#P=0.0022–0.0001 VA+STP vs VA+UCN-01). Data are expressed as mean±s.e.m. of three independent experiments.
Figure 9.

Supra-additive induction of apoptosis following concurrent exposure of cultured thoracic cancer cells to the combinations of VA (1.0 or 5.0 mM) and Parthenolide (30 μM) (+P<0.05–0.01 combination effects of VA(1.0 mM)+Parthenolide vs the sum of individual drug effects and #P<0.0001 combination effects of VA(5.0 mM)+Parthenolide vs the sum of individual drug effects). Data are expressed as mean±s.e.m. of three independent experiments.
DISCUSSION
In this study, we attempted to evaluate the possibility of enhancing the cytotoxic effect of VA, a commonly used antiepileptic drug with HDAC-inhibitory activity, on cultured thoracic cancer cells by combining it with the kinase inhibitor STP or its clinically relevant analogue UCN-01. Valproic acid, by itself, is not a very efficient anticancer agent, at least for thoracic cancers. It exerts a mild growth-inhibitory effect in cultured thoracic cancer cells with the in vitro IC50's ranging from 4.0 to 8.0 mM. This is mainly attributable to cell cycle arrest at the G1/S checkpoint and very weak induction of apoptosis. Similar to other well-established HDACIs like TSA or SAHA, VA significantly stimulated the NF-κB transcriptional activity in cultured thoracic cancer cells. The kinetics of VA-induced NF-κB activation was somewhat delayed with discernible increase of the transcriptional activity being observed only 18–24 h of drug exposure as compared to the rapid NF-κB activation (6–12 h after the onset of drug exposure) following TSA treatment (data not shown). Partial to complete inhibition of VA-induced activation of NF-κB was achieved by concurrent exposure of VA-treated cells to UCN-01, CC or STP. As HDACI-mediated activation of NF-κB transcriptional activity is positively regulated by PKC, it is conceivable that UCN-01 or STP, being potent PKC inhibitor (Figures 1A and 2), suppressed NF-κB activation in VA-treated cells via this mechanism. As a matter of fact, high levels of p-adducin in VA-treated cells were totally abrogated by either STP or UCN-01 (Figure 2). Suppression of NF-κB activation by these kinase inhibitors was paralleled by the supra-additive induction of apoptosis in combination-treated cells, the magnitude of which appeared to correlate with the degrees of suppressed NF-κB transcriptional activity (for instance, STP vs UCN-01). Staurosporine (200 nM) was more efficient than UCN-01 (500 nM) in mediating profound apoptosis of cells concurrently treated with the clinically relevant concentration of VA of 1.0 mM (Figure 8). Inhibition of NF-κB transcriptional activity in VA-treated cells by STP or UCN-01 has functional significance and directly contributed to the synergistic enhancement of apoptosis and not merely a secondary event or a surrogate marker of downregulation of PKC activity. In concordance with previous reports (Rundall et al, 2004), including our own (Maxhimer et al, 2005), direct inhibition of NF-κB by using Parthenolide profoundly potentiated apoptosis of VA-treated cells. Parthenolide, a sesquiterpene lactone that was first isolated from the feverfew herb (Tanacetum parthenium) is a relatively specific small molecule inhibitor of NF-κB (Bork et al, 1997; Hehner et al, 1999). Parthenolide has undergone a phase I clinical trial as an anticancer agent (Curry et al, 2004). Thus, combining clinically applicable HDACIs such as Depsipetide, MS-275 or SAHA with NF-κB inhibitor like Parthenolide to enhance the anticancer effect of HDACIs may have clinically translatable potential. Targeting the NF-κB signalling pathway, similar to targeting PKC, using pharmacologic inhibitors to enhance the cytotoxic effects of VA as well as other HDACIs in cancers may have practical clinical application. This is the current focus of our laboratory investigation.
Staurosporine and UCN-01, in addition to being PKC inhibitors, have also been shown to inhibit the kinase activity of PDK1, leading to the suppression of Akt phosphorylation and activation among other downstream targets (Sato et al, 2002). Sato et al (2004) have also demonstrated that PDK1 may directly phosphorylate and activate MEK and ERK1/2. It is therefore conceivable that STP or UCN-01 can mediate suppression of Akt and/or ERK1/2 activation. Indeed, UCN-01 has been shown to downregulate Akt activation (but concomitantly stimulate ERK1/2) in head and neck squamous cell carcinoma (Amornphimoltham et al, 2004; Kondapaka et al, 2004). Continuous exposure of thoracic cancer cells to UCN-01 (250–1000 nM) in 10% FCS RPMI culture medium (in contrast to low serum conditions as were previously described (Amornphimoltham et al, 2004; Kondapaka et al, 2004)) led to a profound but short-lived reduction of pAkt at 1 h after drug exposure followed by a strong activation of Akt at 24 h time point. On the other hand, there was a profound and durable inhibition of ERK1/2 activation in UCN-01-treated cells. This is in direct contrast to previous studies that described activation of MEK/ERK1/2 by UCN-01 in head/neck squamous cell carcinoma cell lines (Amornphimoltham et al, 2004; Kondapaka et al, 2004) or leukaemia cell lines (Dai et al, 2001, 2002). The mechanism of this discrepancy is not clear and may relate to the intrinsic difference of cell lines and experimental conditions employed. Staurosporine profoundly inhibited ERK1/2 activation and at the same time mediated phosphorylation of Akt in cultured thoracic cancer cells within the similar time interval. This effect of STP on Akt phosphorylation was surprising, given the fact that its closely related analogue UCN-01 suppressed Akt phosphorylation (Sato et al, 2002; Amornphimoltham et al, 2004; Kondapaka et al, 2004; and also our own observation). This was totally unexpected but very reproducible in many independent experiments with our cell lines and the molecular basis of this discrepancy was unclear. Not surprising, however, STP or UCN-01 exerted a potent inhibitory effect on PKC activity indicated by a profound time- and dose-dependent depletion of p-adducin levels in treated H460 or TE12 cells. Grant and co-workers have observed reduction of MEK/ERK1/2 activity in cells treated with combinations of HDACIs and other kinase inhibitors, including perifosine, 17-AAG or LY294002 that mediated substantial apoptosis (Dai et al, 2001; Rahmani et al, 2003b, 2005). Downregulation of MEK/ERK1/2 plays an important role in the synergistic induction of apoptosis as forced expression of constitutively active MEK abrogates the cytotoxic effects of these drug combinations (Dai et al, 2001; Rahmani et al, 2003b, 2005). It was further indicated that LY294002 enhances HDACI cytotoxicity via downregulation of MEK/ERK1/2 activity but not via abrogation of Akt activation (Rahmani et al, 2003b). It, therefore, appears from the literature that abrogation of MEK/ERK1/2 signalling may have more dominant impact on the enhancement of HDACI cytotoxicity in cancer cells than inhibition of Akt-mediated signalling pathway. UCN-01 and STP consistently suppressed MEK/ERK1/2 and PKC activity in VA-treated cells and correlated well with its synergistic interaction with VA to induce massive apoptosis. It is not known if UCN-01- or STP-mediated inhibition of VA-induced NF-κB activation, while conceivably attributable to their PKC-inhibitory effect, can be mechanistically linked to their ability to downregulate ERK1/2 or Akt activity. Inhibition of MEK/ERK1/2 and PKC by STP or UCN-01 can both contribute to their VA sensitisation effect in thoracic cancer cells, yet the relative contribution of each to this effect is not clear at present. Even though we did not focus our investigation on the effect of UCN-01 on PDK1 kinase activity, it is conceivable that via inhibition of PDK1, which occupies an apical position in regulating multiple downstream kinases, UCN-01 can simultaneously inhibit multiple parallel targets including PKC, MEK/ERK1/2 and Akt.
In summary, UCN-01 and VA, at clinically achievable drug concentrations and conditions (for instance 1.0 mM of VA and 500 nM of UCN-01) (Brodie and Dichter, 1996; Sausville et al, 2001), interact to mediate additive and supra-additive induction of apoptosis of cultured thoracic cancer cells. This chemosensitisation effect of UCN-0-1 in VA-treated cells is most likely secondary to its effect on downregulation of essential signal transduction pathways (PKC, ERK1/2, NF-κB) that regulate HDACI-induced apoptosis. Moreover, our study further confirms NF-κB as a valuable target in the ongoing development of clinically applicable strategies to potentiate the anticancer effect of HDACIs. Within this context, direct targeting of NF-κB using clinically relevant pharmacologic inhibitor like Parthenolide appears to be more effective in enhancing the anticancer property of VA, particularly at concentration such as 1.0 mM that can be readily achievable in the clinic.
Acknowledgments
This research was supported by the Intramural Research Programme of the National Cancer Institute, NIH.
References
- Amornphimoltham P, Sriuranpong V, Patel V, Benavides F, Conti CJ, Sauk J, Sausville EA, Molinolo AA, Gutkind JS (2004) Persistent activation of the Akt pathway in head and neck squamous cell carcinoma: a potential target for UCN-01. Clin Cancer Res 10: 4029–4037 [DOI] [PubMed] [Google Scholar]
- Blaheta RA, Cinatl Jr J (2002) Anti-tumor mechanisms of valproate: a novel role for an old drug. Med Res Rev 22: 492–511 [DOI] [PubMed] [Google Scholar]
- Bork PM, Schmitz ML, Kuhnt M, Escher C, Heinrich M (1997) Sesquiterpene lactone containing Mexican Indian medicinal plants and pure sesquiterpene lactones as potent inhibitors of transcription factor NF-kappaB. FEBS Lett 402: 85–90 [DOI] [PubMed] [Google Scholar]
- Brodie MJ, Dichter MA (1996) Antiepileptic drugs. N Engl J Med 334: 168–175 [DOI] [PubMed] [Google Scholar]
- Burgess AJ, Pavey S, Warrener R, Hunter LJ, Piva TJ, Musgrove EA, Saunders N, Parsons PG, Gabrielli BG (2001) Up-regulation of p21(WAF1/CIP1) by histone deacetylase inhibitors reduces their cytotoxicity. Mol Pharmacol 60: 828–837 [PubMed] [Google Scholar]
- Curry III EA, Murry DJ, Yoder C, Fife K, Armstrong V, Nakshatri H, O'Connell M, Sweeney CJ (2004) Phase I dose escalation trial of feverfew with standardized doses of parthenolide in patients with cancer. Invest New Drugs 22: 299–305 [DOI] [PubMed] [Google Scholar]
- Dai Y, Landowski TH, Rosen ST, Dent P, Grant S (2002) Combined treatment with the checkpoint abrogator UCN-01 and MEK1/2 inhibitors potently induces apoptosis in drug-sensitive and -resistant myeloma cells through an IL-6-independent mechanism. Blood 100: 3333–3343 [DOI] [PubMed] [Google Scholar]
- Dai Y, Rahmani M, Grant S (2003) An intact NF-kappaB pathway is required for histone deacetylase inhibitor-induced G1 arrest and maturation in U937 human myeloid leukemia cells. Cell Cycle 2: 467–472 [PubMed] [Google Scholar]
- Dai Y, Yu C, Singh V, Tang L, Wang Z, McInistry R, Dent P, Grant S (2001) Pharmacological inhibitors of the mitogen-activated protein kinase (MAPK) kinase/MAPK cascade interact synergistically with UCN-01 to induce mitochondrial dysfunction and apoptosis in human leukemia cells. Cancer Res 61: 5106–5115 [PubMed] [Google Scholar]
- Gottlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S, Sleeman JP, Lo CF, Nervi C, Pelicci PG, Heinzel T (2001) Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J 20: 6969–6978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunstein M (1997) Histone acetylation in chromatin structure and transcription. Nature 389: 349–352 [DOI] [PubMed] [Google Scholar]
- Gurvich N, Tsygankova OM, Meinkoth JL, Klein PS (2004) Histone deacetylase is a target of valproic acid-mediated cellular differentiation. Cancer Res 64: 1079–1086 [DOI] [PubMed] [Google Scholar]
- Hehner SP, Hofmann TG, Droge W, Schmitz ML (1999) The antiinflammatory sesquiterpene lactone parthenolide inhibits NF-kappa B by targeting the I kappa B kinase complex. J Immunol 163: 5617–5623 [PubMed] [Google Scholar]
- Jaboin J, Wild J, Hamidi H, Khanna C, Kim CJ, Robey R, Bates SE, Thiele CJ (2002) MS-27-275, an inhibitor of histone deacetylase, has marked in vitro and in vivo antitumor activity against pediatric solid tumors. Cancer Res 62: 6108–6115 [PubMed] [Google Scholar]
- Kelly WK, Richon VM, O'Connor O, Curley T, MacGregor-Curtelli B, Tong W, Klang M, Schwartz L, Richardson S, Rosa E, Drobnjak M, Cordon-Cordo C, Chiao JH, Rifkind R, Marks PA, Scher H (2003) Phase I clinical trial of histone deacetylase inhibitor: suberoylanilide hydroxamic acid administered intravenously. Clin Cancer Res 9: 3578–3588 [PubMed] [Google Scholar]
- Kim YK, Han JW, Woo YN, Chun JK, Yoo JY, Cho EJ, Hong S, Lee HY, Lee YW, Lee HW (2003) Expression of p21(WAF1/Cip1) through Sp1 sites by histone deacetylase inhibitor apicidin requires PI 3-kinase-PKC epsilon signaling pathway. Oncogene 22: 6023–6031 [DOI] [PubMed] [Google Scholar]
- Kondapaka SB, Zarnowski M, Yver DR, Sausville EA, Cushman SW (2004) 7-Hydroxystaurosporine (UCN-01) inhibition of Akt Thr308 but not Ser473 phosphorylation: a basis for decreased insulin-stimulated glucose transport. Clin Cancer Res 10: 7192–7198 [DOI] [PubMed] [Google Scholar]
- Kramer OH, Zhu P, Ostendorff HP, Golebiewski M, Tiefenbach J, Peters MA, Brill B, Groner B, Bach I, Heinzel T, Gottlicher M (2003) The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO J 22: 3411–3420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks PA, Jiang X (2005) Histone deacetylase inhibitors in programmed cell death and cancer therapy. Cell Cycle 4: 549–551 [DOI] [PubMed] [Google Scholar]
- Maxhimer JB, Reddy RM, Zuo J, Cole GW, Schrump DS, Nguyen DM (2005) Induction of apoptosis of lung and esophageal cancer cells treated with the combination of histone deacetylase inhibitor (trichostatin A) and protein kinase C inhibitor (calphostin C). J Thorac Cardiovasc Surg 129: 53–63 [DOI] [PubMed] [Google Scholar]
- Mayo MW, Denlinger CE, Broad RM, Yeung F, Reilly ET, Shi Y, Jones DR (2003) Ineffectiveness of histone deacetylase inhibitors to induce apoptosis involves the transcriptional activation of NF-kappa B through the Akt pathway. J Biol Chem 278: 18980–18989 [DOI] [PubMed] [Google Scholar]
- Murphy JJ, Heptinstall S, Mitchell JR (1988) Randomised double-blind placebo-controlled trial of feverfew in migraine prevention. Lancet 2: 189–192 [DOI] [PubMed] [Google Scholar]
- Nguyen DM, Schrump WD, Chen GA, Tsai W, Nguyen P, Trepel JB, Schrump DS (2004) Abrogation of p21 expression by flavopiridol enhances depsipeptide-mediated apoptosis in malignant pleural mesothelioma cells. Clin Cancer Res 10: 1813–1825 [DOI] [PubMed] [Google Scholar]
- Nguyen DM, Schrump WD, Tsai WS, Chen A, Stewart JH, Steiner F, Schrump DS (2003) Enhancement of depsipeptide-mediated apoptosis of lung or esophageal cancer cells by flavopiridol: activation of the mitochondria-dependent death-signaling pathway. J Thorac Cardiovasc Surg 125: 1132–1142 [DOI] [PubMed] [Google Scholar]
- Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS (2001) Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem 276: 36734–36741 [DOI] [PubMed] [Google Scholar]
- Piekarz R, Bates S (2004) A review of depsipeptide and other histone deacetylase inhibitors in clinical trials. Curr Pharm Des 10: 2289–2298 [DOI] [PubMed] [Google Scholar]
- Rahmani M, Reese E, Dai Y, Bauer C, Payne SG, Dent P, Spiegel S, Grant S (2005) Coadministration of histone deacetylase inhibitors and perifosine synergistically induces apoptosis in human leukemia cells through Akt and ERK1/2 inactivation and the generation of ceramide and reactive oxygen species. Cancer Res 65: 2422–2432 [DOI] [PubMed] [Google Scholar]
- Rahmani M, Yu C, Dai Y, Reese E, Ahmed W, Dent P, Grant S (2003a) Coadministration of the heat shock protein 90 antagonist 17-allylamino-17-demethoxygeldanamycin with suberoylanilide hydroxamic acid or sodium butyrate synergistically induces apoptosis in human leukemia cells. Cancer Res 63: 8420–8427 [PubMed] [Google Scholar]
- Rahmani M, Yu C, Reese E, Ahmed W, Hirsch K, Dent P, Grant S (2003b) Inhibition of PI-3 kinase sensitizes human leukemic cells to histone deacetylase inhibitor-mediated apoptosis through p44/42 MAP kinase inactivation and abrogation of p21(CIP1/WAF1) induction rather than Akt inhibition. Oncogene 22: 6231–6242 [DOI] [PubMed] [Google Scholar]
- Richon VM, Emiliani S, Verdin E, Webb Y, Breslow R, Rifkind RA, Marks PA (1998) A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc Natl Acad Sci USA 95: 3003–3007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosato RR, Almenara JA, Cartee L, Betts V, Chellappan SP, Grant S (2002) The cyclin-dependent kinase inhibitor flavopiridol disrupts sodium butyrate-induced p21WAF1/CIP1 expression and maturation while reciprocally potentiating apoptosis in human leukemia cells. Mol Cancer Ther 1: 253–266 [PubMed] [Google Scholar]
- Rosato RR, Almenara JA, Yu C, Grant S (2004) Evidence of a functional role for p21WAF1/CIP1 down-regulation in synergistic antileukemic interactions between the histone deacetylase inhibitor sodium butyrate and flavopiridol. Mol Pharmacol 65: 571–581 [DOI] [PubMed] [Google Scholar]
- Rosato RR, Grant S (2004) Histone deacetylase inhibitors in clinical development. Expert Opin Investig Drugs 13: 21–38 [DOI] [PubMed] [Google Scholar]
- Ruefli AA, Ausserlechner MJ, Bernhard D, Sutton VR, Tainton KM, Kofler R, Smyth MJ, Johnstone RW (2001) The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc Natl Acad Sci USA 98: 10833–10838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rundall BK, Denlinger CE, Jones DR (2004) Combined histone deacetylase and NF-kappaB inhibition sensitizes non-small cell lung cancer to cell death. Surgery 136: 416–425 [DOI] [PubMed] [Google Scholar]
- Sambucetti LC, Fischer DD, Zabludoff S, Kwon PO, Chamberlin H, Trogani N, Xu H, Cohen D (1999) Histone deacetylase inhibition selectively alters the activity and expression of cell cycle proteins leading to specific chromatin acetylation and antiproliferative effects. J Biol Chem 274: 34940–34947 [DOI] [PubMed] [Google Scholar]
- Sato S, Fujita N, Tsuruo T (2002) Interference with PDK1-Akt survival signaling pathway by UCN-01 (7-hydroxystaurosporine). Oncogene 21: 1727–1738 [DOI] [PubMed] [Google Scholar]
- Sato S, Fujita N, Tsuruo T (2004) Involvement of 3-phosphoinositide-dependent protein kinase-1 in the MEK/MAPK signal transduction pathway. J Biol Chem 279: 33759–33767 [DOI] [PubMed] [Google Scholar]
- Sausville EA, Arbuck SG, Messmann R, Headlee D, Bauer KS, Lush RM, Murgo A, Figg WD, Lahusen T, Jaken S, Jing X, Roberge M, Fuse E, Kuwabara T, Senderowicz AM (2001) Phase I trial of 72-h continuous infusion UCN-01 in patients with refractory neoplasms. J Clin Oncol 19: 2319–2333 [DOI] [PubMed] [Google Scholar]
- Schrump DS, Nguyen DM (2005) Targeting the epigenome for the treatment and prevention of lung cancer. Semin Oncol 32: 488–502 [DOI] [PubMed] [Google Scholar]
- Schrump DS, Nguyen DM, Kunst TE, Hancox A, Figg WD, Steinberg SM, Pishchik V, Becerra Y (2002) Phase I study of sequential deoxyazacytidine/depsipeptide infusion in patients with malignancies involving lungs or pleura. Clin Lung Cancer 4: 186–192 [DOI] [PubMed] [Google Scholar]
- Seynaeve CM, Kazanietz MG, Blumberg PM, Sausville EA, Worland PJ (1994) Differential inhibition of protein kinase C isozymes by UCN-01, a staurosporine analogue. Mol Pharmacol 45: 1207–1214 [PubMed] [Google Scholar]
- Struhl K (1998) Histone acetylation and transcriptional regulatory mechanisms. Genes Dev 12: 599–606 [DOI] [PubMed] [Google Scholar]
- Wolffe AP, Kurumizaka H (1998) The nucleosome: a powerful regulator of transcription. Prog Nucleic Acid Res Mol Biol 61: 379–422 [DOI] [PubMed] [Google Scholar]
- Yu X, Guo ZS, Marcu MG, Neckers L, Nguyen DM, Chen GA, Schrump DS (2002) Modulation of p53, ErbB1, ErbB2, and Raf-1 expression in lung cancer cells by depsipeptide FR901228. J Natl Cancer Inst 94: 504–513 [DOI] [PubMed] [Google Scholar]