

Abstract
Random mutagenesis and screening for enzymatic activity has been used to engineer horse heart myoglobin to enhance its intrinsic peroxidase activity. A chemically synthesized gene encoding horse heart myoglobin was subjected to successive cycles of PCR random mutagenesis. The mutated myoglobin gene was expressed in Escherichia coli LE392, and the variants were screened for peroxidase activity with a plate assay. Four cycles of mutagenesis and screening produced a series of single, double, triple, and quadruple variants with enhanced peroxidase activity. Steady-state kinetics analysis demonstrated that the quadruple variant T39I/K45D/F46L/I107F exhibits peroxidase activity significantly greater than that of the wild-type protein with k1 (for H2O2 oxidation of metmyoglobin) of 1.34 × 104 M−1 s−1 (≈25-fold that of wild-type myoglobin) and k3 [for reducing the substrate (2, 2′-azino-di-(3-ethyl)benzthiazoline-6-sulfonic acid] of 1.4 × 106 M−1 s−1 (1.6-fold that of wild-type myoglobin). Thermal stability of these variants as measured with circular dichroism spectroscopy demonstrated that the Tm of the quadruple variant is decreased only slightly compared with wild-type (74.1°C vs. 76.5°C). The rate constants for binding of dioxygen exhibited by the quadruple variant are identical to the those observed for wild-type myoglobin (kon, 22.2 × 10−6 M−1 s−1 vs. 22.3 × 10−6 M−1 s−1; koff, 24.3 s−1 vs. 24.2 s−1; KO2, 0.91 × 10−6 M−1 vs. 0.92 × 10−6 M−1). The affinity of the quadruple variant for CO is increased slightly (kon, 0.90 × 10−6 M−1s−1 vs. 0.51 × 10−6 M−1s−1; koff, 5.08 s−1 vs. 3.51 s−1; KCO, 1.77 × 10−7 M−1 vs. 1.45 × 10−7 M−1). All four substitutions are in the heme pocket and within 5 Å of the heme group.
Heme-containing peroxidases are ubiquitous in living organisms and are one of the most extensively studied classes of enzyme (1). Peroxidases catalyze the oxidation of a variety of substrates at the expense of hydroperoxides, most often H2O2. The peroxidase catalytic cycle may be represented by the following sequence of reactions (1):
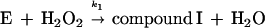 |
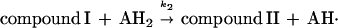 |
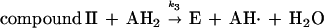 |
The symbol E represents the so-called resting enzyme that possesses Fe(III)-heme, AH2 is a reducing substrate, AH⋅ is the free radical product of the one electron reduction of AH2, and k1, k2 and k3 are rate constants. Compound I is an intermediate form of the enzyme that is oxidized by two equivalents relative to the resting enzyme, and compound II is the one electron reduction product of compound I. Typical heme-containing peroxidases have molecular masses of 30–50 kDa and a ferriprotoporphyrin IX prosthetic group. The structure, function (2), and kinetic properties (3) of peroxidases have been studied extensively.
The catalytic activity and broad substrate specificity of peroxidases has made them widely useful in biochemistry and in diagnostic tests. For this reason, the expression of functional, recombinant peroxidases and the design of novel peroxidases with altered substrate specificity are attractive goals. In the past decade, an increasing number of peroxidases from various sources has been cloned (4–16) and studied by site-directed mutagenesis (17–20). However, peroxidases invariably are expressed in Escherichia coli as either soluble apoproteins or as heme-deficient inclusion bodies (21, 22). This characteristic precludes direct enzymatic assay of peroxidase-producing bacterial colonies.
Myoglobin and hemoglobin are related iron protoheme IX-containing proteins that also have been studied in detail. Although the function of these proteins is the reversible binding of dioxygen, the active sites of hemoglobin and myoglobin of heme proteins share many physical, spectroscopic, and chemical similarities with the active sites of peroxidases. In fact, as demonstrated first in 1900 by Kobert (23), hemoglobin reacts readily with hydrogen peroxide. In 1923, Wu (24) reported the peroxidase activity of hemoglobin, and, in 1938, Polonovski and Jayle (25) first detected the existence of haptoglobin by an increase in the peroxidase activity of hemoglobin caused by interaction of hemoglobin with a preparation that subsequently was shown to contain haptoglobin. The reaction of myoglobin with hydrogen peroxide, on the other hand, apparently was not considered until the report of George and Irvine in 1952 (26), and the ability of myoglobin to catalyze peroxide oxidation of substrates was not reported until the subsequent work of Keilin and Hartree (27). On reaction with H2O2, myoglobin and hemoglobin each form a protein derivative that is similar to compound I formed by peroxidases and that is capable of oxidizing a wide range of reducing substrates such as phenol and aromatic amines. The specific activity of hemoglobin and myoglobin in catalysis of such reactions is, of course, far less than that of peroxidases (28).
Previous work has reported the synthesis of a gene encoding horse heart myoglobin and the use of this gene for efficient expression of native myoglobin in E. coli (29). With the availability of this expression system, the opportunity arose to apply successive cycles of random mutagenesis in conjunction with bacterial colony activity screening (in vitro evolution) to develop variants of myoglobin with enhanced peroxidase activity. A similar strategy has been applied in recent years to identify enzyme variants with increased stability to thermal denaturation (30–32), altered substrate specificity (33, 34), and increased catalytic activity (35–37). Through application of this approach, we now have identified a quadruple variant of horse heart myoglobin with peroxidase activity that is 25-fold greater than that of wild-type myoglobin and that nevertheless retains other essential functional properties of the protein.
MATERIALS AND METHODS
Random Mutagenesis.
The conditions for PCR random mutagenesis (38) were optimized to produce an average of two base pair substitutions per molecule within the gene of horse heart myoglobin: a 100 μl reaction mixture contained 67 mM Tris⋅Cl (pH 8.8), 16.6 mM (NH4)2SO4, 6.7 mM MgSO4, 0.07% β-mercaptoethanol (vol/vol), 0.06 mM MnSO4, 200 μM each of dNTP, 0.1 μM each of oligonucleotide primers, 10 ng template DNA, and 2.5 units Taq DNA polymerase (Life Technologies, Gaithersburg, MD). pGYM, a derivative of plasmid pEMBL18+ containing a synthetic horse heart myoglobin gene (29), was used as the template for the first cycle of PCR random mutagenesis. Subsequent cycles of PCR random mutagenesis used sequentially selected variant plasmid DNA as templates. The forward primer was −21M13 (5′-TGT AAA ACG ACG GCC AGT-3′), and the reverse primer was M13R (5′-CAG GAA ACA GCT ATG ACC-3′), which flank the horse heart myoglobin gene. PCR was carried out with a DNA thermal cycler (Perkin–Elmer/Cetus) with the cycle 96°C, 15 sec; 42°C, 30 sec; 72°C, 45 sec for 25 cycles; then 72°C, 10 min. The PCR reaction was checked by agarose gel electrophoresis, and the amplified DNA fragments were purified with a Wizard PCR Prep kit (Promega). The amplified DNA and pGYM were digested separately with PstI and NcoI, with a Sephaglas BandPrep kit (Pharmacia), and were ligated. The ligation was performed in a total volume of 10 μl containing 0.3 μg PstI- and NcoI-digested pGYM, 0.1 μg amplified DNA, 1 μl 10 mM ATP, 1 μl One-Phor-All PLUS buffer, and 1 μl T4 DNA ligase (Pharmacia, 6.7 Weiss units/μl). The ligation mixture was incubated at 16°C overnight. The ligated plasmid was used to transform E. coli strain LE392 by electroporation with a Gene Pulser (Bio-Rad; Gene Pulser, 2.5 kV, 25 μF; Pulse Controller, 200 Ω) or by the calcium chloride method (39).
Screening Horse Heart Myoglobin Variants for Enhanced Peroxidase Activity.
Transformed E. coli LE392 cells were plated on Luria-Bertani plates containing 100 μg/ml ampicillin (≈100 colonies/plate), and the plates were replicated. E. coli colonies on replica plates were lysed by inverting the plates on chloroform-soaked filter papers for 5 min at room temperature. The plates then were sprayed with solution (4 ml) containing substrate for the peroxidase assay. The freshly prepared stock substrate solution contained 25 mM 2,2′-azino-di-(3-ethyl)benzthiazoline-6-sulfonic acid (ABTS) and 12.5 mM H2O2 in 0.1 M potassium phosphate buffer (pH 6.0). The wild-type horse heart myoglobin has sufficient peroxidase activity to produce a green color with this stock solution but does not produce a signal with successive dilutions. The stock solution was diluted sequentially in successive cycles of screening to increase the stringency of the assay. The dilution of stock substrate solution in each cycle of screening was chosen so that only a few colonies produced a green color. Bacteria producing a green color were isolated from the template plate and were cultured. Plasmid DNA minipreps were made with Qiawell columns (Qiagen, Chatsworth, CA). The resulting plasmid DNAs were used to transform host cells, and the transformants were plated and screened by a second plate assay. Only colonies that gave a positive color reaction in the second screening were used to isolate plasmids that were subjected to DNA sequencing and used as template for the next cycle of PCR random mutagenesis. DNA sequencing was performed by the dye terminator cycle sequencing protocol with a 373 DNA Sequencer (Applied Biosystems) and the −21M13 and M13R primers.
Protein Expression and Purification.
E. coli LE392 freshly transformed with the plasmid containing a horse heart myoglobin variant were inoculated into 5 ml of superbroth containing 200 μg/ml ampicillin and were grown overnight at 37°C. The overnight culture was diluted into superbroth (500 ml) containing 200 μg/ml ampicillin and were grown until A600 = 0.2 ≈ 0.3 (early log phase). This culture was used as inoculum for a 20-liter fermentor. Cells were harvested after growth for 20 h at 37°C. The yield of dark brown cells was ≈80 g. The myoglobin variants were purified by a modification of the protocol of Guillemette et al. (29). The cells were suspended in 400 ml of 20 mM Tris⋅HCl (pH 8.0) (buffer A) and then 400 mg lysozyme, 4 ml 1 M MgC12, 4 mg DNase I, and 2 mg RNase A were added. After stirring for 3 h at 4°C, the cell debris was removed by centrifugation (7,000 × g, 30 min). The supernatant fluid was brought to 50% ammonium sulfate saturation, was equilibrated for 1 h at 4°C, and was centrifuged (7,000 × g, 30 min). The supernatant fluid then was brought to 95% ammonium sulfate saturation and was equilibrated for 3 h at 4°C. After centrifugation (7,000 × g, 30 min), the pellet was redissolved in and dialyzed against buffer A overnight. The protein solution was loaded onto a DEAE-Sepharose CL6B (Pharmacia) column (3 × 15 cm) equilibrated with buffer A. The flow-through fraction was loaded directly onto a Zn-Chelating Fast Flow Sepharose (Pharmacia) column (3 × 10 cm) equilibrated with buffer A. The Zn-chelating column was washed with 0.5 M NaC1 in buffer A before the myoglobin was eluted with 50 mM imidazole in buffer A. (The column was washed with 50 mM EDTA and was regenerated with 0.1 M ZnSO4 in 25 mM acetic acid.) The eluant of partially purified myoglobin was concentrated by centrifugal ultrafiltration (Amicon Centriprep-10) to ≈2 ml and was passed through an FPLC Hiload 26/60 Superdex 75 column (Pharmacia). Fractions with A408/A280 ≥3.5 were pooled. Approximately 2 mg of purified myoglobin were obtained per gram of wet cell pellet. Protein purity was verified by SDS/PAGE (40).
Peroxidase Kinetics of Horse Heart Myoglobin.
The steady-state kinetics of wild-type and variant forms of horse heart myoglobin were analyzed with a Cary 1E UV-visible spectrophotometer (Varian). Initial rates for the oxidation of the substrate ABTS were measured by the change in absorbance at 414 nm over 20 sec at 25°C. The final assay volume was 1 ml, and it contained 0.2 μM horse heart myoglobin, 20 μM ABTS, and varying H2O2 concentrations in 0.1 M Mes (pH 6.0). To choose suitable H2O2 concentrations for initial rate measurements, the H2O2 concentration was varied whereas ABTS concentration was held constant at 20 μM so that the H2O2 concentration at the position of the half-height in the plot of V/[E]0 vs. [H2O2] (V, initial rate; [E]0, myoglobin concentration) could be established. A range from 5× to 1/5 of that H2O2 concentration was used in initial rate measurements. The rate constants k1 and k3 were calculated from the equation for the peroxidase ping-pong mechanism defined by Dunford (2[E]0/V = 1/k1[H2O2] + 1/k3[ABTS]) (3). Myoglobin concentration was determined spectrophotometrically with the pyridine-hemochromogen method (41).
Circular Dichroism Spectroscopy.
Myoglobin samples were prepared in 100 mM sodium phosphate buffer (pH 7.0) and were passed through a 0.22-μm filter to give a final concentration of ≈5 μM. Circular dichroism spectra were recorded with a Jasco Model J-720 spectropolarimeter equipped with a Neslab Model RS-2 remote sensor and a Neslab Model RTE-110 circulating water bath (Neslab Instruments, Portsmouth, NH). The spectropolarimeter was calibrated with ammonium-d-camphor-10-sulfonate (Aldrich). Protein solutions were placed in a cylindrical, water-jacketed quartz cell (0.1-cm path length, volume 200 μl, 25°C), and the average of three scans from 190 to 250 nm was collected. Thermal stability studies were carried out by monitoring the ellipticity at 222 nm over the temperature range 55 to 85°C (heating rate of 50°C/h). The thermal denaturation curves produced in this manner were smoothed with the Jasco software filter function, and the midpoint melting temperature (Tm) was determined from the first derivative of the resulting curve.
Ligand Binding Measurements.
Laser flash photolysis was used for measurement of the rate constants for association of both CO and O2 with wild-type and variant myoglobins. The flash photolysis spectrophotometer was comprised of an optical bench and optical components obtained from OLIS (Bogart, GA) and a Phase-R DL1020 dye laser (LumenX, New Durham, NH) that was operated under computer control (Blue Moon Technical Services, Vancouver). The laser was operated with a methanol solution of Rhodamine 6G (Allied Chemicals, Morristown, NJ). Sample solutions were placed in a modified 10-mm cuvette (3 ml volume).
MbCO samples were prepared by stirring metMb solutions (100 mM sodium phosphate buffer, pH 7.0) under CO for 20 minutes, followed by the addition of sodium dithionite (approximately four equivalents), and stirring under CO for an additional 30 minutes. MbO2 samples were prepared by reducing metMb with sodium dithionite and quickly passing the resulting solution through a Sephadex G-25 column (0.5 × 10 cm) equilibrated with oxygen-saturated sodium phosphate buffer (100 mM, pH 7.0). All samples were prepared immediately before use and to a final protein concentration of ≈10 μM. Bimolecular binding of ligands to myoglobin was followed by monitoring the transient absorbance changes at 436 nm. CO dissociation rates constants were measured by the replacement of CO with NO. NO-saturated buffer was prepared by anaerobically passing NO gas over a column of KOH pellets and then over a stirred solution of deoxygenated sodium phosphate buffer for 30 minutes. A small quantity of a concentrated stock solution of MbCO then was injected, and the reaction was monitored at 424 nm (an absorption maximum in the MbCO and MbNO difference spectrum). Under these conditions, all of the reactions studied followed simple pseudo-first order behavior, and the kinetic data were fitted to a single exponential function (scientist 2.0, Micromath, Orem, Utah).
Rate constants for dissociation of dioxygen from MbO2 were determined by rapid mixing of MbO2 with sodium dithionite in a stopped-flow spectrophotometer. Solutions of MbO2 (10 μM) were mixed with a solution of sodium dithionite prepared under anaerobic conditions to a final concentration of ≈1.5 mg/ml, and the reaction was monitored with a rapid scanning monochromator (OLIS) between 370 and 620 nm. The spectra were analyzed by singular value decomposition and were fitted to a single exponential function with software provided by OLIS. All of the ligand association and dissociation kinetics were studied at 25°C.
RESULTS
Random Mutagenesis.
To probe the effect of the concentration of Mn2+ and the number of cycles on the mutation of horse heart myoglobin amplified by PCR, various protocols were tested to construct mutant pools of plasmid DNA that were used to transform E. coli. Three or four colonies from each mutant pool were picked randomly and were subjected to DNA sequencing. The results showed that the mutation frequency can be adjusted by varying the amount of MnSO4 added to a routine PCR reaction mixture and by varying the number of PCR cycles (Table 1). The PCR conditions can be optimized to achieve an average of one amino acid substitution per molecule. However, all possible mutations were not represented equally in the variants with enhanced peroxidase activity (Table 2). Of the 34 mutations in the sense strand of horse heart myoglobin, 30 resulted from the conversion of A and T, and transitions outnumbered tranversions 24 to 10, with 21 transitions of A → G and T → C.
Table 1.
Optimization of random mutagenesis of horse heart myoglobin by PCR
| PCR protocol, MnSO4 | Nucleotide substitutions | Amino acid substitutions |
|---|---|---|
| 0.50 mM/35 cycles | 6–10 | 4–6 |
| 0.25 mM/25 cycles | 4–6 | 2–4 |
| 0.13 mM/25 cycles | 2–4 | 1–2 |
| 0.06 mM/25 cycles | 2 | 1 |
The PCR protocols for random mutagenesis were modified on the basis of routine PCR [a 100 μl reaction mixture contained 67 mM Tris⋅HCl (pH 8.8), 16.6 mM (NH4)2SO4, 6.7 mM MgSO4, 0.07% β-mercaptoethanol (vol/vol), 200 μM each of dNTP, 0.1 μM each of primers, 10 ng template DNA, and 2.5 units Taq DNA polymerase] by adding varying amounts of MnSO4 and running different number of cycles.
Table 2.
Unique DNA mutations found in the sense strand of all sequenced mutants of the horse heart myoglobin gene after PCR random mutagenesis
| Wild-type | Mutations
|
Types of mutations
|
|||||
|---|---|---|---|---|---|---|---|
| T | C | A | G | Transitions | Transversions | Total | |
| T | — | 9 | 4 | 0 | 9 | 4 | 13 (38%) |
| C | 3 | — | 0 | 0 | 3 | 0 | 3 (9%) |
| A | 4 | 1 | — | 12 | 12 | 5 | 17 (50%) |
| G | 0 | 1 | 0 | — | 0 | 1 | 1 (3%) |
| Total | 34 | 24 (71%) | 10 (29%) | 34 (100%) | |||
Screening.
After the first cycle of mutagenesis, the stock of ABTS solution was diluted 2-fold. At this concentration, bacterial colonies expressing wild-type myoglobin did not produce a color reaction in the plate assay. One-hundred plates (≈10,000 colonies) were screened, and five colonies ultimately were selected after a second plate assay. DNA sequencing revealed that all five of these colonies had a single amino acid replacement, T39I, F46L, K50E, I99T, or I107F (Fig. 1), as a consequence of a single base pair replacement.
Figure 1.

The active sites of (A) horse heart myoglobin (42) and (B) yeast cytochrome c peroxidase (43). The locations of the residues altered by random mutagenesis in the current study are included in the diagram of myoglobin, and critical residues at the active site of cytochrome c peroxidase are indicated. The proximal and distal histidyl residues of both proteins are shown below and above the plane of the heme, respectively.
The five mutants were used individually as templates for a second cycle of PCR random mutagenesis. The amplified DNA fragments were pooled in equal amounts and were subcloned into pEMBL18+. The stock substrate solution was diluted 6-fold, and 130 plates (≈13,000 colonies) were screened. After a second plate assay, one colony finally was selected. The mutant from this colony had two substitutions, F46L/I107F. Other double mutants were constructed by combining the I107F variants with T39I, F46L, or K50E by DNA manipulation through use of a unique Bg1-II restriction site at the position of E59-D60-L61 (29). The resultant double mutants, T39I/I107F, F46L/I107F, and K50E/I107F, had greater apparent peroxidase activity than the single mutants insofar as they all produced a green color in plate assays at one-fourth the concentration of the original screening solution.
The double variants T39I/I107F, F46L/I107F, and K50E/I107F were used individually as templates for the third cycle of PCR random mutagenesis. The amplified DNA fragments of equal amount were pooled and subcloned into pEMBL18+. The stock substrate solution was diluted 30-fold, and 130 plates were screened. Ultimately, one colony with amino acid substitutions of T39I/F46L/I107F was selected (Fig. 1).
The triple variant T39I/F46L/I107F was used as template for the fourth cycle of PCR random mutagenesis, and 180 plates were screened. The stock substrate solution was diluted 100-fold, and one colony with substitutions of T39I/K45D/F46L/I107F was selected (Fig. 1). This quadruple variant was used as template for subsequent PCR random mutagenesis using different PCR protocols with different mutation frequency, and at least 300 plates for each PCR protocol were screened. No mutants that appeared to have improved activity were found. The locations of the amino acid residues that were substituted at the active site of myoglobin in this work are illustrated in Fig. 1.
Peroxidase Kinetics.
As described in the analysis developed by Dunford (3) for the peroxidase ping-pong mechanism, the rate constants k1 and k3 can be determined from steady-state kinetic data, but k2 is indeterminate (3). The three single variants, T39I, F46L, and I107F, have similar rate constant k1, ≈5-fold of its counterpart for wild-type (Table 3). The effect of those substitutions on the increase of k1 is cumulative but not additive. The quadruple variant T39I/K45D/F46L/I107F has the highest k1, 1.34(5) × 104 M−1 s−1, a 24.7-fold increase over the wild-type. The substitutions have little effect on k3. The triple variant T39I/F46L/I107F has the greatest increase, 4.7 × 106 M−1 s−1, 5.3-fold of wild-type, and the quadruple variant, 2.8 × 106 M−1 s−1, is 3.2-fold of wild-type. The promotion of peroxidase activity in the variants was achieved mainly by the increase of k1.
Table 3.
Steady-state kinetic parameters for the peroxidase activity of wild-type and variant forms of horse heart myoglobin [sodium phosphate buffer (0.1 M), pH 6, 25°C]
| Protein |
k1
|
k3
|
||
|---|---|---|---|---|
| (M−1 s−1) | k1(variant)/k1(wild-type) | (M−1 s−1) | k3(variant)/k3(wild-type) | |
| Wild-type Mb | 5.4(2) × 102 | 1 | 8.8(1) × 103 | 1 |
| F46L | 1.32(3) × 103 | 2.4 | 4.0(4) × 103 | 0.5× |
| F46L/I107F | 2.67(2) × 103 | 4.9 | 4.1(3) × 103 | 0.5× |
| T39I/F46L/I107F | 1.02(2) × 104 | 18.9 | 1.9(9) × 104 | 2.2× |
| T39I/K45D/F46L/I107F | 1.34(5) × 104 | 24.7 | 1.4(3) × 104 | 1.6× |
Circular Dichroism Spectroscopy.
The ultraviolet CD spectra of the four variants (data not shown) are all similar to that of wild-type horse heart myoglobin, which is consistent with minimal perturbation of secondary structure by the amino acid substitutions. The melting temperatures obtained from the thermal denaturation curves for the four variants are given in Table 4. Despite the number of substitutions involved, the Tm values obtained for the variants are virtually unchanged from that of the wild-type protein. Only the quadruple variant exhibited slightly reduced stability (ΔTm = −2.4°).
Table 4.
Thermal stability of wild-type and variant forms of horse heart myoglobin in sodium phosphate buffer (100 mM, pH 7.0)
| Protein | Tm, °C |
|---|---|
| Wild-type | 76.5 |
| F46L | 76.1 |
| F46L/I107F | 76.4 |
| T39I/F46L/I107F | 76.2 |
| T39I/K45D/F46L/I107F | 74.1 |
Ligand Binding Measurements.
Representative absorbance traces for O2 and CO recombination with wild-type myoglobin and the quadruple variant are illustrated in Fig. 2. The kinetic parameters derived from these experiments and similar studies of the other variants are provided in Table 5. The second order rate constants (kon) were determined on the basis of solubility constants reported for O2 and CO (44, 45). The values obtained for wild-type myoglobin are in agreement with those reported (41). Relative to wild-type Mb, the F46L variant exhibited a decrease in O2 association rate constant (kon = 1.71 × 107 M−1 s−1) and an increase in O2 dissociation rate constant (koff = 44.3 s−1) so that the affinity of the protein for dioxygen binding was decreased by a factor of 2.3. As additional substitutions were introduced to produce the double, triple and quadruple variants, the affinity of the variants for dioxygen increased incrementally, so that the affinity of the quadruple variant for dioxygen was nearly identical to that of wild-type Mb (9.1 × 10−5 M−1 vs. 9.2 × 10−5 M−1). The mutations also influenced the related parameters for CO binding, but in a less consistent fashion. Again, the F46L variant exhibited a reduced affinity for CO binding, but this behavior resulted from an increased rate constant for dissociation that more than compensated for an increased association rate constant. With sequential introduction of additional mutations, the kinetic parameters for CO binding were affected variably, so that the quadruple variant exhibited kon and koff values that were 1.8-fold and 1.5-fold greater than the corresponding values for the wild-type protein, respectively, and the net effect of the four amino acid substitutions on CO binding affinity was relatively small.
Figure 2.

Transient absorbance at 436 nm after flash photolysis. A shows absorbance traces for the association of deoxy myoglobin with O2, and B shows absorbance traces for the association of deoxy myoglobin with CO. Thin line, wild-type; thick line, quadruple variant T39I/K45D/F46L/I107F.
Table 5.
Parameters for O2 and CO binding to wild-type and variant forms of horse heart myoglobin
| Protein | O2
|
CO
|
||||
|---|---|---|---|---|---|---|
| kon × 10−6 (M−1 s−1) | koff (s−1) | KO2 × 10−6 (M−1) | kon × 10−6 (M−1 s−1) | koff × 102 (s−1) | KCO × 10−7 (M−1) | |
| Wild-type Mb | 22.3 | 24.2 | 0.92 | 0.51 | 3.51 | 1.45 |
| F46L | 17.1 | 44.3 | 0.39 | 0.62 | 5.98 | 1.04 |
| F46L/I107F | 17.1 | 25.2 | 0.68 | 0.55 | 3.31 | 1.66 |
| T39I/F46L/I107F | 19.6 | 23.5 | 0.83 | 0.76 | 4.8 | 1.58 |
| T39I/K45D/F46L/I107F | 22.2 | 24.3 | 0.91 | 0.9 | 5.08 | 1.77 |
DISCUSSION
Myoglobin and hemoglobin are members of the same globin superfamily and are believed to have evolved from a common ancestral or primordial globin over a period of 500–800 million years (46). Evolution conferred on myoglobin and hemoglobin the ability to bind dioxygen reversibly in muscle and blood while some other heme proteins evolved peroxidase activity. The structures of the active sites of horse heart myoglobin and yeast cytochrome c peroxidase (CcP) provided in Fig. 1 permit comparison of selected structural elements of these proteins that demonstrate the resemblance of these proteins while providing insight into the structural basis for their functional differences.
As noted above, both proteins possess a protoheme IX prosthetic group, a proximally coordinated imidazole ligand and a distal His residue. However, the heme iron of myoglobin is always hexacoordinate except for the reduced, deoxygenated derivative, and peroxidases are pentacoordinate except in the presence of exogenous ligands or the addition of peroxides. The hydrogen bonding interactions of the proximal histidyl ligand differ in the two proteins, and the distal histidyl residue of myoglobin is sufficiently close to the distal ligand binding site that it can stabilize coordination of dioxygen, water, or other ligands through hydrogen-bonding interactions. The distal histidyl residue of CcP, however, is sufficiently distant from the heme iron that it does not promote the coordination of a water molecule to the iron atom. In general, the distal heme pocket of CcP is relatively hydrophilic in character whereas the distal heme pocket of myoglobin is relatively hydrophobic. These characteristics are reflected in the divergent functional properties of the two proteins. The structural comparison of the active sites provided in Fig. 1 tends to engender the naive expectation that the functional properties of the two proteins may be interconverted simply by replacing residues observed in one protein with those observed in the other. Preliminary attempts in our laboratories to implement this simplistic approach with site-directed mutagenesis have led to the production of several interesting myoglobin variants that, in fact, exhibit lower peroxidase activity than does the wild-type protein and occasional variants that exhibit no >5- to 6-fold enhancement in activity (unpublished results). Although this approach may eventually produce catalytically effective variants, we turned to random mutagenesis combined with an activity screen as a more efficient method of identifying useful variants.
In Vitro Random Mutagenesis.
The expression of native myoglobin by E. coli combined with the convenience of the plates assays after lysis of bacterial colonies with chloroform permitted efficient screening of a large number of variants. The relatively large change in molar absorptivity that occurs on oxidation of ABTS provides a reasonably sensitive qualitative assay for colonies expressing useful variants. However, correlation of the ABTS concentration required to visualize colonies with the activity of the variant expressed is, of course, compromised by the efficiency with which the variant proteins were expressed by the transformants. As a result, it is essential that any potentially interesting variants identified in this manner be isolated and evaluated quantitatively.
Circular Dichroism Spectroscopy and Thermal Stability.
Both the ultraviolet CD spectra and the identity of the Tm values exhibited by the first three variants (±0.5°C) with the Tm for wild-type Mb suggest that the secondary structures of the four variants are essentially unperturbed relative to that of wild-type horse heart myoglobin. The 2.4°C decrease in Tm for the quadruple variant presumably results from elimination of the hydrogen bond between the heme propionate and K45 in this protein. Elimination of this hydrogen bond destabilizes the binding of heme to apomyoglobin (47), and decreased stability of the heme–apomyoglobin complex is known to be correlated with decreased thermal stability of the holoprotein (48).
Ligand Binding.
After anaerobic reduction, peroxidases readily are oxidized to the ferriheme derivative on addition of dioxygen, but, if carbon monoxide is added instead, relatively stable carbonyl complexes are formed. On the other hand, ferromyoglobin forms stable complexes with both carbon monoxide and dioxygen. This differential ligand binding behavior of myoglobin and peroxidases made it of interest to consider the ligand binding properties of the myoglobin variants selected for increased peroxidase activity. Although introduction of the F46L substitution appears to decrease the affinity of the protein for oxygen slightly, it is interesting that sequential introduction of the other three substitutions returns the parameters for dioxygen binding to those of the wild-type protein as observed for the quadruple variant. Although this pattern is not reproduced by the parameters that define binding of CO, it is nonetheless remarkable that replacement of four residues in the distal heme binding pocket of myoglobin apparently exerts so little influence on the apparent ability of this protein to bind either gaseous ligand. Detailed mechanistic interpretation of the initial results summarized in Table 5 is prevented by our lack of structural information concerning these variants and the relatively small changes in ligand binding parameters observed. Moreover, any meaningful discussion of this type requires consideration of similar data collected as a function of temperature and pH.
Peroxidase Kinetics.
To quantify the effects of the amino acid substitutions replaced by random mutagenesis on the peroxidase activity of horse heart myoglobin, the rate constants k1 and k3 defined by Dunford for the peroxidase ping-pong mechanism were determined under steady-state conditions (3). For the wild-type protein, the rate of H2O2 reduction is slower than that for oxidation of ABTS, which means that the formation of compound I is the rate limiting step. The increase of peroxidase activity observed for each of the variants results primarily from an increase in the rate of reaction with peroxide, k1. On the other hand, the mutations have relatively little effect on the reaction of myoglobin with the substrate ABTS, so that k3 is virtually the same for the wild-type protein and each of the variants. These observations presumably reflect the fact that the amino acid substitutions all occur in the general region of the distal heme binding pocket and that the surface of the protein with which the substrate probably interacts is not changed greatly in any of the variants from that of the wild-type protein. Of interest, a recent analysis of the peroxidase activity of an antibody–heme complex resulted primarily from improved interaction with the substrate (increased k3) with little change in reactivity with peroxide (49)
Molecular Design Considerations.
The present study establishes the feasibility of enhancing the peroxidase activity of myoglobin by in vitro evolution. Perhaps the most intriguing result obtained in this work is that the amino acid residues identified by the strategy of in vitro evolution to promote the peroxidase activity of myoglobin are primarily residues that have attracted minimal attention previously. The proximal and distal histidyl residues and their hydrogen-bonding interactions are apparently unaffected by these changes, and no residues resembling notable residues in the active sites of peroxidases are involved. In short, the new residues introduced at these positions are not residues that normally would be expected to result in significant alteration of protein function. The productive but ill-defined perturbations of the active site that result from the substitutions identified in the present study are sufficiently subtle that they defy interpretation even in retrospect.
Ultimately, the combined application of chance and reason may overcome the limitations inherent in each approach. While this manuscript was in preparation, Watanabe and colleagues reported the construction of two double variants of myoglobin, L29H/H64L and F43H/H64L, by site-directed mutagenesis in an effort to mimic the active site of CcP by effectively moving the distal histidyl residue to a location that is removed sufficiently from the heme iron atom to prevent stabilization of a coordinated water molecule (50, 51). The peroxidase activity of the L29H/H64L variant was found to be lower than that of the wild-type protein (50), and this variant did not form a compound I-like intermediate on addition of hydrogen peroxide. The authors suggested that this variant is not a good mimic of a peroxidase because the distance separating the imidazole group and the heme iron is too great in this variant. Although the peroxidase activity of the F43H/H64L variant was not reported, both variants exhibited significantly greater peroxygenase activity than the wild-type protein (50, 51). Again, the F43H/H64L variant does not form a compound I-like intermediate on addition of hydrogen peroxide, but this behavior results from the fact that this protein exhibits significant catalase activity (51). However, addition of m-chloroperbenzoic acid to the F43H/H64L variant did result in the transient formation of an intermediate with the electronic spectrum of a compound I-like species that was returned to the ferric protein on addition of thioanisole or styrene (52). On the basis of these observations, it seems likely that introduction of the F43H/H64L substitutions into the quadruple variant identified in the present work might result in conversion of myoglobin into a truly effective peroxidase. Another course of in vitro evolution for which the F43H/H64L variant is the starting point also could lead to a variant with the desired properties. Finally, a greater exploration of sequence space, possibly achieved through the strategy of DNA-shuffling (53), almost certainly would result in the generation of myoglobin variants with still greater peroxidase activities.
Acknowledgments
We thank Professor H. Brian Dunford for helpful discussions regarding analysis of peroxidase kinetics and Ms. Kakoli Mistra for assistance in developing the screening assay. This work was supported by the National Centers of Excellence of Canada program in Protein Engineering funded through the Medical Research Council and the Natural Sciences and Engineering Research Council. M. Smith is a Career Investigator of the Medical Research Council of Canada.
ABBREVIATION
- ABTS
2,2′-azido-di-(3-ethyl)-benzthiazoline-6-sulfonic acid)
References
- 1. Everse J, Everse K E, Grisham M B. Peroxidases in Chemistry and Biology, Vols. I and II. Boca Raton, FL: CRC; 1991. [Google Scholar]
- 2.Welinder K G. Curr Opin Struct Biol. 1992;2:388–393. [Google Scholar]
- 3.Dunford H B. In: Peroxidases in Chemistry and Biology. Everse J, Everse K E, Grisham M B, editors. II. Boca Raton: CRC; 1991. pp. 1–24. [Google Scholar]
- 4.Bartonek-Roxà E, Eriksson H, Mattiasson B. Biochim Biophys Acta. 1991;1088:245–250. doi: 10.1016/0167-4781(91)90060-y. [DOI] [PubMed] [Google Scholar]
- 5.Buffard D, Breda C, van Huystee R B, Asemota O, Pierre M, Dang Ha Duc B, Esnault R. Proc Natl Acad Sci USA. 1990;87:8874–8878. doi: 10.1073/pnas.87.22.8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Intapruk C, Highshimura N, Yamamoto K, Okada N, Shinmyo A, Takano M. Gene. 1991;98:237–241. doi: 10.1016/0378-1119(91)90179-f. [DOI] [PubMed] [Google Scholar]
- 7.Rasmussen S K, Welinder K G, Hejgaard J. Plant Mol Biol. 1991;16:317–327. doi: 10.1007/BF00020562. [DOI] [PubMed] [Google Scholar]
- 8.Johansson A, Rasmussen S K, Harthill J E, Welinder K G. Plant Mol Biol. 1992;18:1151–1161. doi: 10.1007/BF00047718. [DOI] [PubMed] [Google Scholar]
- 9.Morgens P H, Callahan A M, Dunn L J, Abeles F B. Plant Mol Biol. 1990;14:715–725. doi: 10.1007/BF00016504. [DOI] [PubMed] [Google Scholar]
- 10.Rebmann G, Hertig C, Bukk J, Mauch F, Dudler R. Plant Mol Biol. 1991;16:329–331. doi: 10.1007/BF00020563. [DOI] [PubMed] [Google Scholar]
- 11.Lagrimini L M, Burkhart W, Moyer M, Rothstein S. Proc Natl Acad Sci USA. 1987;84:7542–7546. doi: 10.1073/pnas.84.21.7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y Z, Reddy C A, Rasooly A. Gene. 1991;97:191–198. doi: 10.1016/0378-1119(91)90051-c. [DOI] [PubMed] [Google Scholar]
- 13.Esworthy R S, Doan K, Doroshow J H, Chu F F. Gene. 1994;144:317–318. doi: 10.1016/0378-1119(94)90400-6. [DOI] [PubMed] [Google Scholar]
- 14.Forkl H, Vandekerckhove J, Drews G, Tadros M H. Eur J Biochem. 1993;214:251–258. doi: 10.1111/j.1432-1033.1993.tb17918.x. [DOI] [PubMed] [Google Scholar]
- 15.Mittler R, Zilinskas B A. J Biol Chem. 1992;267:21802–21807. [PubMed] [Google Scholar]
- 16.Ross R P, Claiborne A. J Mol Biol. 1991;221:857–871. doi: 10.1016/0022-2836(91)80180-3. [DOI] [PubMed] [Google Scholar]
- 17.Wang J, Mauro J M, Edwards S L, Oatley S J, Fishel L A, Ashford V A, Xuong N H, Kraut J. Biochemistry. 1990;29:7160–7173. doi: 10.1021/bi00483a003. [DOI] [PubMed] [Google Scholar]
- 18.Smulevich G, Mauro J M, Fishel L A, English A M, Kraut J, Spiro T G. Biochemistry. 1988;27:5477–5485. doi: 10.1021/bi00415a014. [DOI] [PubMed] [Google Scholar]
- 19.Fishel L A, Villafranca J E, Mauro J M, Kraut J. Biochemistry. 1987;26:351–360. doi: 10.1021/bi00376a004. [DOI] [PubMed] [Google Scholar]
- 20.Newmyer S L, Ortiz de Montellano P R. J Biol Chem. 1995;270:19430–19438. doi: 10.1074/jbc.270.33.19430. [DOI] [PubMed] [Google Scholar]
- 21.Smith A T, Santama N, Dacey S, Edwards M, Bray R C, Thorneley R N F, Burke J F. J Biol Chem. 1990;265:13335–13343. [PubMed] [Google Scholar]
- 22.Fishel L A, Mauro J M, Kraut J. Biochemistry. 1987;26:351–360. doi: 10.1021/bi00376a004. [DOI] [PubMed] [Google Scholar]
- 23.Kobert R. Pflügers Arch Gesamte Physiol Menschen Tiere. 1900;82:603–630. [Google Scholar]
- 24.Wu H. J Biochem (Tokyo) 1923;2:189–194. [Google Scholar]
- 25.Polonovski M, Jayle M F. C R Hebd Seances Mem Soc Biol Ses Fil. 1938;129:457–460. [Google Scholar]
- 26.George P, Irvine D H. Biochem J. 1952;52:511–517. doi: 10.1042/bj0520511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keilin D, Hartree E F. Biochem J. 1955;60:310–325. doi: 10.1042/bj0600310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grisham M B, Everse J. In: Peroxidases in Chemistry and Biology. Everse J, Everse K E, Grisham M B, editors. I. Boca Raton: CRC; 1991. pp. 335–344. [Google Scholar]
- 29.Guillemette J G, Matsushima-Hibiya Y, Atkinson T, Smith M. Protein Eng. 1991;4:585–592. doi: 10.1093/protein/4.5.585. [DOI] [PubMed] [Google Scholar]
- 30.Yamashita M, Kinoshita T, Ihara M, Mikawa T, Murooka Y. J Biochem. 1994;116:1233–1240. doi: 10.1093/oxfordjournals.jbchem.a124669. [DOI] [PubMed] [Google Scholar]
- 31.Rellos P, Scopes R K. Protein Expression Purif. 1994;5:270–277. doi: 10.1006/prep.1994.1041. [DOI] [PubMed] [Google Scholar]
- 32.Kwon K S, Kim J, Shin H S, Yu M H. J Biol Chem. 1994;269:9627–9631. [PubMed] [Google Scholar]
- 33.Olesen K, Kielland-Brandt M C. Protein Eng. 1993;6:409–415. doi: 10.1093/protein/6.4.409. [DOI] [PubMed] [Google Scholar]
- 34.Widersten M, Mannervik B. J Mol Biol. 1995;250:115–122. doi: 10.1006/jmbi.1995.0362. [DOI] [PubMed] [Google Scholar]
- 35.Chen K, Arnold F H. Proc Natl Acad Sci USA. 1993;90:5618–5622. doi: 10.1073/pnas.90.12.5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moore J C, Arnold F H. Nat Biotechnol. 1996;14:458–467. doi: 10.1038/nbt0496-458. [DOI] [PubMed] [Google Scholar]
- 37.Gulick A M, Fahl W E. Proc Natl Acad Sci USA. 1995;92:8140–8144. doi: 10.1073/pnas.92.18.8140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leung D W, Chen E, Goeddel D V. Technique (Philadelphia) 1989;1:11–15. [Google Scholar]
- 39.Cohen S N, Chang A C Y, Hsu L. Proc Natl Acad Sci USA. 1972;69:2110–2114. doi: 10.1073/pnas.69.8.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Laemmli N K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 41.Antonini E, Brunori M. In: Hemoglobin and Myoglobin in Their Reaction with Ligands. Neuberger A, Tatum E L, editors. Amsterdam: North–Holland; 1971b. pp. 222–227. [Google Scholar]
- 42.Evans S V, Brayer G D. J Mol Biol. 1990;213:885–897. doi: 10.1016/S0022-2836(05)80270-0. [DOI] [PubMed] [Google Scholar]
- 43.Finzel B C, Poulos T L, Kraut J. J Biol Chem. 1984;257:13027–13036. [PubMed] [Google Scholar]
- 44.Gordon A J, Ford R A. The Chemists Companion. New York: Wiley Interscience; 1972. [Google Scholar]
- 45.Wiesenburg D A, Guinasso N L. J Am Chem Soc. 1979;97:2927–2928. [Google Scholar]
- 46.Czelusniak J, Goodman M, Hewett-Emmett D, Weiss M L, Venta P J, Tashian R E. Nature (London) 1982;298:297–300. doi: 10.1038/298297a0. [DOI] [PubMed] [Google Scholar]
- 47.Hunter C L, Lloyd E, Eltis L D, Rafferty S P, Lee H, Smith M, Mauk A G. Biochemistry. 1997;36:1010–1017. doi: 10.1021/bi961385u. [DOI] [PubMed] [Google Scholar]
- 48.Hargrove M S, Olson J S. Biochemistry. 1996;35:11310–11318. doi: 10.1021/bi9603736. [DOI] [PubMed] [Google Scholar]
- 49.Kawamura-Konishi Y, Asano A, Yamazaki M, Tashiro H, Suzuki H. J Mol Catal. 1998;B4:181–190. [Google Scholar]
- 50.Ozaki S, Matsui T, Watanabe Y. J Am Chem Soc. 1996;118:9784–9785. [Google Scholar]
- 51.Ozaki S, Matsui T, Watanabe Y. J Am Chem Soc. 1997;119:6666–6667. [Google Scholar]
- 52.Matsui T, Ozaki S, Watanabe Y. J Biol Chem. 1997;272:32735–32738. doi: 10.1074/jbc.272.52.32735. [DOI] [PubMed] [Google Scholar]
- 53.Crameri A, Raillard S-A, Bermudez E, Stemmer W. Nature (London) 1998;391:288–291. doi: 10.1038/34663. [DOI] [PubMed] [Google Scholar]