

Abstract
Neuritic retraction represents a prominent feature of the degenerative phenotype associated with mutations in leucine rich repeat kinase 2 (LRRK2) that are implicated in autosomal dominant and some cases of sporadic Parkinson's disease. Alterations in macroautophagy, the vacuolar catabolism of cytoplasmic constituents, have been described in Parkinson's disease. In this study, we utilized retinoic-acid differentiated SH-SY5Y cells to determine whether autophagy contributes to mutant LRRK2-associated neurite degeneration. Transfection of pre-differentiated SH-SY5Y cells with LRRK2 cDNA containing the common G2019S mutation resulted in significant decreases in neurite length, which were not observed in cells transfected with wild type LRRK2 or its kinase-dead K1906M mutation. G2019S LRRK2 transfected cells also exhibited striking increases in autophagic vacuoles in both neuritic and somatic compartments, as demonstrated by fluorescence and western blot analysis of the autophagy marker green fluorescent protein-tagged microtubule-associated protein Light Chain 3 and by transmission electron microscopy. RNA interference knockdown of LC3 or Atg7, two essential components of the conserved autophagy machinery, reversed the effects of G2019S LRRK2 expression on neuronal process length, whereas rapamycin potentiated these effects. The mitogen activated protein kinase/extracellular signal regulated protein kinase (MAPK/ERK) kinase (MEK) inhibitor 1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio]butadiene (U0126) reduced LRRK2-induced neuritic autophagy and neurite shortening, implicating MAPK/ERK-related signaling. These results indicate an active role for autophagy in neurite remodeling induced by pathogenic mutation of LRRK2.
Keywords: autophagy, extracellular signal regulated protein kinase, leucine-rich repeat kinase 2, neurite degeneration, Parkinson's disease
Genetic models of Parkinsonian neurodegeneration involving leucine rich repeat kinase 2 (LRRK2) have emerged as targets of intense research after several mutations in LRRK2 were shown to segregate with disease in autosomal dominant, PARK8-linked Parkinsonism (Paisan-Ruiz et al. 2004; Zimprich et al. 2004). Studies have demonstrated that the G2019S substitution in the kinase domain confers enhanced kinase activity to LRRK2 that results in cell toxicity (Smith et al. 2005, 2006; West et al. 2005, 2007; Gloeckner et al. 2006; Luzon-Toro et al. 2007). Moreover, engineered kinase-inactive LRRK2 mutants reveal that the pathologic effects of LRRK2 over-expression are dependent upon kinase activity (Gloeckner et al. 2006; Greggio et al. 2006). Mutant LRRK2 expression is associated with progressive neurite shortening that precedes gradual cell death in vitro and in vivo (Macleod et al. 2006). The similarities in the clinicopathologic features of LRRK2-associated familial Parkinsonism and sporadic Parkinson's disease (PD) further support the potential relevance of understanding mechanisms related to LRRK2-associated neurodegeneration (Farrer 2006).
Recent studies from our laboratory demonstrate that macroautophagy, the catabolism of cytoplasmic molecules and organelles following sequestration in an autophagic vacuole (AV), plays a significant role in the MPP+ model of Parkinsonian injury (Zhu et al. 2007). While neurons undergo degeneration when basal autophagic degradation is disrupted (Hara et al. 2006; Komatsu et al. 2006, 2007), dysregulated autophagy can also contribute actively to neurodegeneration (Matyja et al. 2005; Nixon et al. 2005; Chu 2006; Wang et al. 2006; Kiselyov et al. 2007). Inhibition of autophagy induction through knockdown of core autophagy proteins confers protection from MPP+ neurotoxicity (Zhu et al. 2007) and degenerative stimuli in other cell types (Li et al. 2006; Xu et al. 2006). The potential role(s) of autophagy in mediating neuritic injury and remodeling remain to be delineated.
In this study, we demonstrate an active role for autophagy in neurite withdrawal associated with expression of mutant G2019S-LRRK2 in SH-SY5Y neuroblastoma cells that were previously differentiated using retinoic acid (RA). Mutant LRRK2 expression in differentiated SH-SY5Y cells caused neurite shortening with concomitant increases in neuritic and somatic AV content, as demonstrated by the autophagosome marker green fluorescent protein-tagged microtubule-associated protein Light Chain 3 (GFP-LC3) and electron microscopy. Furthermore, RNA interference (RNAi) knockdown of two distinct proteins necessary for autophagy induction significantly reversed these neuritic alterations. These results indicate that autophagy plays an active role in neurite shortening associated with G2019S-LRRK2 expression.
Materials and methods
Cell Culture
SH-SY5Y cells were purchased from ATCC (Manassas, VA, USA). Cells were maintained in a humidified incubator with 5% CO2 at 37°C in antibiotic-free Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal calf serum (BioWhittaker, Walkersville, MD, USA), 2 mM glutamine (BioWhittaker) and 10 mM HEPES (BioWhittaker). For all experiments, cells were plated at a density of 5 × 104/cm2 in LabTek II coverglass culture dishes (Nalge Nunc International/Thermo Fisher (Pittsburgh, PA, USA)) or 6-well plastic culture dishes with the same media formulation supplemented with 10 μM RA to induce differentiation for 72 h prior to transfection. In some experiments, differentiated cell cultures were treated with rapamycin (2 μM; Sigma, St. Louis, MO, USA), 3-methyladenine (3-MA; 5 mM; Sigma) or 1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio]butadiene (U0126); (10 μM; Promega, Madison, WI, USA).
DNA Transfection
Full length, triple hemaglutinin (HA)-tagged wild-type human LRRK2 cDNA was subcloned into the pcDNA3.1 vector. The constructs of K1906M- and G2019S-LRRK2 were engineered via site-directed mutagenesis using the wild type construct as the template. The engineered K1906M mutation has been previously shown to be effective in inhibiting LRRK2 kinase activity (Gloeckner et al. 2006; West et al. 2007). All LRRK2 construct sequences were verified by DNA sequencing and restriction analysis. pEGFP-LC3 was received as a gift from Tamotsu Yoshimori (Osaka University, Japan). EGFP-Cl was purchased from ClonTech (Mountain View, CA, USA) and pmaxGFP was purchased from Amaxa Biosystems (Gaithersburg, MD, USA). Previously differentiated SH-SY5Y cells were co-transfected with a LRRK2 construct and either pEGFP-LC3 or pMAX-GFP in a 15 : 1 mass ratio with 0.1% Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA). Cells were fixed 48-h post-transfection with 4% paraformaldehyde.
RNA interference
Inhibition of the autophagy machinery via RNAi was performed as previously described (Zhu et al. 2007). Briefly, small interfering RNA (siRNA) sequences targeting human LC3 (5′-GAAGGCGCUUACAGCUCAA-3′; 40 nM; Invitrogen) or one of two independent siRNA preparations targeting human Atg7 (5′-GGAGUCACAGCUCUUCCUU-3′, 20 nM, Ambion, Austin, TX, USA; and Dharmacon smartpool Atg7 siRNA, 20 nM, Dharmacon, Lafayette, CO, USA) were introduced using 0.1% Lipofectamine 48 h prior to transfection with respective LRRK2/GFP plasmids. A fluorescent siRNA oligonucleotide (DY-547 siGLO Lamin A/C; 20 nM; Dharmacon) was used to confirm that these conditions result in efficient delivery of siRNA to 95% ± 3.9 (SD) of cells. Non-targeted RNA oligonucleotide pool (Dharmacon) was used as control. Efficacy and specificity of protein knockdown was assessed by western blotting.
Transmission electron microscopy
Cells were grown and treated as above in 6-well plates. At 48 h after LRRK2 transfection, cell monolayers were fixed in 2.5% glutaraldehyde for 1 h and post-fixed for 1-h in 1%OsO4/1%K3Fe(CN)6. Cells were then dehydrated in serial dilutions of ethanol and embedded in Polybed 812 epoxy resin (Polysciences, Warrington, PA, USA). Ultrathin sections (60 nm) were stained and photographed as previously described (Zhu et al. 2007).
Western blotting
Cell lysates were obtained with buffer containing protease inhibitors as previously described (Zhu et al. 2007). Equal amounts of protein per treatment condition, determined by Coomassie Plus Protein Assay (Pierce, Rockford, IL, USA), were electrophoresed through 5–15% gradient polyacrylamide gels and transferred to Immobilon-P membranes (Millipore, Bedford, MA, USA). Membranes were blocked with 5% non-fat milk and subsequently incubated in mouse anti-LC3 IgG (1 : 500, Nanotools), mouse anti-p44/42 mitogen activated protein kinase (MAPK) [p-extracellular signal regulated kinase (ERK), 1 : 1000; Cell Signaling, Danvers, MA, USA), rabbit anti-GFP (1 : 5000; Invitrogen) or mouse anti-HA Tag (1 : 1000, Covance, Princeton, NJ, USA) overnight at 4°C. Immunoreactivity was revealed by probing with horseradish peroxidase conjugated anti-mouse or anti-rabbit IgG (Amersham, Piscataway, NJ, USA) followed by exposure to ECL detection reagents (Amersham). Following membrane stripping, equal protein loading per treatment condition was confirmed by probing with mouse anti-β-actin IgG (1 : 5000; Sigma) or rabbit anti-ERK (t-ERK, 1 : 30 000; Upstate). Densitometric analysis was performed using electrophoresis analysis software (Kodak, Rochester, NY, USA).
Indirect immunofluorescence
Fixed cultures were permeabilized with 0.1% Triton-X in phosphate buffered saline and blocked with 5% donkey serum. Cells were incubated in mouse anti-HA Tag (1 : 1000, Covance) overnight at 4°. Immunoreactivity was revealed via incubation with Cy3-conjugated donkey anti-mouse IgG (1 : 1000; Vector, Burlingame, CA, USA). Nuclei were stained with 4′,6-diamidino-2-phenylindole (1 : 500 dilution; Invitrogen). Cells were imaged on an Olympus IX71 microscope with Microsuite Five imaging software (Olympus America, Melville, NY, USA).
Quantitative image analysis and statistics
Forty random fields of HA-tag labeled cells were photographed by an individual that was blinded to the experimental conditions. The channels were extracted to greyscale and neurite lengths traced and measured using the public domain NIH Image J Software supplemented with Neuron J plug-in (Meijering et al. 2004). GFP-LC3 granule dimensions and counts were analyzed using the threshold function of Image J. GFP-LC3 puncta were defined as sharply demarcated structures that exceeded two standard deviations over the adjacent background of diffuse cytosolic GFP intensity. The nuclei were excluded from analysis and multiple filter sets used to exclude autofluorescent lipofuscin granules, which can be seen in mouse brain sections but was not observed in the SH-SY5Y cell system. Transfected cells with overlapping neurites or cell bodies were excluded from quantitative analysis. Data are expressed as means ± SEM. Statistically significant differences were detected by analyses of variance (ANOVA); groups were subsequently compared using unpaired t-tests with Bonferroni corrections. Values of p < 0.05 were considered statistically significant.
Results
Previously differentiated SH-SY5Y cells were co-transfected with EGFP, to allow visualization of individual cell processes, and one of the following: vector control, wild type-, G2019S- or K1906M- LRRK2 plasmids. Transfection of LRRK2 constructs in differentiated SH-SY5Y cells resulted in diffuse cytosolic LRRK2 expression as demonstrated by immunofluorescent staining for the HA-tag (Fig. 1a). Under our transfection conditions, no aggregation of GFP nor of any of the over-expressed LRRK2 proteins was apparent at the time points studied. Expression of the full length LRRK2 protein was confirmed by western blot demonstration of specific HA-tagged protein bands at 280 kDa for the wild type and mutant LRRK2 constructs (Fig. 1b). Merging the channels for EGFP and Cy3-labeled HA-tag confirmed absence of HA immunoreactivity in vector-transfected cells and successful co-transfection of cells with EGFP and the three LRRK2 plasmids (Fig. 1c). Transfection with G2019S-LRRK2 caused shortened neuritic processes in contrast to cells transfected with vector, wild-type LRRK2 or kinase-dead K1906M-LRRK2 (Fig. 1c and d). Cell body morphology was unaffected by these treatments (Fig. 1e).
Fig. 1.
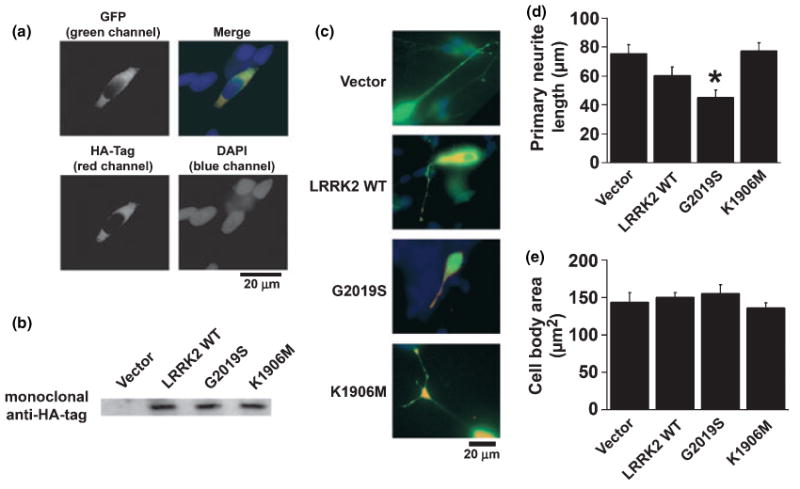
Expression of G2019S-leucine rich repeat kinase 2 (LRRK2)-hemaglutinin (HA) cDNA is associated with blunted neuritic processes in differentiated SH-SY5Y cells. (a) Co-transfection of EGFP-C1 vector and G2019S-LRRK2-HA in differentiated SH-SY5Y cells resulted in diffuse cytosolic expression as analyzed by green fluorescent protein (GFP) fluorescence and immunofluorescence to the HA-tag. The merged image demonstrates efficient co-transfection; nuclei are either associated with expression of both plasmids or neither plasmid. Note the absence of EGFP and LRRK2 aggregates or granules under these conditions. (b) Western blot demonstrating expression of HA-tagged wild type and mutant LRRK2 proteins (280 kDa) in SH-SY5Y cultures. (c) Representative merged EGFP (green) and LRRK2-HA (red) overlay photos showing blunted neuritic processes with G2019S-LRRK2 expression. (d) Quantitative data demonstrating shortened neuritic processes, but (l) preserved cell body dimensions in G2019S-LRRK2 transfected cells. *p < 0.05 vs. vector control.
As alterations in autophagy have been implicated in neuritic pathology (Larsen et al. 2002; Wang et al. 2006), we investigated the potential role of autophagy in LRRK2-mediated alterations. The autophagy marker protein LC3, a mammalian homolog of the yeast Atg8 protein, is covalently modified and redistributes to AVs during induction of autophagy, and this process can be followed by the appearance of LC3 puncta or ring-like structures (Kabeya et al. 2000), with covalent lipidation detected by mobility shift from LC3-I to LC3-II by SDS-PAGE. Cytoplasmic GFP-LC3 puncta were quantified in cells co-transfected with GFP-LC3 and LRRK2 constructs as an index of AV content (Fig. 2). pcDNA vector-transfected cells demonstrated diffuse GFP-LC3 distribution with only very rare GFP-LC3 granules (Fig. 2a and b). In contrast, increases in AV number and size were observed in cells transfected with G2019S-LRRK2 in both neuritic and somatic compartments. Wild-type LRRK2 expression was associated with a trend towards increased AV numbers (Fig. 2b–e). No increase in AV content was observed in cells transfected with kinase dead K1906M-LRRK2.
Fig. 2.
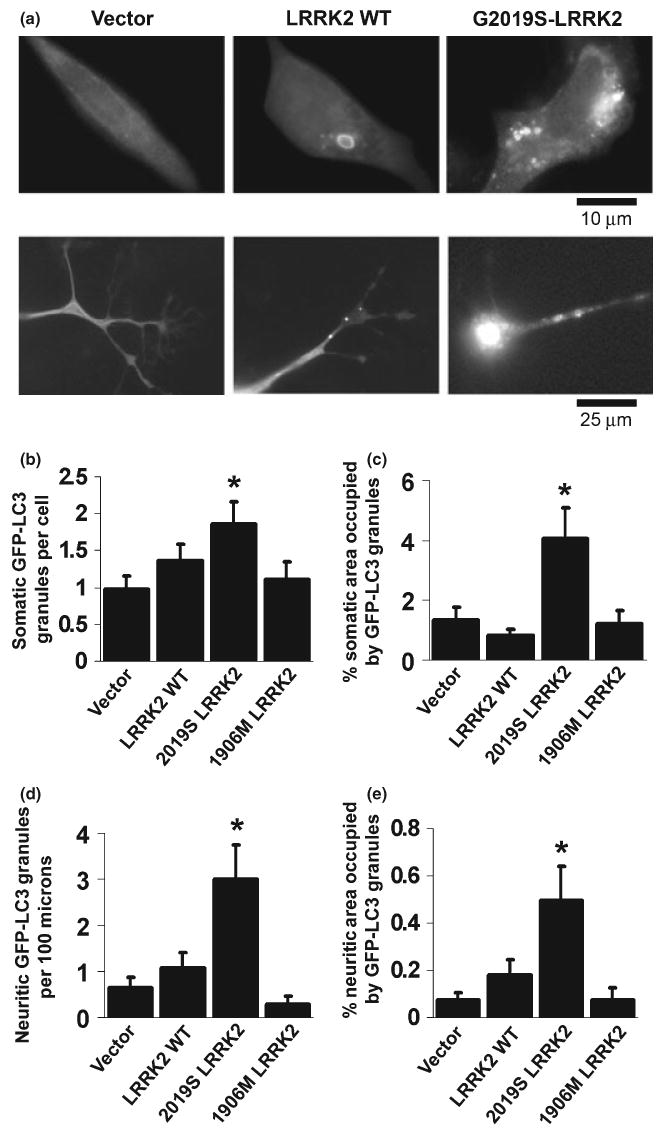
G2019S-leucine rich repeat kinase 2 (LRRK2) expression is associated with increased autophagic vacuoles (AVs) in both somatic and neuritic compartments. (a) Representative photos of LC3 puncta and rings in the soma (upper row) and neurites (lower row) of differentiated SH-SY5Y cells co-transfected with green enhanced fluorescent protein-tagged microtubule-associated protein Light Chain 3 (GFP-LC3) and LRRK2 cDNAs or vector control. Quantitative image analysis demonstrates increases in both the number of AVs and the % cytoplasmic area occupied by AVs in somatic (b, c) and neuritic (d, e) compartments. *p < 0.05 vs. vector control.
Depending on the cell type, protein aggregates have been reported in 5% or more of transfected cells, with the greatest number observed with the Y1699C mutation (Greggio et al. 2006). To verify that the observed changes in LC3 reflected increased AV content rather than protein aggregation (Bjorkoy et al. 2005), differentiated SH-SY5Y cells transfected with G2019S-LRRK2 were studied by transmission electron microscopy (Fig. 3). AVs were uncommon in pcDNA vector treated control cultures (Fig 3a and c). In contrast, G2019S-LRRK2 treated cultures exhibited increased AVs at various stages of maturation in somatic and neuritic compartments (Fig 3b and d). In accord with fluorescence studies of HA-tagged LRRK2, protein aggregates were not readily observed in the electron micrographs.
Fig. 3.
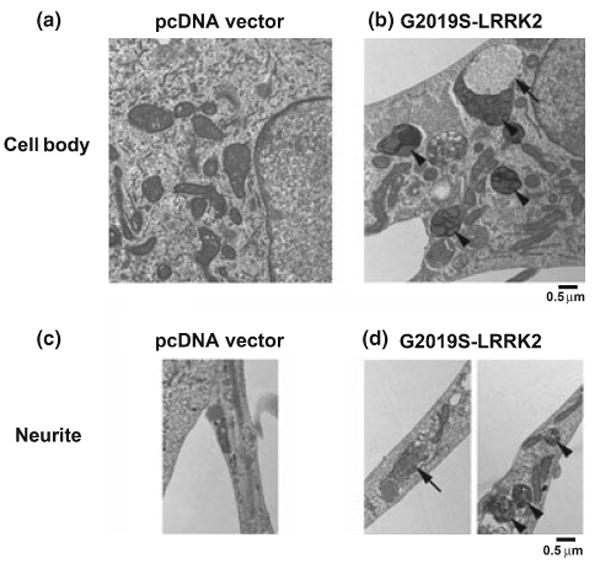
Transmission electron micrographs demonstrating increased autophagic vacuoles (AVs) in differentiated SH-SY5Y cells transfected with G2019S-leucine rich repeat kinase 2 (LRRK2). Compared to vector-transfection (a and c), cultures transfected with G2019S-LRRK2 (b and d) exhibit increases in both early (arrows) and late AVs (arrowheads) in somatic (a, b) and neuritic (c, d) compartments. Earlier AVs were observed more frequently in neurites, while predominantly late AVs accumulated in the soma.
RNA-interference knockdown of core autophagy proteins was performed to test the hypothesis that autophagy plays an active role in neurite shortening associated with G2019S-LRRK2 expression. In contrast to the neurite shortening observed with G2019S-LRRK2 expression following control siRNA treatment, knockdown of LC3/Atg8 expression using a previously described siRNA sequence (Zhu et al. 2007) attenuated G2019S-LRRK2-induced neurite shortening (Fig. 4a). Protection from G2019S-LRRK2-induced neurite shortening was also observed with RNAi knockdown of Atg7, using either a commercially available siRNA preparation (not illustrated) or a previously published siRNA sequence (Fig. 4c) (Yu et al. 2004). RNAi knockdown of Atg 7 or LC3 was confirmed by western blot analysis (Fig. 4b and c, inset panels). Reversal of G2019S-LRRK2-induced neurite autophagy by RNAi knockdown of Atg7 was established by monitoring GFP-LC3 puncta in Atg7 siRNA treated cells (Fig. 4d) and by inhibition of the GFP-LC3 mobility shift on western blot (Fig. 4d, inset panel). Greater than 90% siRNA transfection efficiency of cells, including cells over-expressing HA-tagged 2019S-LRRK2, was demonstrated using a fluorescent tagged short double-stranded RNA (Fig. 4e).
Fig. 4.
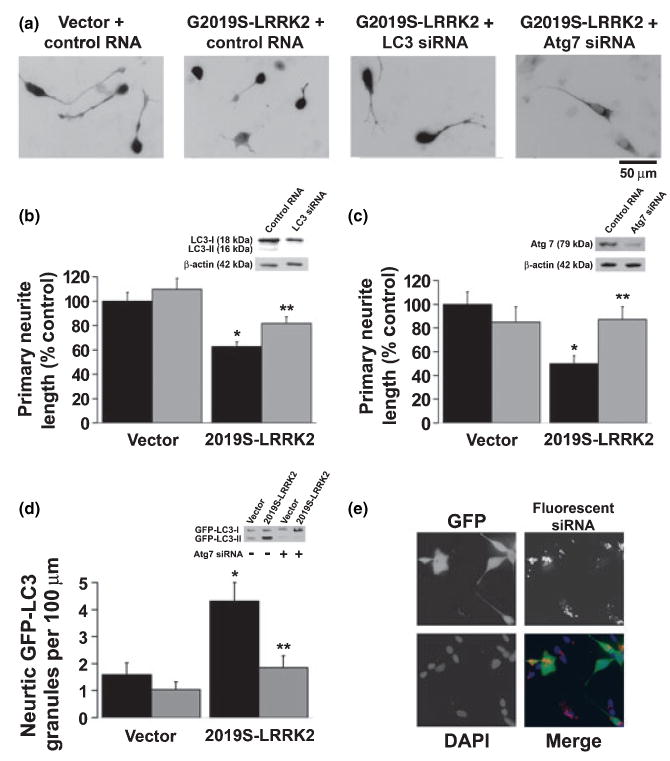
Inhibition of core autophagy machinery conferred protection from neurite blunting associated with G2019S-leucine rich repeat kinase 2 (LRRK2). (a) Representative photos demonstrating shorter neuritic processes in G2019S-LRRK2 transfected cells compared to vector transfected cells and G2019S-LRRK2 transfected cells which have undergone RNA interference (RNAi) knockdown of Light Chain 3 (LC3) or of Atg7. (b) Quantitative data demonstrate that LC3 RNAi conferred protection from G2019S-LRRK2 associated neurite shortening. Black bars indicate cultures pre-treated with control small interfering RNA (siRNA) and grey bars indicate cultures pre-treated with siRNA for LC3. (c) Quantitative data demonstrate that Atg7 RNAi prevented G2019S-LRRK2 associated neurite shortening. Black bars indicate cultures pre-treated with control siRNA and grey bars indicate cultures pre-treated with siRNA targeting Atg7. Insets, (b) and (c): western blot demonstrating LC3 and Atg7 knockdown, respectively. (d) Analysis of green fluorescent protein-tagged microtubule-associated protein Light Chain 3 (GFP-LC3) granules confirm that Atg7 RNAi effectively inhibited G2019S-LRRK2 mediated increases in neuritic autophagic vacuole (AV) content. Black bars indicate cultures pre-treated with control siRNA and grey bars indicate cultures pre-treated with siRNA targeting Atg7. Inset (d): western blot demonstrating GFP-LC3 mobility shift. Increases in lipidated GFP-LC3-II was observed in 2019S-LRRK2 expressing cells in contrast to vector transfected cells (left lanes). RNA interference knockdown of Atg7, which is necessary for LC3 lipidation, blocked the conversion of GFP-LC3-I to GFP-LC3-II in 2019S-LRRK2 co-expressing cells. (e) Representative figure of differentiated SH-SY5Y cells co-transfected with pmaxGFP (green) and 2019S-LRRK2 following pre-treatment with fluorescently-tagged siRNA (red). Greater than 90% of all cells, including 93–96% of GFP/2019S-LRRK2 co-transfected cells, demonstrated cytoplasmic siRNA. *p < 0.05 vs. vector control; **p < 0.05 vs. control siRNA.
Several pharmacologic treatments were performed to gain insight into potential mechanisms by which mutant LRRK2 induces autophagic stress in neurites. Co-treatment of differentiated SH-SY5Y cells with rapamycin (2 μM) potentiated the increase in neuritic AVs and neuritic shortening observed with over-expression of 2019S-LRRK2 (Fig. 5a). However, rapamycin did not induce significant shortening in wild-type LRRK2 or vector transfected cells (Fig. 5a). Co-treatment with 3-MA (5 mM) did not prevent mutant LRRK2 induced neurite shortening (Fig. 5b). In contrast, co-treatment of 2019S-LRRK2 transfected cells with the MAPK/ERK kinase (MEK) inhibitor U0126 (10 μM) attenuated the increase in neuritic AVs and significantly reduced 2019S-LRRK2-induced neurite shortening (Fig. 5c).
Fig. 5.
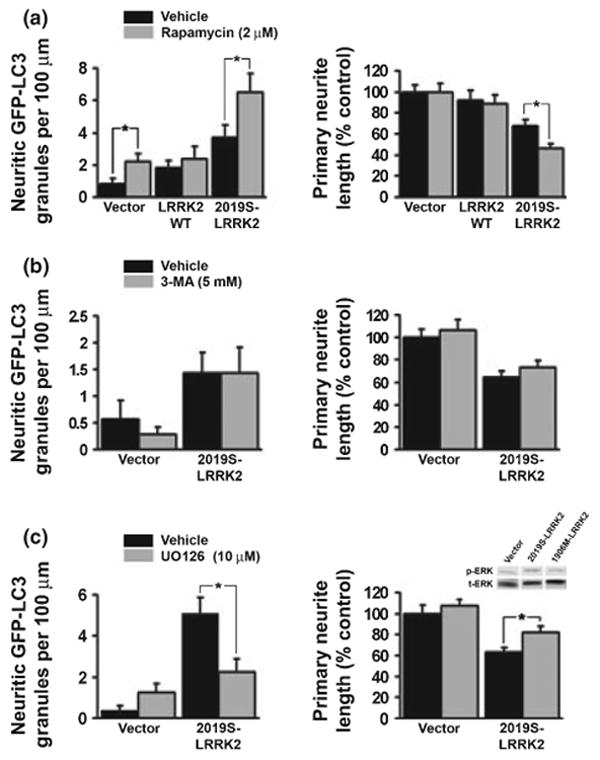
G2019S-leucine rich repeat kinase 2 (LRRK2) induced neuritic autophagy and neurite shortening are potentiated with rapamycin treatment and attenuated with U0126 treatment. (a) Quantitative data showing that 2 μM rapamycin increases neuritic autophagic vacuoles (AVs) in Vector and 2019S-LRRK2 expressing cells (left panel) but exacerbates neuritic shortening in 2019S-LRRK2 expressing cells only (right panel). (b) 3-Methyladenine (3-MA, 5 mM) did not reduce 2019S-LRRK2 induced neurite autophagy (left panel) or neurite shortening (right panel). (c) U0126 (10 μM) attenuated the 2019S-LRRK2 induced increase in neuritic AVs (left panel) and reduced the degree of 2019S-LRRK2 induced neurite shortening (right panel) in RA-differentiated SH-SY5Y cells. Inset (c): western blot demonstrating increased extracellular signal regulated kinase (ERK) phosphorylation in undifferentiated SH-SY5Y cells expressing 2019S-LRRK2 in comparison to vector transfected and kinase dead LRRK2 expressing cells. *p < 0.05 by ANOVA followed by t-test with Bonferroni correction.
Discussion
Expression of LRRK2 cDNAs bearing PD associated mutations results in shortening of neuritic processes (Smith et al. 2005, 2006; Greggio et al. 2006; Macleod et al. 2006). Several disease-associated mutations appear to confer enhanced kinase activity to LRRK2, suggesting that increased phosphorylation of LRRK2 targets stimulates pathways leading to cell toxicity (West et al. 2005, 2007; Smith et al. 2006; Guo et al. 2007; Jaleel et al. 2007; Lewis et al. 2007; Li et al. 2007). Although activation of the apoptotic cascade has been suggested to contribute to cell death (Smith et al. 2005, 2006; Macleod et al. 2006; Iaccarino et al. 2007), little is known concerning mechanisms of neurite shortening.
Our studies indicate that autophagy plays an active role in mutant LRRK2-mediated neurite shortening. As observed in cortical neurons (Macleod et al. 2006), expression of the PD-associated G2019S mutation in LRRK2 resulted in shortened neuritic processes in RA-differentiated SH-SY5Y cell cultures. Although cells were pre-differentiated with RA, neuronal processes continue to extend in the time frame of the study; therefore it is not possible to conclude whether this shortening results from neurite retraction, reduced extension, or a combination of mechanisms. There was a concurrent increase in GFP-LC3 puncta or granules within neuritic and somatic compartments in cells co-transfected with G2019S-LRRK2, which were not observed in cells co-transfected with control vector or kinase dead K1906M-LRRK2. Cells transfected with wild-type LRRK2 expression showed trends toward decreased neurite length and increased AV numbers that were not statistically significant. Elevated somatic and neuritic AV content in G2019S-LRRK2 transfected cells was also observed using transmission electron microscopy. To determine whether autophagy plays an active role in neurite blunting, we utilized RNAi mediated knockdown of the essential autophagy proteins LC3/Atg8 and Atg7 (Ohsumi 2001). Inhibition of 2019S-LRRK2-induced AV induction by Atg protein knockdown, which was confirmed by reduced GFP-LC3 puncta formation and attenuated GFP-LC3 mobility shift (Chan et al. 2007), conferred protection from neurite retraction induced by mutant LRRK2.
Leucine-rich repeat kinase 2 is expressed widely in the normal brain, with strong expression in the substantia nigra, basal ganglia, cortex, hippocampus and cerebellum (Biskup et al. 2006; Higashi et al. 2007; Melrose et al. 2007). Interestingly, LRRK2 exhibits structural homology to upstream kinases in MAPK pathways, and is associated with microtubules, synaptic vesicles and organelle membranes (Biskup et al. 2006; Hatano et al. 2007), suggesting a possible role in intracellular membrane trafficking. LRRK2 has a MAPKKK-like kinase domain as well as a Roco G-protein domain (Ito et al. 2007). The disease-associated mutations of residue 1441 in the GTPase domain cause decreased GTPase activity, which would be predicted to prolong retention in an active conformation (Lewis et al. 2007; Li et al. 2007). G proteins play important roles in both neurite extension and retraction (Koh 2006), and moesin, a potential substrate of LRRK2, is implicated in neurite outgrowth (Jaleel et al. 2007). As the net balance between protein synthesis/antero-grade transport and autophagic degradation/retrograde transport plays a determining factor in axonal homeostasis (Hollenbeck 1993), autophagy could represent another mechanism by which LRRK2 regulates neurite length.
Mitogen activated protein kinases can drive activation of autophagy in toxin-induced models of Parkinsonian injury (Zhu et al. 2007). We previously found that autophagy plays a significant role in the MPP+-model of Parkinsonian injury (Zhu et al. 2007). Inhibition of MPP+-induced MAPK activation reduces autophagy and mitochondrial degradation despite the persistence of toxin-induced mitochondrial damage, suggesting that MAPKs regulate autophagy in this toxin model of PD (Zhu et al. 2007). Ultrastructural studies of human PD brain tissues also reveal increased AVs (Anglade et al. 1997), some of which are associated with activated ERK (Zhu et al. 2002, 2003). It is interesting that G2019S LRRK2, which shows increased kinase activity mediated by a MAPKKK-like domain, also induces neurite remodeling by activating autophagy, and this is accompanied by increases in phospho-ERK. Further studies will be necessary to determine if involvement of MEK/ERK signaling is a direct or indirect consequence of elevated LRRK2 activity.
Interestingly, while physiologic induction of autophagy as part of a starvation- or trophic factor deprivation-response is regulated by class III phosphoinositide 3-kinase (PI3K) signaling, autophagy and mitochondrial degradation induced in the MPP+ model are unaffected by PI3K inhibitors such as 3-MA, but are regulated by MAPK signaling (Chu et al. 2007; Zhu et al. 2007). Interestingly, treatment with the MEK inhibitor U0126, but not 3-MA, reduced mutant LRRK2-induced neurite autophagy and shortening. Taken together, the current and previously published data (Zhu et al. 2003, 2007) suggest a common contribution of ERK-related autophagy dysregulation in toxic and genetic models of Parkinsonian neurodegeneration.
Either insufficient or excessive/dysregulated autophagy can contribute to neurodegeneration (Boland and Nixon 2006; Chu 2006), and robust autophagy induction can contribute to a state of autophagic stress in susceptible cell types (Chu 2006). Consistent with this, the autophagy inducing drug rapamycin further potentiates neurite shortening in 2019S-LRRK2 expressing cells, but does not cause neurite shortening in vector transfected cells. It is unclear whether this may reflect threshold differences, as increases in neuritic AV content induced by either mutant LRRK2 or combined 2019S-LRRK2/rapamycin are greater than that observed with rapamycin in control transfected cells. Alternatively, there may be fundamental differences in pathways involved in autophagic degeneration induced by pathologic stimuli, as suggested by differences in sensitivity to PI3K inhibitors. Finally, it is possible that neurite degeneration proceeds as a result of imbalances between the degree of autophagy induction and the ability of the cell to degrade and regenerate new cellular constituents, and rapamycin does not cause this type of autophagic stress in the absence of mutant LRRK2.
There are several potential mechanisms by which LRRK2 expression may affect AV formation, trafficking and/or maturation. In addition to potential regulation of autophagy induction and membrane trafficking through MAPKKK-like and G-protein related signaling (Dachsel et al. 2007), mutant LRRK2 has been reported to associate with endolysosomal and mitochondrial membranes (Biskup et al. 2006; Hatano et al. 2007), which may be related to regulation of the lysosomal system or of mitochondrial distribution, another factor in neuritic remodeling (Mattson 2007). Mutant LRRK2 may also affect microtubules indirectly by inducing tau phosphorylation (Macleod et al. 2006), which would result in disruption of AV/lysosomal fusion, impaired clearance of protein aggregates and toxicity (Boland and Nixon 2006). Thus, it is possible that mutant LRRK2 can induce autophagic stress through effects on both autophagy induction and maturation/fusion.
While basal autophagy is essential for neuronal and axonal maintenance (Hara et al. 2006; Komatsu et al. 2007), dysregulated autophagy is increasingly implicated in models of neurite pathology. Prominent AVs are observed in dystrophic neurites following exposure to methamphetamine in VMAT2 deficient cells (Larsen et al. 2002), a model that causes neurite degeneration in the absence of cell death. We have also noted neuritic AVs in SH-SY5Y cells exposed to low-dose or short-duration MPP+ treatments [unpublished data, see also (Chu et al. 2007)]. Axonal AVs represent a prominent feature of excitotoxic Purkinje neuron degeneration in the Lurcher mouse (Wang et al. 2006), and have been observed in large numbers in Alzheimer's disease (Nixon et al. 2005). However, as increased AV content can represent both increased induction and decreased trafficking/degradation (Chu 2006), the mechanism and role of neuritic AVs in these models is still unclear. Most recently, it has been shown that autophagy actively mediates neurite retraction following nerve growth factor withdrawal in superior cervical ganglion neurons in culture (Yang et al. 2007). The current study of mutant LRRK2-mediated neurite shortening is the first to utilize molecular knockdown of specific Atg proteins to demonstrate that autophagy can play an active role in neurite degeneration in a genetic model of PD.
To summarize, AVs have been identified in vulnerable neurons of PD and related Lewy body disease patients (Anglade et al. 1997; Zhu et al. 2003). In this study, we found that expression of G2019S-LRRK2 in differentiated SH-SY5Y cells resulted in neuritic shortening accompanied by up-regulation of neuritic and somatic AVs. MEK/ERK-related signaling contributes to autophagy regulation downstream of mutant LRRK2, and rapamycin potentiates LRRK2-mediated neurite shortening. RNAi knockdown of essential core autophagy proteins prevented the AV increases elicited by G2019S-LRRK2 expression, conferring protection from neurite shortening. These results indicate that autophagy plays an active role in neurite remodeling mediated by mutant LRRK2 expression. Identification of a role for autophagy as a mediator of LRRK2-induced alterations may provide further insights into both physiologic and pathologic neurite remodeling in PD-relevant pathways of neurodegeneration.
Acknowledgments
We thank Matthew Farrer (Mayo Clinic, Jacksonville, FL, USA) for the wild-type LRRK2 construct, Tamotsu Yoshimori (Osaka University, Japan) for the pEGFP-LC3 plasmid, and Simon Watkins and the Center for Biology Imaging (University of Pittsburgh, PA, USA) for assistance with electron microscopy. This work was supported in part by the National Institutes of Health (R01 AG026389). EDP was supported in part by the Pathologist Investigator Residency/Research Training (PIRT) Program (Department of Pathology, University of Pittsburgh School of Medicine).
Abbreviations used
- 3-MA
3-methyladenine
- AV
autophagic vacuole
- ERK
extracellular signal regulated kinase
- GFP-LC3
green fluorescent protein-tagged microtubule-associated protein Light Chain 3
- HA
hemaglutinin
- LRRK2
leucine-rich repeat kinase 2
- MAPK
mitogen activated protein kinase
- MEK
MAPK/ERK kinase
- PD
Parkinson's disease
- PI3K
phosphoinositide 3-kinase
- RA
retinoic acid
- RNAi
RNA interference
- siRNA
small interfering RNA
- U0126
1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio]butadiene
References
- Anglade P, Vyas S, Javoy-Agid F, et al. Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease. Histol Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- Biskup S, Moore DJ, Celsi F, et al. Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann Neurol. 2006;60:557–569. doi: 10.1002/ana.21019. [DOI] [PubMed] [Google Scholar]
- Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland B, Nixon RA. Neuronal macroautophagy: from development to degeneration. Mol Aspects Med. 2006;27:503–519. doi: 10.1016/j.mam.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Chan EYW, Kir S, Tooze SA. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J Biol Chem. 2007;282:25464–25474. doi: 10.1074/jbc.M703663200. [DOI] [PubMed] [Google Scholar]
- Chu CT. Autophagic stress in neuronal injury and disease. J Neuropathol Exp Neurol. 2006;65:423–432. doi: 10.1097/01.jnen.0000229233.75253.be. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CT, Zhu J, Dagda R. Beclin 1-independent pathway of damage-induced mitophagy and autophagic stress: implications for neurodegeneration and cell death. Autophagy. 2007;3:663–666. doi: 10.4161/auto.4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dachsel JC, Taylor JP, Mok SS, et al. Identification of potential protein interactors of Lrrk2. Parkinsonism Relat Disord. 2007 doi: 10.1016/j.parkreldis.2007.01.008. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer MJ. Genetics of Parkinson disease: paradigm shifts and future prospects. Nature Reviews. 2006;7:306–318. doi: 10.1038/nrg1831. [DOI] [PubMed] [Google Scholar]
- Gloeckner CJ, Kinkl N, Schumacher A, Braun RJ, O'Neill E, Meitinger T, Kolch W, Prokisch H, Ueffing M. The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum Mol Genet. 2006;15:223–232. doi: 10.1093/hmg/ddi439. [DOI] [PubMed] [Google Scholar]
- Greggio E, Jain S, Kingsbury A, et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 2006;23:329–341. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Guo L, Gandhi PN, Wang W, Petersen RB, Wilson-Delfosse AL, Chen SG. The Parkinson's disease-associated protein, leucine-rich repeat kinase 2 (LRRK2), is an authentic GTPase that stimulates kinase activity. Exp Cell Res. 2007;313:3658–3670. doi: 10.1016/j.yexcr.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Hatano T, Kubo S, Imai S, Maeda M, Ishikawa K, Mizuno Y, Hattori N. Leucine-rich repeat kinase 2 associates with lipid rafts. Hum Mol Genet. 2007;16:678–690. doi: 10.1093/hmg/ddm013. [DOI] [PubMed] [Google Scholar]
- Higashi S, Biskup S, West AB, et al. Localization of Parkinson's disease-associated LRRK2 in normal and pathological human brain. Brain Res. 2007;1155:208–219. doi: 10.1016/j.brainres.2007.04.034. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ. Products of endocytosis and autophagy are retrieved from axons by regulated retrograde organelle transport. J Cell Biol. 1993;121:305–315. doi: 10.1083/jcb.121.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iaccarino C, Crosio C, Vitale C, Sanna G, Carri MT, Barone P. Apoptotic mechanisms in mutant LRRK2-mediated cell death. Hum Mol Genet. 2007;16:1319–1326. doi: 10.1093/hmg/ddm080. [DOI] [PubMed] [Google Scholar]
- Ito G, Okai T, Fujino G, Takeda K, Ichijo H, Katada T, Iwatsubo T. GTP binding is essential to the protein kinase activity of LRRK2, a causative gene product for familial Parkinson's disease. Biochemistry. 2007;46:1380–1388. doi: 10.1021/bi061960m. [DOI] [PubMed] [Google Scholar]
- Jaleel M, Nichols RJ, Deak M, Campbell DG, Gillardon F, Knebel A, Alessi DR. LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson's disease mutants affect kinase activity. Biochem J. 2007;405:307–317. doi: 10.1042/BJ20070209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselyov K, Jennigs JJ, Jr, Rbaibi Y, Chu CT. Autophagy, mitochondria and cell death in lysosomal storage diseases. Autophagy. 2007;3:259–262. doi: 10.4161/auto.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh CG. Rho GTPases and their regulators in neuronal functions and development. Neurosignals. 2006;15:228–237. doi: 10.1159/000101527. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Wang QJ, Holstein GR, Friedrich VL, Jr, Iwata J, Kominami E, Chait BT, Tanaka K, Yue Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci USA. 2007;104:14489–14494. doi: 10.1073/pnas.0701311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen KE, Fon EA, Hastings TG, Edwards RH, Sulzer D. Methamphetamine-induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J Neurosci. 2002;22:8951–8960. doi: 10.1523/JNEUROSCI.22-20-08951.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PA, Greggio E, Beilina A, Jain S, Baker A, Cookson MR. The R1441C mutation of LRRK2 disrupts GTP hydrolysis. Biochem Biophys Res Commun. 2007;357:668–671. doi: 10.1016/j.bbrc.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Capan E, Zhao Y, Zhao J, Stolz D, Watkins SC, Jin S, Lu B. Autophagy is induced in CD4+ T cells and important for the growth factor-withdrawal cell death. J Immunol. 2006;177:5163–5168. doi: 10.4049/jimmunol.177.8.5163. [DOI] [PubMed] [Google Scholar]
- Li X, Tan YC, Poulose S, Olanow CW, Huang XY, Yue Z. Leucine-rich repeat kinase 2 (LRRK2)/PARK8 possesses GTPase activity that is altered in familial Parkinson's disease R1441C/G mutants. J Neurochem. 2007;103:238–247. doi: 10.1111/j.1471-4159.2007.04743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzon-Toro B, de la Torre ER, Delgado A, Perez-Tur J, Hilfiker S. Mechanistic insight into the dominant mode of the Parkinson's disease-associated G2019S LRRK2 mutation. Hum Mol Genet. 2007;16:2031–2039. doi: 10.1093/hmg/ddm151. [DOI] [PubMed] [Google Scholar]
- Macleod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006;52:587–593. doi: 10.1016/j.neuron.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Mitochondrial regulation of neuronal plasticity. Neurochem Res. 2007;32:707–715. doi: 10.1007/s11064-006-9170-3. [DOI] [PubMed] [Google Scholar]
- Matyja E, Taraszewska A, Naganska E, Rafalowska J. Autophagic degeneration of motor neurons in a model of slow glutamate excitotoxicity in vitro. Ultrastruct Pathol. 2005;29:331–339. doi: 10.1080/01913120500214333. [DOI] [PubMed] [Google Scholar]
- Meijering E, Jacob M, Sarria JC, Steiner P, Hirling H, Unser M. Design and validation of a tool for neurite tracing and analysis in fluorescence microscopy images. Cytometry A. 2004;58:167–176. doi: 10.1002/cyto.a.20022. [DOI] [PubMed] [Google Scholar]
- Melrose HL, Kent CB, Taylor JP, et al. A comparative analysis of leucine-rich repeat kinase 2 (LRRK2) expression in mouse brain and Lewy body disease. Neuroscience. 2007;147:1047–1058. doi: 10.1016/j.neuroscience.2007.05.027. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–122. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol. 2001;2:211–216. doi: 10.1038/35056522. [DOI] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Jain S, Evans EW, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Smith WW, Pei Z, Jiang H, Moore DJ, Liang Y, West AB, Dawson VL, Dawson TM, Ross CA. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc Natl Acad Sci USA. 2005;102:18676–18681. doi: 10.1073/pnas.0508052102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci. 2006;9:1231–1233. doi: 10.1038/nn1776. [DOI] [PubMed] [Google Scholar]
- Wang QJ, Ding Y, Kohtz DS, et al. Induction of autophagy in axonal dystrophy and degeneration. J Neurosci. 2006;26:8057–8068. doi: 10.1523/JNEUROSCI.2261-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci USA. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Moore DJ, Choi C, et al. Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet. 2007;16:223–232. doi: 10.1093/hmg/ddl471. [DOI] [PubMed] [Google Scholar]
- Xu Y, Kim SO, Li Y, Han J. Autophagy contributes to caspase-independent macrophage cell death. J Biol Chem. 2006;281:19179–19187. doi: 10.1074/jbc.M513377200. [DOI] [PubMed] [Google Scholar]
- Yang Y, Fukui K, Koike T, Zheng X. Induction of autophagy in neurite degeneration of mouse superior cervical ganglion neurons. Eur J Neurosci. 2007;25:2979–2988. doi: 10.1111/j.1460-9568.2007.05914.x. [DOI] [PubMed] [Google Scholar]
- Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–1502. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- Zhu JH, Kulich SM, Oury TD, Chu CT. Cytoplasmic aggregates of phosphorylated extracellular signal-regulated protein kinases in Lewy body diseases. Am J Pathol. 2002;161:2087–2098. doi: 10.1016/S0002-9440(10)64487-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JH, Guo F, Shelburne J, Watkins S, Chu CT. Localization of phosphorylated ERK/MAP kinases to mitochondria and autophagosomes in Lewy body diseases. Brain Pathol. 2003;13:473–481. doi: 10.1111/j.1750-3639.2003.tb00478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JH, Horbinski C, Guo F, Watkins S, Uchiyama Y, Chu CT. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am J Pathol. 2007;170:75–86. doi: 10.2353/ajpath.2007.060524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal-dominant Parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]