

Abstract
Integral membrane proteins are synthesized on the cytoplasmic face of the endoplasmic reticulum (ER). After being translocated or inserted into the ER, they fold and undergo post-translational modifications. Within the ER, proteins are also subjected to quality control checkpoints, during which misfolded proteins may be degraded by proteasomes via a process known as ER-associated degradation. Molecular chaperones, including the small heat shock protein αA-crystallin, have recently been shown to play a role in this process. We have now found that αA-crystallin is expressed in cultured mouse collecting duct cells, where apical Na+ transport is mediated by epithelial Na+ channels (ENaC). ENaC-mediated Na+ currents in Xenopus oocytes were reduced by co-expression of αA-crystallin. This reduction in ENaC activity reflected a decrease in the number of channels expressed at the cell surface. Furthermore, we observed that the rate of ENaC delivery to the cell surface of Xenopus oocytes was significantly reduced by co-expression of αA-crystallin, whereas the rate of channel retrieval remained unchanged. We also observed that αA-crystallin and ENaC co-immunoprecipitate. These data are consistent with the hypothesis that small heat shock proteins recognize ENaC subunits at ER quality control checkpoints and can target ENaC subunits for ER-associated degradation.
Like most other integral membrane proteins, newly synthesized epithelial Na+ channel (ENaC)4 subunits translocate co-translationally into the endoplasmic reticulum (ER), where folding and post-translational modifications occur. Within the ER, proteins are also subjected to quality control checkpoints to ensure that only properly folded proteins mature beyond the ER. If folding is inefficient, the misfolded protein may be degraded by proteasomes via a process known as ER-associated degradation (ERAD). This prevents the accumulation of abnormal proteins in the ER, which, left unchecked, may form toxic protein aggregates. Molecular chaperones, which can bind to exposed, hydrophobic motifs in unfolded proteins, play a key role in selecting substrates for this process.
ENaC is expressed at the apical membranes of Na+ absorptive epithelia. There, in conjunction with the basolateral Na+/K+ ATPase, ENaC facilitates transepithelial Na+ transport (1). ENaC is found in a variety of tissues, including the lung airway and alveoli and the distal nephron, where ENaC influences mucociliary clearance and extracellular fluid Na+ and volume regulation, respectively (1–3). ENaC is comprised of three homologous subunits, α, β, and γ, although the stoichiometry of the functional channel remains controversial (4–7). Each subunit has short cytoplasmic amino- and carboxyl-terminal domains (50–110 residues), two transmembrane segments, and a large extracellular domain (∼450 residues) (8–10). Like most other molecular chaperones, small heat shock proteins (sHsps) can bind unfolded, aggregation-prone substrates to retain them in solution. In addition, sHsps have been implicated in proteasome function and the ubiquitin-proteosome pathway. We recently observed that α-crystallin domain-containing sHsps facilitate the ERAD of CFTR (11). Although the structure and assembly of CFTR and ENaC are distinct, we hypothesized that these chaperones may also be involved in ENaC quality control. To this end, we established an ENaC expression system in yeast and discovered that sHsps facilitate ENaC α subunit degradation. We also found that the sHsp αA-crystallin is expressed in a cortical collecting duct (CCD) cell line and that αA-crystallin and the ENaC α subunit form stable complexes. Finally, we determined the functional effect of αA-crystallin expression on ENaC in a heterologous system and found that αA-crystallin lowered the number of functional channels because of a decrease in the rate of channel delivery to the cell surface. These data demonstrate a broader role for sHsps in ERAD and represent only the second chaperone class that has been found to impact ENaC biogenesis.
Experimental Procedures
Vectors and Antibodies
Mouse ENaC α subunits with either carboxyl-terminal HA or V5 epitope tags (αHA, αV5), amino-terminal HA and carboxyl-terminal V5 epitope tags (HAαV5), β subunit with a carboxyl-terminal FLAG epitope tag (β), and γ subunit with a carboxyl-terminal Myc epitope tag (γ) were previously characterized (12). The antibodies used were: horseradish peroxidase-conjugated rat monoclonal anti-HA (Roche Applied Sciences), mouse monoclonal anti-HA (Covance, Berkeley, CA), rabbit anti-αA-crystallin (Assay Designs, Ann Arbor, MI), mouse monoclonal anti-V5 and anti-V5 antibody conjugated to horseradish peroxidase (Invitrogen), and rabbit anti-Sec61p (13).
Protein Degradation Assays in Yeast
The cDNA encoding HAαV5 (12) was inserted into the yeast pGPD426 constitutive expression vector at the HindIII and ClaI restriction sites. Plasmids were introduced into the indicated strains using lithium acetate-mediated transformation (14). Transformants were selected by growth on Sc–Ura medium containing glucose (15). To assess the rate of ENaC α subunit ERAD, cycloheximide chase experiments were performed as described previously (16). The immunoblots were incubated with an anti-V5 antibody conjugated to horseradish peroxidase (Invitrogen) to detect ENaC α subunit and with a rabbit anti-Sec61p antibody (13) as a loading control. The blots were visualized by chemiluminescence as described below and quantified with a Versadoc (Bio-Rad).
Reverse Transcription-PCR
Mouse kidney total RNA was from Clontech Laboratories, Inc. (Mountain View, CA). Total RNA from cultured CCD cells was isolated using an RNAqueous 4PCR kit (Ambion, Austin, TX). One microgram total RNA was reverse transcribed using either random hexamers or oligo(dT) as primers using SuperScript II reverse transcriptase (Invitrogen). Nested PCR was performed on control “no reverse transcriptase” template and random-primed or oligo(dT)-primed cDNA templates.
Immunoblotting
CCD and MDCK cells were maintained as previously described (17, 18) and transiently transfected with ENaC subunits and αA-crystallin as indicated using Lipofectamine 2000 (Invitrogen) for use the following day (12). The immunoblotting methods were adapted from Hughey et al. (12). Briefly, cytoplasmic extracts of CCD cells were produced by scraping cells into phosphate-buffered saline (137 mm NaCl, 2.6 mm KCl, 15.2 mm Na2HPO4, 1.47 mm KH2PO4) with protease inhibitor mixture set III (EMD Biosciences, San Diego, CA) and passing them 20 times through a 22-gauge needle. Whole cell lysates were produced by scraping cells into 100 mm NaCl, 5 mm EDTA, 50 mm triethanolamine, pH 8.6, and protease inhibitor mixture set III followed by the addition of SDS (Bio-Rad) to 0.5% (w/v) and incubation at 95 °C for 5 min as described previously (19). Triethanolamine (final concentration, 75 mm) and Triton X-100 (final concentration, 1.25% (v/v)) were then added, after which samples were agitated for 15 min at 4 °C. MDCK cells were extracted into 0.3 ml of lysis buffer (0.4% sodium deoxycholate, 1% Nonidet P-40, 63 mm EDTA, and 50 mm Tris-HCl, pH 8) supplemented with protease inhibitor set III and phosphatase inhibitor set I (Calbiochem, San Diego, CA). The extracts were centrifuged to remove insoluble material. For immunoprecipitation experiments, the supernatants were incubated overnight at 4 °C on a rotating wheel with antibodies as indicated and 25 μl of protein A immobilized on Sepharose 4B beads (Zymed Laboratories Inc., S. San Francisco, CA). Immunoprecipitates on beads were washed and then subjected to SDS-PAGE (4–15% gradient gel) and transferred to an Immobilon-NC membrane. The membranes were incubated with antibody as indicated overnight at 4 °C, followed by peroxidase-conjugated secondary antibody when needed. Purified bovine αA-crystallin (Assay Designs) and Precision Protein Standards (Bio-Rad) were used as markers. The blots were developed using Western Lightning Chemiluminescence reagent (PerkinElmer Life Science) and visualized with either BioMax MR film (Kodak, New Haven, CT) or using a Versadoc (Bio-Rad).
ENaC Activity Measurements
ENaC mediated Na+ currents were measured in Xenopus oocytes by the two-electrode voltage clamp technique (TEV). Stage V and VI oocytes free of follicle cell layers were injected with cRNA encoding wild type α, β, and γ ENaC subunits and varying amounts of cRNA encoding αA-crystallin (11), Hsc70 (20), or γ-glutamyl transpeptidase (21), as indicated. The oocytes were maintained in modified Barth's saline (MBS; 88 mm NaCl, 1 mm KCl, 2.4 mm NaHCO3, 15 mm HEPES, 0.3 mm Ca(NO3)2, 0.41 mm CaCl2, 0.82 mm MgSO4, 10 μg/ml sodium penicillin, 10 μg/ml streptomycin sulfate, 100 μg/ml gentamicin sulfate, pH 7.4). TEV was performed 24 h after injection using a DigiData 1320A interface and a GeneClamp 500B Voltage Clamp amplifier (Axon Instruments, Foster City, CA). In experiments with MG-132 (Calbiochem), oocytes were incubated in MBS supplemented with 6 μm MG-132 for 3–4 h prior to current measurements (22). Data acquisition and analyses were performed using pClamp 8.2 software (Axon Instruments). The pipettes were pulled from borosilicate glass capillaries (World Precision Instruments, Sarasota, FL) with a micropipette puller (Sutter Instrument Co., Novato, CA) and had resistance of 0.5–5 megaohms when filled with 3 m KCl and inserted into the bath solution. The oocytes were maintained in a recording chamber (Automate Scientific, San Francisco, CA) with 20 μl of bath solution and continuously perfused with bath solution at a flow rate of 4–5 ml/min. The bath solution contained 110 mm NaCl, 2 mm KCl, 2 mm CaCl2, 10 mm HEPES, pH 7.4. All of the experiments were performed at 20–24 °C.
ENaC exocytosis rates were determined as previously described (23, 24) from oocytes injected with cRNAs for αS583C mutant and wild type β and γ ENaC subunits either in the presence or absence of 6 ng of cRNA for αA-crystallin. 24–36 h after injection, channels at the cell surface were blocked with 1 mm MTSET for 4 min, resulting in a covalent modification of the channel that causes an ∼80% reduction of current. After removal of MTSET from the bath, Na+ current was measured by TEV at −100 mV every 30 s for 10 min to monitor current recovery. The initial rates of reappearance of amiloride-sensitive currents were determined from the linear portion of the current recovery curve (0–2 min).
ENaC endocytosis rates were determined from oocytes injected with cRNA for wild type α, β, and γ ENaC subunits either in the presence or absence of 6 ng of cRNA for αA-crystallin. 24–36 h after injection, amiloride-sensitive currents were determined by TEV before and after 2, 4, and 6 h of incubation with 5 μm brefeldin A. Amiloride-sensitive currents were expressed relative to the initial amiloride-sensitive current, and data for the current declines were compared.
ENaC Surface Expression Measurements
The experiments were performed essentially as described (25). Xenopus oocytes were injected with varying amounts of cRNA encoding αA-crystallin and 2 ng each of an extracellular FLAG epitope-tagged β ENaC subunit (βF) and wild type α and γ ENaC subunits. Negative controls utilized wild type ENaC β subunit with no epitope tag (no tag). Two days after injection, the oocytes were blocked for 30 min in MBS supplement with 10 mg/ml bovine serum albumin (MBS/BSA) and then incubated for 1 h with MBS/BSA supplement with 1 μg/ml of a mouse monoclonal anti-FLAG antibody (M2) (Sigma-Aldrich) at 4 °C. The oocytes were then washed at 4 °C for 1 h in MBS/BSA and incubated with MBS/BSA supplemented with 1 μg/ml secondary antibody for 1 h at 4 °C (peroxidase-conjugated AffiniPure F(ab'2) fragment goat anti-mouse IgG; Jackson ImmunoResearch, West Grove, PA). The oocytes were extensively washed (12 times over 2 h) at 4 °C and transferred to MBS without BSA. Individual oocytes were placed in 100 μl of SuperSignal Elisa Femto maximum sensitivity substrate (Pierce) and incubated at room temperature for 1 min. Chemiluminescence was quantified in a TD-20/20 luminometer (Turner Designs, Sunnyvale, CA).
Statistical Analysis
Normalized data from oocytes were normalized within an individual batch of oocytes as previously described (26). All of the data are presented as the means ± S.E. p values were determined by a Student's t test performed with Excel 2003 (Microsoft Corp., Redmond, WA) or a one-way ANOVA with a Newman-Keuls post hoc test performed with Igor Pro 5.05A (Wavemetrics, Lake Oswego, OR), as indicated. A p value ≤0.05 was considered significant.
Results
Hsp26 and Hsp42 Facilitate ENaC α Subunit Degradation in Yeast
We recently demonstrated that α-crystallin domain-containing sHsps select aberrant forms of CFTR in both yeast and mammalian cells for ERAD (11). This chloride channel is co-expressed with ENaC in lung epithelia. Together these channels regulate lung fluid volume, which affects mucociliary clearance (27). We therefore hypothesized that chaperones with α-crystallin domains may also be involved in ENaC quality control. To test this hypothesis, we inserted the α subunit of mouse ENaC into a yeast vector engineered for constitutive expression. Degradation in both wild type yeast and a variety of yeast mutants was measured by a cycloheximide chase, and the results were quantified after Western blot analysis. The first mutant examined lacked two members of the sHsp family, Hsp26 and Hsp42. Although these two proteins largely recognize the same target proteins, Hsp42 is constitutively present at high levels, whereas Hsp26 expression is temperature-sensitive (28, 29). We found that degradation of α-ENaC was reduced in the hsp26Δhsp42Δ mutant strain compared with wild type yeast (Fig. 1, p < 0.05 at 20 and 60 min). To verify that the ENaC α subunit is an ERAD substrate, we examined the effect on ENaC α subunit degradation when Ufd1p was mutated. Ufd1p is a component of the Cdc48p/Ufd1p/Npl4p AAA ATPase complex that actively transfers ubiquitinated substrates from the ER to the proteasome (30). The absence of functional Ufd1p significantly decreased α subunit degradation (Fig. 1, p < 0.05 at 60 min). To confirm this observation, we examined the effect of mutating a component of the proteasome “cap,” Cim3p, on ENaC α subunit degradation. Cim3p (also known as Rpt6p) is also a AAA ATPase, and the proteasome cap is thought to drive substrates into the proteasome core for degradation (31). Indeed, we found that the degradation of the ENaC α subunit in the cim3-1 mutant was significantly reduced compared with wild type yeast (Fig. 1, p < 0.05 at 90 min). These results indicate that the ENaC α subunit is a bona fide ERAD substrate in yeast and that the sHsps promote ENaC α subunit clearance in this organism.
FIGURE 1. ERAD of ENaC α subunit in yeast requires sHsps for maximal efficiency.
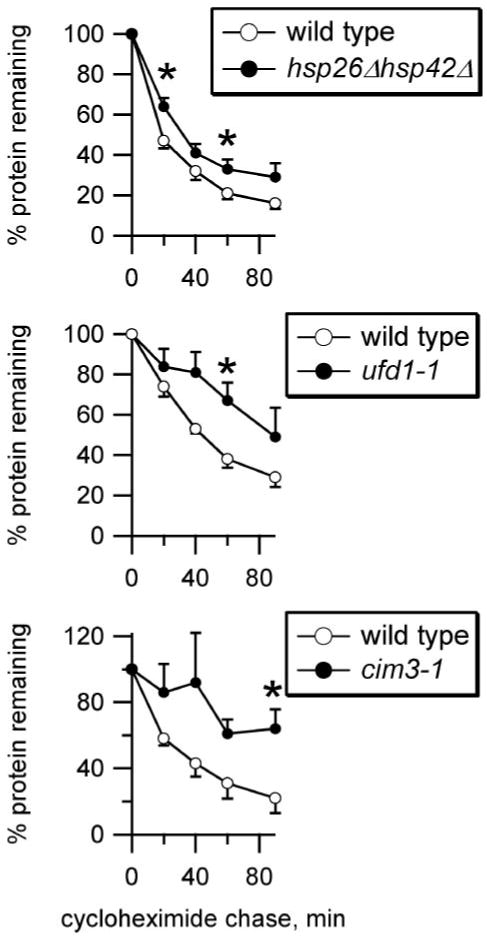
ENaC HAαV5 degradation was measured by lysing cells at various times during a chase following cycloheximide addition, and protein levels were quantified by Western analysis and normalized to Sec61 levels. ENaC α subunit degradation was measured for both wild type (○) and mutant (●) yeast, as indicated. n = 6 (hsp26Δhsp42Δ) or 4 (ufd1-1, cim3-1). *, p < 0.05 versus wild type yeast, determined by Student's t test.
αA-crystallin Is Expressed in the Kidney and in Cultured Collecting Duct Cells
In addition to its function in the lung, the role of ENaC in the distal nephron of the kidney remains a focus of research and clinical interest. Because ENaC is expressed in the CCD of the distal nephron (32) and because αA-crystallin is the closest mammalian homolog to Hsp26 in yeast, we determined whether αA-crystallin is also expressed in mouse kidney and specifically in the CCD. Reverse transcription-PCR using RNA isolated from both whole mouse kidney and a mouse CCD cell line demonstrated a product at the predicted size of 175 bp using nested αA-crystallin primers (Fig. 2, A and B). The identity of the 175-bp product was confirmed by nucleotide sequencing. We confirmed that αA-crystallin protein was present in CCD cells by Western blot analysis. Cell lysates of cultured murine CCD cells were probed with anti-αA-crystallin, and a product at ∼20 kDa was evident (Fig. 2C). This species was similar to the molecular masses of purified bovine αA-crystallin protein alone or when added to lysate. This result confirms that CCD cells endogenously express αA-crystallin.
FIGURE 2. αA-crystallin is expressed in CCD cells and co-immunoprecipitates with ENaC α subunit.
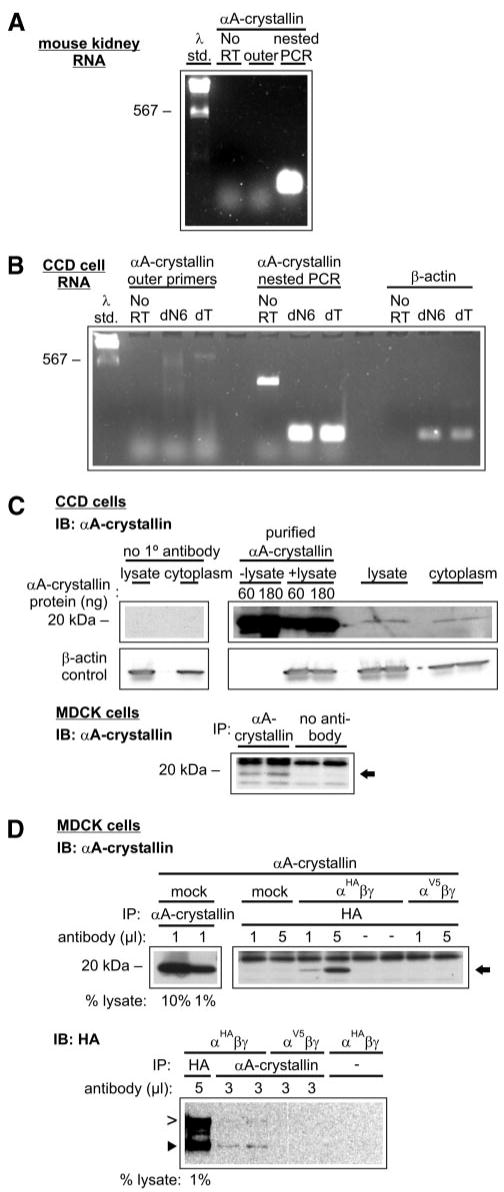
αA-crystallin is expressed in mouse kidney tissue and cultured CCD cells (A and B). One microgram of total RNA isolated from native mouse kidney tissue (A) or cultured CCD cells (B) was reverse transcribed using oligo dT (dT) or random hexamers (dN6, CCD cells only) as primers (n = 2). Negative controls were performed in reactions lacking reverse transcriptase (No RT). Predicted reverse transcription-PCR product sizes were 224 bp for αA-crystallin outer primers, 175 bp for αA-crystallin nested primers, and 174 bp for β-actin primers. Note that only nested primers yielded a strong signal of the expected size. C, cultured CCD cell lysates and cytoplasmic extracts were immunoblotted (IB) with mouse anti-αA-crystallin (n = 2). Purified bovine αA-crystallin protein was added as indicated. MDCK cell lysates were immunoprecipitated and immunoblotted with anti-αA-crystallin (n = 3). αA-crystallin is indicated with an arrow. D, ENaC α subunit and αA-crystallin co-immunoprecipitate. MDCK cell extracts were transfected with vectors engineered for the expression of αA-crystallin, ENaC αHA or αV5 subunit, and ENaC β and γ subunits as indicated. Extracts were immunoprecipitated with the indicated amounts of either anti-HA antibody or anti-αA-crystallin antibody. IB: αA-crystallin, n = 4; IB: HA, n = 2. αA-crystallin is indicated with an arrow. The furin-cleaved (65 kDa, ►) and uncleaved (95 kDa, >) ENaC α subunit products are also indicated.
αA-crystallin and ENaC Form Complexes
To determine whether αA-crystallin and ENaC subunits interact, we next performed co-immunoprecipitation experiments. MDCK cells were used for these experiments because of the absence of endogenously expressed ENaC (18). In experiments where untransfected cells were both immunoprecipitated and immunoblotted with an anti-αA-crystallin antibody, we found that MDCK cells endogenously expressed αA-crystallin (Fig. 2C). We performed experiments where αA-crystallin along with the β, γ, and HA epitope-tagged α (αHA) ENaC subunits were co-transfected. When lysates of these cells were immunoprecipitated with anti-HA antibody and immunoblotted with anti-αA-crystallin antibody, we observed that αA-crystallin co-immunoprecipitated with the αHA ENaC subunit (Fig. 2C). In control experiments in which either (i) ENaC subunits were not co-transfected, (ii) antibody was omitted from the immunoprecipitation, or (iii) a V5 epitope-tagged α subunit was used, little or no αA-crystallin was apparent in the precipitate. We also examined whether endogenous αA-crystallin co-immunoprecipitated with the ENaC αHA subunit. When lysates of cells co-transfected with β, γ, and αHA ENaC subunits were immunoprecipitated with anti-αA-crystallin antibody and subsequently immunoblotted with anti-HA antibody, we found that the αHA ENaC subunit co-immunoprecipitated with endogenously expressed αA-crystallin (Fig. 2D). In control experiments where a V5-epitope tagged α subunit was used or antibody was omitted from the immunoprecipitation step, no ENaC α subunit was observed in the precipitate. Both the furin-mediated cleaved and uncleaved bands of the αHA subunit were co-immunoprecipitated with αA-crystallin. We have recently shown that proteolytic processing of ENaC by furin in the trans-Golgi network is required for channel activation (12, 33). This result suggests that αA-crystallin could associate with the α subunit of ENaC before and after trans-Golgi network-dependent processing events.
αA-crystallin Reduces ENaC Expression in Oocytes
To determine whether αA-crystallin affects the functional expression of ENaC, we examined the effect of co-expressing human αA-crystallin with mouse ENaC on channel activity in Xenopus oocytes. Like other sHsps, αA-crystallin associates with unfolded proteins to prevent their aggregation and maintain solubility, but continued association may target the bound protein for ERAD (11). We injected cRNAs encoding the α, β, and γ mouse ENaC subunits into Xenopus oocytes along with water or various amounts of cRNA encoding αA-crystallin or with cRNA encoding γ-glutamyl transpeptidase as a control. 24 h after injection, we measured the amiloride-sensitive Na+ currents for each group by TEV (Fig. 3A). We found that co-expression of αA-crystallin with ENaC in oocytes reduced ENaC-mediated Na+ currents in a dose-dependent manner, whereas γ-glutamyl transpeptidase cRNA had no effect.
FIGURE 3. Effect of αA-crystallin co-expression on the functional expression of ENaC in Xenopus oocytes.
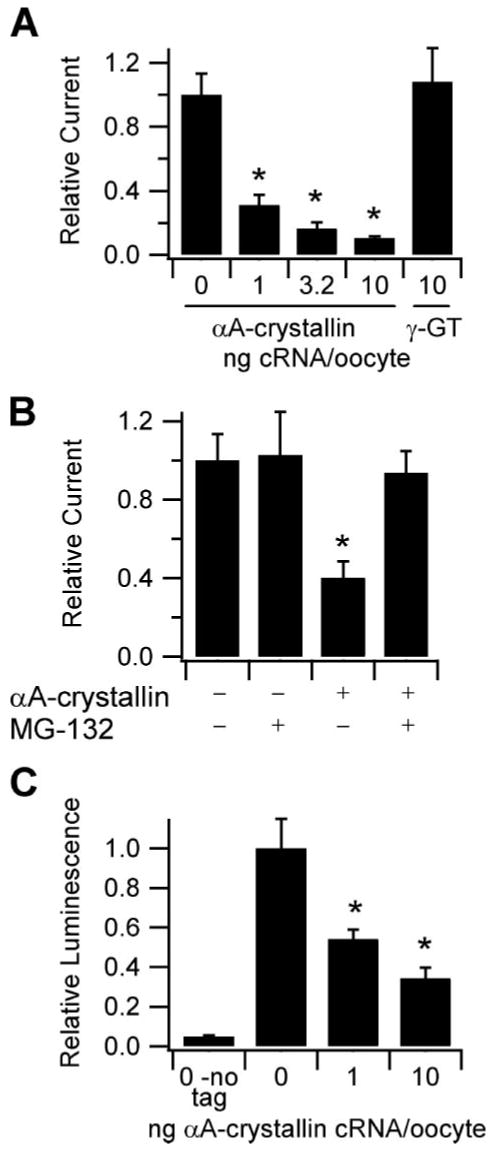
A, TEV measurements were performed at −100 mV with oocytes injected with 1 ng each of cRNA encoding the α, β, and γ ENaC subunits and the indicated amounts of cRNA encoding either αA-crystallin or γ-glutamyl transpeptidase (γ-GT). Base-line current was −2.5 μA (n ≥ 16). *, p < 0.0001 versus 0 αA-crystallin, determined by ANOVA. B, effect of proteasomal inhibition on the reduction of ENaC-mediated currents by αA-crystallin. The oocytes were injected with 1 ng of cRNAs encoding each of α, β, and γ ENaC subunits and either water or 6 ng of αA-crystallin cRNA, as indicated. 3 h prior to TEV measurements at −100 mV, half of the oocytes in each group were incubated with 6 μm MG-132. Base-line current was −5.5 μA (n = 15). *, p < 0.05 versus all other groups, determined by ANOVA. C, surface expression of oocytes co-expressing various amounts of αA-crystallin along with wild type α, γ, and βF subunits was determined using anti-FLAG antibodies and a chemiluminescence assay. The oocytes expressing wild type α, β, and γ subunits (no tag) were used to measure background (n ≥ 20 for all groups). *, p < 0.001 versus 0 αA-crystallin, determined by ANOVA.
We then tested our hypothesis that αA-crystallin inhibition of ENaC currents results from enhanced ERAD in Xenopus oocytes. Using the proteasomal inhibitor MG-132 (34), we determined whether proteasomal activity was required for the inhibitory effect of αA-crystallin on ENaC currents (Fig. 3B). We found that currents from oocytes expressing ENaC alone were not affected by MG-132. However, in oocytes expressing ENaC and αA-crystallin, MG-132 abolished the inhibitory effect of αA-crystallin on ENaC current. This result demonstrates that the inhibitory effect of αA-crystallin on ENaC current depends on the activity of the proteasome, an essential ERAD component.
Because of the role of αA-crystallin in facilitating the ERAD of CFTR, we hypothesized that αA-crystallin reduced ENaC current in oocytes by decreasing the number of channels at the cell surface. We therefore measured ENaC surface expression with β subunit having a FLAG epitope tag in the extracellular loop (βF) co-expressed with wild type α and γ subunits using a chemiluminescence-based assay (24, 25). We found that co-expression of αA-crystallin with ENaC reduced channel surface expression in a dose-dependent manner. Moreover, the extent of ENaC surface expression reduction when each amount of the injected αA-crystallin was used paralleled the extent of the reduction in ENaC-mediated Na+ currents (Fig. 3C). These results are consistent with αA-crystallin inhibiting ENaC function by reducing the number of channels at the cell surface rather than affecting single channel properties.
αA-crystallin Retards the Rate of ENaC Delivery to the Cell Surface
A reduction in the number of channels at the cell surface can be achieved either by a decrease in the rate of channel delivery, by an increase in the rate of channel retrieval, or by a combination of both mechanisms. We hypothesized that αA-crystallin reduces ENaC surface expression by reducing the number of channels available for delivery to the membrane, which would be consistent with enhanced ERAD (Fig. 1). We tested this hypothesis by measuring both the rate of channel delivery and the rate of channel retrieval.
To measure the rate of channel delivery to the cell surface, we took advantage of the αS583C mutant. This mutation is located at the outer “mouth” of the channel pore, where covalent modification by the water-soluble sulfhydryl reactive reagent MTSET blocks ∼80% of the current originating from ENaC at the cell surface (35). Recovery of amiloride-sensitive current subsequent to MTSET washout can be attributed to the delivery of new, unblocked channels to the cell surface. 24 h after injecting oocytes with cRNAs encoding αS583C, wild type β and γ subunits and either water or cRNA for αA-crystallin, we measured ENaC current recovery by TEV after applying 1 mm MTSET for 4 min (Fig. 4A). We observed that current recovery was significantly faster in oocytes injected with water than those injected with αA-crystallin.
FIGURE 4. Effect of αA-crystallin co-expression on the rates of ENaC cell surface delivery and retrieval in Xenopus oocytes.
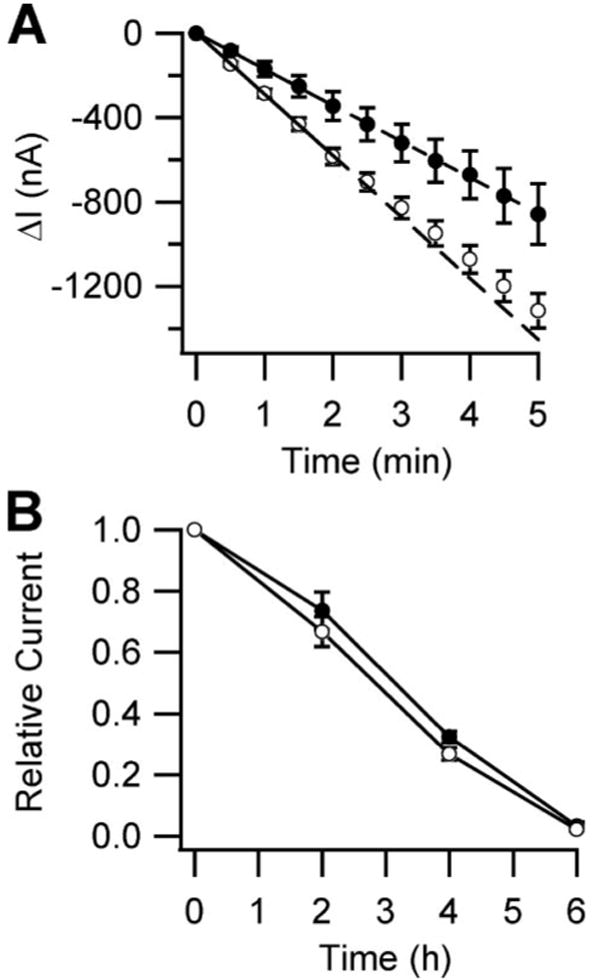
A, surface delivery rates were determined from oocytes injected with cRNA encoding αS583C, wild type β and γ ENaC subunits, and either water (○) or 6 ng of αA-crystallin cRNA (●). 24 h after injection, the oocytes were treated with MTSET for 4 min, and the currents were measured by TEV at −100 mV every 30 s for 5 min (n = 10). The initial rates were determined by linear regression from the first 2 min for each oocyte. The rates were −290 ± 19 and −170 ± 33 nA/min in the absence and presence of αA-crystallin, respectively (p < 0.05, determined by Student's t test). Wild type base-line current was −7.3 μA. B, surface retrieval rates were determined from oocytes injected with cRNA encoding wild type α, β, and γ ENaC subunits and either water (○) or 6 ng of αA-crystallin cRNA (●). The oocytes were incubated in bath solution alone (data not shown) or bath solution supplemented with 5 μm brefeldin A. The currents were measured every 2 h by TEV at −100 mV (n = 8). The wild type base-line current was −3.3 μA.
To measure the rate of cell surface channel retrieval, we halted the delivery of newly synthesized ENaC to the cell surface using brefeldin A. This antibiotic inhibits the formation of transport vesicles that mediate protein transport from the ER to the Golgi apparatus, thus inhibiting integral membrane protein transport to the plasma membrane (36). Having prevented new channel delivery, it becomes possible to measure the half-lives of channels at the cell surface. We therefore injected oocytes with cRNAs encoding wild type ENaC and either water or αA-crystallin. Two days after injection, each group was incubated in a buffer supplemented with 5 μm brefeldin A, and the current was measured every 2 h (Fig. 4B). After the 6-h time point, amiloride was added to determine whether a leak had developed over the course of the experiment. In both the presence or absence of αA-crystallin, the current decreased with a half-life of ∼3 h. Control experiments performed in parallel without the addition of brefeldin A showed that the ENaC current remained stable over 4 h, with a ∼20% decrease at 6 h (data not shown). Together, these results indicate that αA-crystallin decreased ENaC functional expression by reducing the rate of channel delivery to the cell surface but did not affect the rate of channel retrieval from the surface.
Overexpression of Hsc70 Potentiates the Effect of αA-crystallin in Reducing ENaC-mediated Current
We recently reported that overexpression of Hsc70 reduces ENaC expression and current in oocytes (26). Because eukaryotic sHsps have been suggested to release their substrates to ATP-dependent chaperones, such as Hsc70, and may function in the same pathway (37, 38), we hypothesized that αA-crystallin overexpression would exacerbate the Hsc70-mediated reduction in ENaC expression. We tested this hypothesis by measuring the amiloride-sensitive currents of oocytes expressing ENaC in the presence of messages encoding either one or both of these chaperones (Fig. 5). We found that Hsc70 and αA-crystallin reduced ENaC currents by 44 and 38%, respectively. When expressed together, these chaperones reduced ENaC current by 64%, a greater reduction in current than observed with either chaperone alone. Because the combined effect of these two chaperones was not significantly different from the additive effect calculated from these chaperones acting independently, we cannot differentiate between these chaperones acting in independent pathways or acting cooperatively within the same pathway. Nevertheless, these data verify that the αA-crystallin and Hsc70 chaperones play an important role during ENaC biogenesis.
FIGURE 5. Effect of Hsc70 on the inhibition of ENaC-mediated current by αA-crystallin.
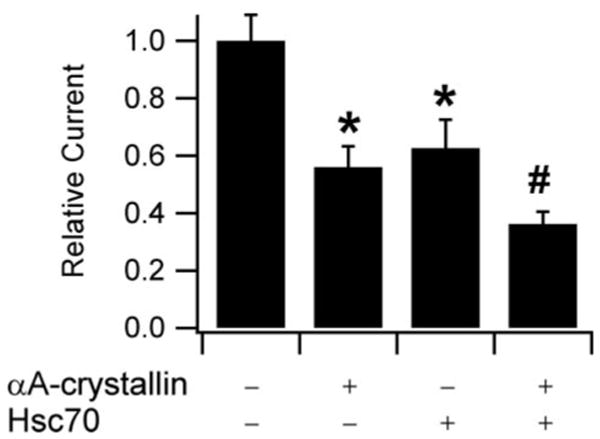
TEV measurements were performed at −100 mV. All of the oocytes were injected with 0.5 ng of cRNAs encoding each of α, β, and γ ENaC subunits. 2 ng of cRNA for αA-crystallin and 10 ng of cRNA for Hsc70 were injected as indicated. n = 20 for each experiment. The base-line current was −2.5 μA. *, p < 0.01 versus ENaC alone. #, p < 0.0001 versus ENaC alone, p < 0.05 versus ENaC + Hsc70. p values were determined by ANOVA.
Discussion
Many of the events during ENaC biogenesis have been described, including Asn-linked glycosylation in the ER, proteolytic cleavage in the trans-Golgi network, and Nedd4-2 mediated ubiquitination at or near the cell surface leading to channel retrieval and degradation (12, 33, 39, 40). In this work, we have begun to characterize the quality control mechanisms to which ENaC subunits are subjected at early steps during their biosynthesis. We have shown that mutation of genes required for ERAD in yeast decreased ENaC α subunit degradation and that the deletion of genes encoding two functionally redundant sHsps, Hsp26 and Hsp42, attenuated ENaC α subunit degradation. These α-crystallin domain-containing chaperones help maintain the solubility of aggregation-prone proteins in yeast (28). We have also shown that the sHsp αA-crystallin is expressed in mouse kidney tissue and in cortical collecting ducts cells derived from mouse kidney that express ENaC. Others have found that αA-crystallin is expressed in the liver and lung where ENaC subunits also reside (41, 42). Further, we have demonstrated that αA-crystallin and the ENaC α subunit co-immunoprecipitate, indicating direct or indirect physical interaction between the two proteins. A functional interaction can be inferred by reduced channel activity when human αA-crystallin and wild type mouse ENaC are co-expressed in oocytes. This effect depends on an active proteasome and was due to a decrease in the number of channels at the cell surface caused by a reduction in the rate of channel insertion.
Based on our data, we propose that ENaC subunits are subject to ER quality control via ERAD and that this process is facilitated by α-crystallin domain-containing sHsps. It was previously observed that sHsps selectively accelerate the degradation of ΔF508-CFTR and that these sHsps may distinguish terminally misfolded forms of ΔF508-CFTR from the wild type protein (11). In this study, we have utilized wild type ENaC α, β, and γ subunits. The question then arises: why are sHsps targeting wild type ENaC for ERAD? In yeast expressing endogenous sHsps, we observed that deletion of these sHsps resulted in a modest but significant reduction of ENaC α subunit degradation. In Xenopus oocytes where human αA-crystallin and mouse α, β, and γ ENaC subunits were overexpressed, we observed a dramatic and dose-dependent decrease in ENaC expression. The simplest explanation for these data is an increased degradation rate. Here we note that unlike CFTR, which is comprised of a single polypeptide chain, ENaC is comprised of three different subunits. We also note that ENaC likely exits the ER as an assembled channel (43). Thus, prior to and during ENaC subunit assembly, there may be solvent-exposed hydrophobic subunit interfaces with the potential to nonspecifically aggregate. This may account for the co-immunoprecipitation of uncleaved ENaC α subunit with αA-crystallin. We therefore propose that sHsps maintain ENaC subunit solubility during intermediate stages of folding or during subunit assembly. We also observed that the cleaved form of the ENaC α subunit co-immunoprecipitated with αA-crystallin. Because the α subunit is cleaved only after the channel has been assembled and transits through the trans-Golgi network, this result suggests that αA-crystallin also interacts with the α subunit in other compartments. Although this phenomenon may be related to ENaC overexpression in these experiments, it is evidence that αA-crystallin may play additional roles in ENaC α subunit biogenesis or trafficking.
Small heat shock proteins interact with a wide spectrum of cellular proteins (28), and thus the question arises as to the specificity of action of distinct members in this family of molecular chaperones. In other words, it is reasonable to ask whether the overexpression of αA-crystallin will affect the biogenesis of every polytopic membrane protein that ultimately resides at the cell surface. We recently showed that αA-crystallin overexpression in HEK293 cells only decreases the stability of the ΔF508 mutant form of CFTR but had no effect on the maturation of wild type CFTR (11). Although the nature of this specificity remains unclear, the data indicate that this sHsp exhibits some specificity of action; thus, not every membrane protein that transits through the secretory pathway is altered by αA-crystallin overexpression.
Given our recent finding that Hsc70 reduced ENaC functional expression (26), we tested the hypothesis that Hsc70 augments the αA-crystallin effect on ERAD. We found that co-expressed Hsc70 and αA-crystallin attenuated ENaC-mediated current to a larger extent than did αA-crystallin alone. These data support our model that sHsps function at an ER quality control branch point, where sHsp binding and stabilization leads either to native channel folding and assembly or to ERAD. We propose further that ENaC subunits are stabilized in the pre-native state by sHsps and that Hsp70 catalyzes conversion to the native state, consistent with our recent observation that moderate overexpression of Hsp70 increases ENaC currents (26). These processes depend on the ability of sHsps to efficiently disassemble and release the target protein, which has been shown to be required for effective chaperone activity (44–47). Although sHsps must dissociate from their targets at a finite rate, it remains unclear whether dissociation is spontaneous or is catalyzed by the binding of other chaperones. In either case, sHsp overexpression would drive sHsp·target complex formation, which we suggest accounts for the dose-dependent decrease in ENaC functional expression we observed in oocytes.
It has been shown that 80–99% of synthesized ENaC subunits do not reach the cell surface where they can participate in transepithelial Na+ transport (43, 48). Our results suggest that the ERAD pathway, as one component of ER quality control, probably accounts for a significant fraction of the degraded ENaC subunits. Although ENaC biosynthesis seems inherently wasteful, the “waste” attributed to ERAD results in a higher quality product and blocks the formation of potentially toxic species. In the future, it will be critical to identify other modulators of ENaC ERAD, a pursuit that might have therapeutic consequences.
Acknowledgments
CCD cells (mpkCCDc14) were a gift from Alain Vandewalle. MDCK type 1 cells were a gift from Daniela Rotin.
Footnotes
This work was supported in part by Grants GM75061 (to J. L B.), DK065161 (to T. R. K. and R. P. H.), and DK58046 (to R. C. R.) from the National Institutes of Health.
The abbreviations used are: ENaC, epithelial Na+ channel; ER, endoplasmic reticulum; ERAD, ER-associated degradation; CFTR, cystic fibrosis transmembrane conductance regulator; sHsp, small heat shock protein; CCD, cortical collecting duct; MTSET, [2-(trimethylammonium) ethyl] methanethiosulfonate Bromide; TEV, two-electrode voltage clamp; MBS, modified Barth's saline; BSA, bovine serum albumin; MDCK, Madin-Darby canine kidney; HA, hemagglutinin; ANOVA, analysis of variance.
References
- 1.Garty H, Palmer LG. Physiol Rev. 1997;77:359–396. doi: 10.1152/physrev.1997.77.2.359. [DOI] [PubMed] [Google Scholar]
- 2.Rossier BC, Pradervand S, Schild L, Hummler E. Annu Rev Physiol. 2002;64:877–897. doi: 10.1146/annurev.physiol.64.082101.143243. [DOI] [PubMed] [Google Scholar]
- 3.Mall M, Grubb BR, Harkema JR, O'Neal WK, Boucher RC. Nat Med. 2004;10:487–493. doi: 10.1038/nm1028. [DOI] [PubMed] [Google Scholar]
- 4.Kosari F, Sheng S, Li J, Mak DO, Foskett JK, Kleyman TR. J Biol Chem. 1998;273:13469–13474. doi: 10.1074/jbc.273.22.13469. [DOI] [PubMed] [Google Scholar]
- 5.Firsov D, Gautschi I, Merillat AM, Rossier BC, Schild L. EMBO J. 1998;17:344–352. doi: 10.1093/emboj/17.2.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Snyder PM, Cheng C, Prince LS, Rogers JC, Welsh MJ. J Biol Chem. 1998;273:681–684. doi: 10.1074/jbc.273.2.681. [DOI] [PubMed] [Google Scholar]
- 7.Staruschenko A, Medina JL, Patel P, Shapiro MS, Booth RE, Stockand JD. J Biol Chem. 2004;279:27729–27734. doi: 10.1074/jbc.M404169200. [DOI] [PubMed] [Google Scholar]
- 8.Kellenberger S, Schild L. Physiol Rev. 2002;82:735–767. doi: 10.1152/physrev.00007.2002. [DOI] [PubMed] [Google Scholar]
- 9.Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, Rossier BC. Nature. 1994;367:463–467. doi: 10.1038/367463a0. [DOI] [PubMed] [Google Scholar]
- 10.Canessa CM, Merillat AM, Rossier BC. Am J Physiol. 1994;267:C1682–C1690. doi: 10.1152/ajpcell.1994.267.6.C1682. [DOI] [PubMed] [Google Scholar]
- 11.Ahner A, Nakatsukasa K, Zhang H, Frizzell RA, Brodsky JL. Mol Biol Cell. 2006;18:806–814. doi: 10.1091/mbc.E06-05-0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hughey RP, Mueller GM, Bruns JB, Kinlough CL, Poland PA, Harkleroad KL, Carattino MD, Kleyman TR. J Biol Chem. 2003;278:37073–37082. doi: 10.1074/jbc.M307003200. [DOI] [PubMed] [Google Scholar]
- 13.Stirling CJ, Rothblatt J, Hosobuchi M, Deshaies R, Schekman R. Mol Biol Cell. 1992;3:129–142. doi: 10.1091/mbc.3.2.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ito H, Kamei K, Iwamoto I, Inaguma Y, Garcia-Mata R, Sztul E, Kato K. J Biochem (Tokyo) 2002;131:593–603. doi: 10.1093/oxfordjournals.jbchem.a003139. [DOI] [PubMed] [Google Scholar]
- 15.Adams A, Gottschling DE, Kaiser CA, Stearns T. Methods in Yeast Genetics. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1997. [Google Scholar]
- 16.Zhang Y, Michaelis S, Brodsky JL. Methods Mol Med. 2002;70:257–265. doi: 10.1385/1-59259-187-6:257. [DOI] [PubMed] [Google Scholar]
- 17.Bens M, Vallet V, Cluzeaud F, Pascual-Letallec L, Kahn A, Rafestin-Oblin ME, Rossier BC, Vandewalle A. J Am Soc Nephrol. 1999;10:923–934. doi: 10.1681/ASN.V105923. [DOI] [PubMed] [Google Scholar]
- 18.Morris RG, Schafer JA. J Gen Physiol. 2002;120:71–85. doi: 10.1085/jgp.20018547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Breitfeld PP, Casanova JE, Harris JM, Simister NE, Mostov KE. Methods Cell Biol. 1989;32:329–337. doi: 10.1016/s0091-679x(08)61178-4. [DOI] [PubMed] [Google Scholar]
- 20.Rubenstein RC, Zeitlin PL. Am J Physiol. 2000;278:C259–C267. doi: 10.1152/ajpcell.2000.278.2.C259. [DOI] [PubMed] [Google Scholar]
- 21.Bruns JB, Hu B, Ahn YJ, Sheng S, Hughey RP, Kleyman TR. Am J Physiol. 2003;285:F600–F609. doi: 10.1152/ajprenal.00095.2003. [DOI] [PubMed] [Google Scholar]
- 22.Malik B, Schlanger L, Al-Khalili O, Bao HF, Yue G, Price SR, Mitch WE, Eaton DC. J Biol Chem. 2001;276:12903–12910. doi: 10.1074/jbc.M010626200. [DOI] [PubMed] [Google Scholar]
- 23.Snyder PM, Olson DR, Bucher DB. J Biol Chem. 1999;274:28484–28490. doi: 10.1074/jbc.274.40.28484. [DOI] [PubMed] [Google Scholar]
- 24.Carattino MD, Hill WG, Kleyman TR. J Biol Chem. 2003;278:36202–36213. doi: 10.1074/jbc.M300312200. [DOI] [PubMed] [Google Scholar]
- 25.Zerangue N, Schwappach B, Jan YN, Jan LY. Neuron. 1999;22:537–548. doi: 10.1016/s0896-6273(00)80708-4. [DOI] [PubMed] [Google Scholar]
- 26.Goldfarb SB, Kashlan OB, Watkins JN, Suaud L, Yan W, Kleyman TR, Rubenstein RC. Proc Natl Acad Sci U S A. 2006;103:5817–5822. doi: 10.1073/pnas.0507903103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knowles MR, Olivier KN, Hohneker KW, Robinson J, Bennett WD, Boucher RC. Chest. 1995;107:71S–76S. doi: 10.1378/chest.107.2_supplement.71s. [DOI] [PubMed] [Google Scholar]
- 28.Haslbeck M, Braun N, Stromer T, Richter B, Model N, Weinkauf S, Buchner J. EMBO J. 2004;23:638–649. doi: 10.1038/sj.emboj.7600080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haslbeck M, Ignatiou A, Saibil H, Helmich S, Frenzl E, Stromer T, Buchner J. J Mol Biol. 2004;343:445–455. doi: 10.1016/j.jmb.2004.08.048. [DOI] [PubMed] [Google Scholar]
- 30.Jentsch S, Rumpf S. Trends Biochem Sci. 2007;32:6–11. doi: 10.1016/j.tibs.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 31.Adams J. Cancer Treat Rev. 2003;29 1:3–9. doi: 10.1016/s0305-7372(03)00081-1. [DOI] [PubMed] [Google Scholar]
- 32.Ciampolillo F, McCoy DE, Green RB, Karlson KH, Dagenais A, Molday RS, Stanton BA. Am J Physiol. 1996;271:C1303–C1315. doi: 10.1152/ajpcell.1996.271.4.C1303. [DOI] [PubMed] [Google Scholar]
- 33.Hughey RP, Bruns JB, Kinlough CL, Harkleroad KL, Tong Q, Carattino MD, Johnson JP, Stockand JD, Kleyman TR. J Biol Chem. 2004;279:18111–18114. doi: 10.1074/jbc.C400080200. [DOI] [PubMed] [Google Scholar]
- 34.Lee DH, Goldberg AL. Trends Cell Biol. 1998;8:397–403. doi: 10.1016/s0962-8924(98)01346-4. [DOI] [PubMed] [Google Scholar]
- 35.Sheng S, Li J, McNulty KA, Kieber-Emmons T, Kleyman TR. J Biol Chem. 2001;276:1326–1334. doi: 10.1074/jbc.M008117200. [DOI] [PubMed] [Google Scholar]
- 36.Dinter A, Berger EG. Histochem Cell Biol. 1998;109:571–590. doi: 10.1007/s004180050256. [DOI] [PubMed] [Google Scholar]
- 37.Haslbeck M, Miess A, Stromer T, Walter S, Buchner J. J Biol Chem. 2005;280:23861–23868. doi: 10.1074/jbc.M502697200. [DOI] [PubMed] [Google Scholar]
- 38.Cashikar AG, Duennwald M, Lindquist SL. J Biol Chem. 2005;280:23869–23875. doi: 10.1074/jbc.M502854200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Snyder PM, McDonald FJ, Stokes JB, Welsh MJ. J Biol Chem. 1994;269:24379–24383. [PubMed] [Google Scholar]
- 40.Snyder PM. Endocrinology. 2005;146:5079–5085. doi: 10.1210/en.2005-0894. [DOI] [PubMed] [Google Scholar]
- 41.Srinivasan AN, Nagineni CN, Bhat SP. J Biol Chem. 1992;267:23337–23341. [PubMed] [Google Scholar]
- 42.McDonald FJ, Price MP, Snyder PM, Welsh MJ. Am J Physiol. 1995;268:C1157–C1163. doi: 10.1152/ajpcell.1995.268.5.C1157. [DOI] [PubMed] [Google Scholar]
- 43.Valentijn JA, Fyfe GK, Canessa CM. J Biol Chem. 1998;273:30344–30351. doi: 10.1074/jbc.273.46.30344. [DOI] [PubMed] [Google Scholar]
- 44.Shashidharamurthy R, Koteiche HA, Dong J, McHaourab HS. J Biol Chem. 2005;280:5281–5289. doi: 10.1074/jbc.M407236200. [DOI] [PubMed] [Google Scholar]
- 45.Sun Y, MacRae TH. Cell Mol Life Sci. 2005;62:2460–2476. doi: 10.1007/s00018-005-5190-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lentze N, Aquilina JA, Lindbauer M, Robinson CV, Narberhaus F. Eur J Biochem. 2004;271:2494–2503. doi: 10.1111/j.1432-1033.2004.04180.x. [DOI] [PubMed] [Google Scholar]
- 47.Pasta SY, Raman B, Ramakrishna T, Rao Ch M. Mol Vis. 2004;10:655–662. [PubMed] [Google Scholar]
- 48.Weisz OA, Wang JM, Edinger RS, Johnson JP. J Biol Chem. 2000;275:39886–39893. doi: 10.1074/jbc.M003822200. [DOI] [PubMed] [Google Scholar]