

Abstract
The Sec61 protein translocation complex in the endoplasmic reticulum (ER) membrane is composed of three subunits. The α-subunit, called Sec61p in yeast, is a multispanning membrane protein that forms the protein conducting channel. The functions of the smaller, carboxyl-terminally tail-anchored β sub-unit Sbh1p, its close homologue Sbh2p, and the γ subunit Sss1p are not well understood. Here we show that co-translational protein translocation into the ER is reduced in sbh1Δ sbh2Δ cells, whereas there is a limited reduction of post-translational tranlocation and no effect on export of a mutant form of α-factor precursor for ER-associated degradation in the cytosol. The translocation defect and the temperature-sensitive growth phenotype of sbh1Δ sbh2Δ cells were rescued by expression of the transmembrane domain of Sbh1p alone, and the Sbh1p transmembrane domain was sufficient for coimmunoprecipitation with Sec61p and Sss1p. Furthermore, we show that Sbh1p co-precipitates with the ER transmembrane protein Rtn1p. Sbh1p-Rtn1p complexes do not appear to contain Sss1p and Sec61p. Our results define the transmembrane domain as the minimal functional domain of the Sec61β homologue Sbh1p in ER translocation, identify a novel interaction partner for Shb1p, and imply that Sbh1p has additional functions that are not directly linked to protein translocation in association with the Sec61 complex.
During biosynthesis proteins destined for secretion or for intracellular membranes of the secretory or the endocytic pathways are first translocated into the endoplasmic reticulum (ER)7 (1). The channel mediating protein translocation is conserved in evolution from bacteria to eukaryotic cells (2, 3). This channel is formed by the Sec61 complex in eukaryotic cells and the SecY complex in eubacteria and Archaea. The largest of the Sec61/SecY complex subunit is Sec61α in eukaryotes and SecY in eubacteria and Archaea. This subunit forms the actual protein conducting channel. The β subunit is termed Sec61β in mammals, and SecG in eubacteria and Archaea. In the yeast Saccharomyces cerevisiae two homologous β subunits, Sbh1p and Sbh2p, exist. The γ subunit is called Sec61γ in mammalian cells, Sss1p in S. cerevisiae, and SecE in eubacteria and Archaea. In S. cerevisiae two homologous trimeric Sec61 complexes exist. The Sec61 complex is composed of Sec61p, Sbh1p, and Sss1p (4–7). This complex together with Sec63p and Kar2p functions in co-translational translocation (8). The Sec61 complex mediates also post-translational protein translocation in association with Kar2p, Sec62p, Sec63p, Sec71p, and Sec72p (6, 9). This heptameric complex is called the Sec complex. In addition to its role in co- and post-translational translocation the Sec61 complex is also involved in retro-translocation of misfolded proteins to the cytosol for degradation (10). The second S. cerevisiae translocation complex, the Ssh1 complex, consists of the Sec61p homologue Ssh1p, the Sbh1p homologue Sbh2p, and Sss1p (2, 8). The Ssh1 complex has been shown to contribute to co- and post-translational translocation and ER-associated degradation (ERAD) (4, 11).
The role of the Sec61α subunit as the protein conducting channel is well established, whereas the functions of the β and γ subunits remain largely unclear. The β subunit is not essential but has a facilitating role in translocation in mammalian cells (12). In addition to its interactions with Sec61p, the mammalian β subunit has been shown to interact with signal peptidase (12), and the ribosome (13) and the yeast β subunit, Sbh1p, was shown to act as a guanine nucleotide exchange factor for signal recognition particle receptor (14). The two yeast β subunits, Sbh1p and Sbh2p (previously also called Seb1p and Seb2p), are encoded by non-essential genes. Deletion of either gene alone has no effect on growth, whereas the deletion of both SBH1 and SBH2 was shown to result in temperature sensitivity for growth at 38 °C (4, 7) and a slight defect in translocation of the α-mating pheromone precursor (ppαf) and Kar2p in vivo (4). In a heterologous in vitro translation-translocation system a stronger translocation defect was observed in cells lacking both Sbh1p and Sbh2p (4).
Previously, a stabilizing role in the translocation complex has been suggested for both β and γ subunits (15). Both in mammalian and yeast cells, β subunits have been shown to interact with proteins that are not obviously functionally linked with protein translocation. The β subunit has been shown to co-immunoprecipitate with Sec15p and Sec8p, two components of the exocyst complex (16, 17) whose main function is thought to be tethering of transport vesicles to the plasma membrane (18). The functional relevance of the translocon-exocyst interaction is presently unknown. Overexpression of one of the exocyst subunits, SEC3, results in accumulation of Sec61p in the emerging bud in yeast (19). These results have led to the proposal that translocon-exocyst interactions may play a role in ER inheritance during cytokinesis (18, 20).
We have analyzed the function of Sbh1 protein using a set of SBH1 mutants. We show that the transmembrane (TM) domain of Sbh1p on its own can functionally substitute the full-length protein in the ER. We demonstrate that the TM domain of Sbh1p is sufficient for interaction with Sec61p and Sss1p. In addition, we identify Rtn1p, an ER membrane protein, as a novel interaction partner for Sbh1p. These results define a functionally important domain in the Sec61 β subunit Sbh1p and reveal novel interactions of this protein that support the idea that Sbh1p has functions outside the Sec61 complex.
EXPERIMENTAL PROCEDURES
Yeast Strains and Culture Conditions
The yeast strains used in this study are shown in supplementary Table S1. All strains excluding WCG4a (From D. Wolf, University of Stuttgart), H956, H1107, and H1109 (S. Ferro-Novick (Yale University) are congenic to H304 or H973 (obtained from P. Novick (Yale University)). Gene deletions and carboxyl-terminal tagging of proteins were achieved with the PCR-based methods using pFA6a-kanMX, pFA6a-natNT2, pYM-hphNT1, and pYM1 as templates (from M. Knop, EMBL, Heidelberg) as previously described (21, 22). H3384, H3386, and H3387 were obtained by integration of the ClaI cut YIpαa-L, encoding Bacillus amyloliquefaciens α-amylase gene, in the leu2 locus. To generate sbh1Δ (H3386), sbh2Δ (H3387), and sbh1Δ sbh2Δ (H3388) strains for analysis of α-amylase secretion and to ensure that the same number of the α-amylase expression cassette is presents in these integrant strains, H3384 was crossed with H3235. The resulting diploid strain was sporulated, dissected, and spores were analyzed to obtain strains for α-amylase expression in combination of sbh1Δ, sbh2Δ, and sbh1Δ sbh2Δ. Strain H3543 was generated by deletion of the TRP1 gene from H3232 with a PCR cassette generated using pFA6a-natNT2 as a template. The yeast growth media were prepared essentially as previously described (23). Synthetic media lacking uracil or appropriate amino acids were used for plasmid selection. G418 (Invitrogen) was used at a concentration of 200 μg/ml, nourseothricin at 100 μg/ml (Werner BioAgents, Germany), and hygromycin (EMD Biosciences, Inc.) at 300 μg/ml. Yeast was cultivated aerobically on plates or in shaker flasks at 30 °C if not stated otherwise. Complementation and suppression tests on a dilution series of yeast strains were performed essentially as described (17). Sporulation and tetrad analysis were done as described (24).
Plasmids
Standard DNA methods were used. Oligonucleotides were purchased from Sigma Genosys. The sequences of oligonucleotides used are available on request. The plasmids used for gene expression in yeast are listed in supplementary Table S2. Construction of the full-length YEpSBH1-(1–82) and plasmids encoding the truncated forms of Sbh1p, full-length Sss1p, and Sss1p-TM domain were constructed with PCR. Briefly, DNA fragments encoding the corresponding genes were generated by oligonucleotides containing XbaI and XhoI restriction enzyme recognition sites and cloned into the XbaI/XhoI cut pVT102U (25) or p425 (26). SBH2 or sbh2-tm domain (amino acids 57–82) encoding sequences were generated by oligonucleotides containing XbaI and XhoI restriction enzyme recognition sites and cloned into the XbaI/XhoI cut pVT102U. The YIpαa-L was constructed by cloning the α-amylase expression cassette with ADH1 promoter and terminator as a BamHI/SalI fragment from YIpαa6 (27) into BlueScript SK(−). From there the same fragment was cloned as the BamHI/SalI fragment into pRS405 (28). To generate YEpBIO-SBH1-(1–82) and YEpBIO-SBH1-(50–75), a DNA fragment encoding biotin acceptor peptide (BIO-tag) derived from Propionibacterium shermanii transcarboxylase was added in front of SBH1 or SBH1(50–75) by nested PCR using pFA6a-HBH-kanMX6 (29) (gift from Peter Kaiser, University of California) as the template for BIO-tag with oligonucleotides containing BamHI/XhoI sites and then cloned into vector p426ADH (26). All constructs generated by PCR were sequenced.
Antibodies
The α-amylase antibodies have been described before (30). Antibodies against Sss1p were a gift from Randy Schekman (31). Antibodies to Sec61p, Sbh1p, and Sso1/2 have been described previously (17, 32, 33). Monoclonal anti-HA (12CA5) antibodies were from Roche Applied Science. Antibody dilutions used in Western blotting were 1:1000 for α-amylase and anti-Sbh1p, 1:50000 for anti-Sso1/2p, 1:1000 for anti-Sss1p, and 1:2000 for anti-Sec61p. HRP-conjugated goat anti-mouse and anti-rabbit antibodies (Bio-Rad) were diluted 1:2000. Quantification of the band intensities was done by scanning the films with GS-710 densitometer (Bio-Rad) using the Quantity One program, version 4.0 (Bio-Rad).
Preparation of Microsomes and Cytosol and in Vitro ERAD Assays
Wild type and mutant cells were grown in YPD at 30 °C to early logarithmic phase and microsomes were prepared by spheroplasting and hypotonic lysis as described previously (34). Cytosol was prepared from exponentially growing WCG4a by liquid nitrogen lysis as described previously (34). In vitro ERAD of the non-glycosylated mutant form of pro-α-factor (Δgpαf) was assayed as in Pilon et al. (34) using 2 μl of microsomes OD280 = 30, and 2 μl of in vitro translated, 35S-labeled pΔgpαf (500,000 cpm) per sample.
Immunoprecipitations and Pulldowns
For 35S labeling of α-factor, cells were grown in complete minimal medium without methionine and cysteine at 30 °C. Labeling, lysis, and immunoprecipitation were performed as in Gillece et al. (35) using 1.5 A600 of cells and 10 μl of polyclonal anti-Sbh1p antiserum per sample. For pulldown of BIO-tagged or immunoprecipitation of HA-tagged proteins, a membrane protein-enriched fraction was isolated as described (6). Briefly, yeast cells were grown to mid-log phase (A600 ∼ 1). After centrifugation, the cells were resuspended in lysis buffer A (50 mm HEPES-KOH (pH 7.5), 25 mm KAC, 5 mm MgAC, 1 mm EDTA, 2 mm dithiothreitol, 1 mm phenylmethylsulfonyl fluoride, 10% glycerol) supplemented with Complete protease inhibition mixture (Roche), and homogenized by vortexing with acid-washed glass beads. The lysates were centrifuged for 10 min at 400 × g and the supernatant subjected to centrifugation at 72,000 × g for 35 min. The membrane pellets were resuspended in buffer B (50 mm HEPES-KOH (pH 7.5), 800 mm KAC, 16 mm MgAC, 10% glycerol, 7 mmβ-mercaptoethanol) with 2.5% saponin supplemented with Complete protease inhibition mixture and incubated on ice for 30 min and then centrifuged at 126,000 × g for 60 min. The pellets were resuspended in buffer C (50 mm HEPES-KOH (pH 7.5), 400 mm KAC, 16 mm MgAC, 10% glycerol, 7 mmβ-mercaptoethanol) with 3% digitonin and Complete protease inhibition mixture and incubated on ice for 30 min and then centrifuged at 304,000 × g for 30 min, thereafter the pellets were resuspended in buffer B and used for pull-downs or immunoprecipitations. Streptavidin or anti-HA (12CA5) antibody-coupled Dynabeads (Dynal) were used for precipitation of Rtn1p-3HA, BIO-Sbh1p, or BIO-Sbh1-TM. Coupling of anti-HA antibodies to Protein G-coupled magnetic beads was carried out according to guidelines of the manufacturer (Dynal). Incubation with the lysate was carried out at 4 °C by end over rotation for 1 h followed by 5 washes at 4 °C by rotation, 15 min each. The bound proteins were eluted twice with 0.1% SDS at 95 °C. The eluted samples were pooled and used for SDS-PAGE and Western blotting. The protein bands were detected with specific antibodies or HRP-conjugated streptavidin (Molecular Probes) diluted 1:5000 and visualized with the ECL detection system (Amersham Biosciences).
Post-translational Translocation in Vitro and N-Glycosidase F Treatment
In vitro translated ppαf was translocated into wild type or mutant microsomes at the indicated temperatures in the presence of ATP and an ATP-regenerating system as described (36) using 0.3 eq of membranes per sample (1 eq = 1 μl of microsomes OD280 = 50). At the end of the incubation, samples were precipitated with trichloroacetic acid and proteins resolved by 15% SDS-PAGE gels. For N-glycopeptidase F treatment, membranes were lysed and glycosylated gpαf precipitated with concanavalin A-Sepharose as reported previously (37). Washed beads were subsequently incubated for 2 h at 37 °C with or without 2 units of N-glycosidase F (Roche) in 40 μl of 100 mm sodium phosphate (pH 7.5). Beads were heated in 2× SDS sample buffer (100 mm Tris-HCl, pH 6.8, 200 mm dithiothreitol, 4% SDS, 0.2% bromphenol blue, 10% glycerol) and proteins analyzed on 18% polyacrylamide, 2 m urea gels. For inhibition of glucosidase or mannosidase activities, membranes were preincubated with the indicated concentrations of castanospermine and/or 1-deoxymannojirimycin (Calbiochem) for 15 min at 20 °C, and the drugs present at the same concentrations during the translocation reactions.
Co-translational Translocation in Vitro
Microsomes (0.3 eq) were incubated with 2.3 μg in vitro transcribed ppαf mRNA (using SP6 polymerase, (38)) at the indicated temperatures in a final volume of 10 μl containing 8 μCi (=0.3 MBq) of translation grade [35S]methionine (Amersham Biosciences), 3 μl of nuclease-treated, gel-filtered yeast cytosol, 7 units of RNase inhibitor (Amersham Biosciences), 3.3 μg of creatine phosphokinase, 3.3 μl of 3× translation buffer (66 mm HEPES, pH 7.4, 75 mm creatine phosphokinase, 2.25 mm ATP, 300 μm GTP, 120 μm amino acid mixture without methionine (Amersham Biosciences), 360 mm potassium acetate, 6 mm magnesium acetate, 5.1 mm dithiothreitol) and diethylpyrocarbonate-treated water. At the end of the incubation, membranes were sedimented and washed once in B88 (20 mm HEPES-KOH, pH 6.8, 250 mm sorbitol, 150 mm potassium acetate, 5 mm magnesium acetate), before resuspension in 2× SDS samples buffer, heating to 65 °C for 10 min, and analysis by 15% SDS-PAGE and autoradiography.
Secretion and Translocation of α-Amylase
To analyze intracellular and secreted forms of α-amylase, yeast strains H3384, H3386, H3387, and H3388 were grown overnight in YPD medium at 24 °C. The overnight cultures were diluted with fresh medium to A600 = 0.3 and re-grown to A600 = 2. Cultures (3 ml) were centrifuged at 3000 × g and the pellets were washed once with water. Cells and culture medium were subjected to SDS-PAGE and Western blotting with anti-α-amylase antibodies. To analyze α-amylase translocation, H304, H956, H1107, H1109, and H3232 cells containing YEpαa6 expression plasmid, or H3384 and H3388 cells transformed with different Sbh1p mutant encoding plasmids, were grown overnight at 24 °C. Cultures were diluted with fresh medium to A600 = 2, shifted to 37.5 °C, and grown for the indicated times. Cells were collected, washed once with water followed by resuspension in 2% SDS containing protease inhibitors (Complete, Roche), and a 5-min incubation at 95 °C. Thereafter cells were vortexed in the presence of glass beads and 2% SDS and protease inhibitors (Complete, Roche). The protein concentration of the lysates was measured with BCA protein assay kit (Pierce) and equal amounts of lysates were subjected to 10% SDS-PAGE and Western blotting. Purified B. amyloliquefaciens α-amylase (Sigma A7595) was used as a control for the non-glycosylated form of α-amylase.
Ubiquitin-assisted Translocation Assay (UTA)
Strain H3543 was transformed with plasmids encoding Suc223, Sec2277, Dap2300, or the empty vector pRS314 (39, 40) (gifts from N. Johnsson, University of Muenster, Germany, and J. Brown, Newcastle University, United Kingdom). Patches of transformants grown on SCD-Trp-Leu were replicated on SCD-Trp-Leu-Ura plates and their growth was followed for 3 days at 24 °C.
RESULTS
Sbh1p and Sbh2p Participate in Protein Import into the ER, but Are Dispensable for ERAD
The function of the non-essential β-subunit Sbh1p in association with Sec61p and Sss1p is poorly understood. We have previously shown that overexpression of SBH1 has a positive effect on production of secreted bacterial α-amylase in S. cerevisiae (17). Therefore, we studied possible effects of deletion of the SBH genes on secretion of this reporter protein. The wild type (H3384), sbh1Δ (H3386), sbh2Δ (H3387), and sbh1Δ sbh2Δ (H3388) strains containing an integrated copy of the α-amylase gene were grown in liquid culture and the secreted and intracellularly retained α-amylase were detected by Western blotting with α-amylase-specific antibodies. Compared with the wild type cells, cells with single deletions of SBH1 or SBH2 secreted 59 and 56% of α-amylase, respectively, whereas in the sbh1Δ sbh2Δ strain substantially less (33%) of the enzyme was secreted into the growth medium (Fig. 1A). In the sbh1Δ sbh2Δ cell lysate, the majority of the intracellular α-amylase migrated at the position of the unglycosylated bacterial enzyme (Fig. 1A). This faster migrating form of α-amylase co-migrated with the form of α-amylase that accumulated in translocation mutants sec61-2 and sec63-1 cells shifted to 37.5 °C for 2 h (Fig. 1B). To verify that the faster migrating band represents untranslocated α-amylase, sbh1Δ sbh2Δ and wild type cells were lysed and fractionated by differential centrifugation. The faster migrating band was still detected in the supernatant of sbh1Δ sbh2Δ cells after pelleting the membranes for 1 h at 100,000 × g (supplementary Fig. S1) indicating that it represents the untranslocated form of the protein. Together these results suggest that translocation of α-amylase is defective in vivo in sbh1Δ sbh2Δ cells at restrictive temperatures. Previously, only a moderate defect was observed in in vivo translocation of the α-factor precursor and Kar2p in sbh1Δ sbh2Δ cells (4). A more pronounced defect was, however, evident in in vitro experiments using a heterologous translation-translocation system (4). Because of the differences in the results, we re-examined the in vitro translocation of yeast α-factor using a homologous yeast translation-translocation system.
FIGURE 1. Sbh1p and Sbh2p participate in protein import into the ER in vivo.
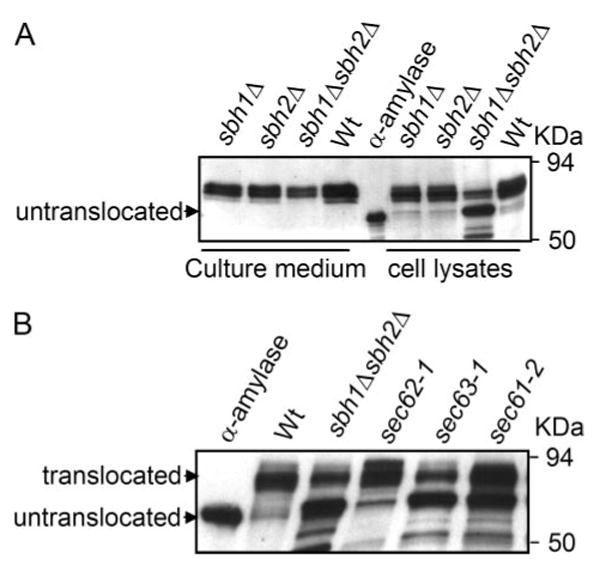
A, Western blot analysis of secreted and intracellular α-amylase. sbh1Δ (H3386), sbh2Δ (H3387), and sbh1Δ sbh2Δ (H3388) cells expressing α-amylase were grown at the permissive temperature and shifted to 37.5 °C for 9 h. Proteins present in the culture medium and cell lysate were resolved by SDS-PAGE, transferred to nitrocellulose membrane, and blots probed with antibodies against α-amylase. Untranslocated indicates the position of purified bacterial α-amylase. B, Western blot analysis of intracellular bacterial α-amylase in WT (H304), sbh1Δ sbh2Δ (H3232), sec62-1 (H1109), sec63-1 (H1107), and sec61-2 (H956) cells. Cells transformed with YEpαa6 were grown at 37.5 °C for 1 h, collected, and processed for SDS-PAGE and Western blotting with anti-α-amylase antibodies. α-Amylase indicates the lane loaded with purified bacterial α-amylase.
For post-translational translocation 0.3 eq yeast microsomes were incubated with ATP, an ATP-regenerating system, and 300,000 cpm of ppαf translated in vitro in a yeast cell extract for the indicated periods of time at 10, 30, or 37 °C. At the end of the incubations, samples were precipitated with trichloroacetic acid and analyzed by SDS-PAGE. Translocation was quantified and expressed as the ratio of N-glycosylated protein (3gpαf) to total radiolabeled protein in each lane (3gpαf + ppαf). In contrast to previous results (4), we observed only a modest post-translational import defect into the ER for ppαf at 37 °C (Fig. 2, A and B). The import defect was more pronounced when co-translational import was examined (Fig. 2B, lower right).
FIGURE 2. Effect of Sbh1p and Sbh2p on in vitro protein import into the ER.
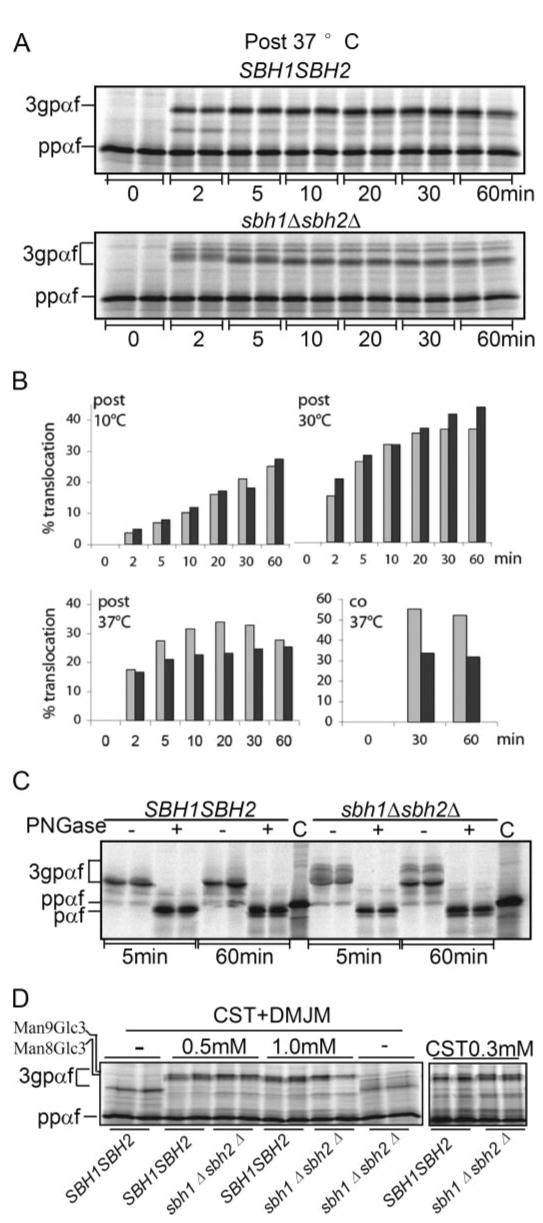
A, post-translational in vitro translocation of α-factor precursor (ppαf) into sbh1Δ sbh2Δ microsomes. Yeast microsomes (0.3 eq) were incubated with 300,000 cpm of ppαf translated in vitro in a yeast cell extract in the presence of ATP, and an ATP-regenerating system for the indicated periods of time at 37 °C. At the end of the incubation, samples were precipitated with trichloroacetic acid and analyzed by SDS-PAGE. B, quantitation of the post- and co-translational translocation of ppαf into sbh1Δ sbh2Δ microsomes in vitro at 10, 30, and 37 °C. Translocation was quantified and expressed as the percent of N-glycosylated protein (3gpαf) to total radiolabeled protein in each lane (3gpαf + ppαf). For co-translational import, 0.3 eq of membranes were used per 10 μl of in vitro translation/translocation reaction containing 2.3 μg of ppαf mRNA. Light gray, SBH1 SBH2; dark gray, sbh1Δ sbh2Δ. C, analysis of signal peptide cleavage of α-factor precursor in sbh1Δ sbh2Δ cells. In vitro translated ppαf was translocated into wild type and mutant membranes for 5 or 60 min, the membranes were lysed and glycosylated 3gpαf precipitated with concanavalin A-Sepharose. Precipitated 3gpαf was incubated with N-glycanase to remove N-linked glycans or mock-digested, and deglycosylated, translocated proteins were analyzed by SDS-PAGE on 18% 2 m urea gels, aliquots of in vitro translated ppαf containing the signal peptide were loaded as controls (C). D, effect of castanospermine (CST) and 1-deoxymannojirimycin (DMJM) on migration of 3gpαf. Membranes from sbh1Δ sbh2Δ and WT cells were preincubated with the indicated concentrations of castanospermine and/or 1-deoxymannojirimycin and during the translocation reactions followed by gel analysis. Positions of 3gpαf with Man9Glc3 and Man8Glc3 glycans are indicated on the left.
In our in vitro translocation experiments, we observed that 3gpαf initially migrated as a group of bands at a higher molecular weight in sbh1Δ sbh2Δ membranes at all temperatures (Fig. 2A and not shown). After 10–20 min the most prominent of these bands migrated at the same position as 3gpαf in wild type membranes (Fig. 2A). Because Sec61β in mammalian cells has been shown to anchor signal peptidase to the translocon (12) we asked if the abnormal size of 3gpαf in sbh1Δ sbh2Δ membranes was due to delayed signal cleavage. We translocated in vitro translated ppαf into wild type and mutant membranes for 5 or 60 min, lysed the membranes, and precipitated glycosylated 3gpαf with concanavalin A-Sepharose. Precipitated 3gpαf was incubated with N-glycanase to remove N-linked glycans, and deglycosylated, translocated proteins were analyzed by SDS-PAGE on 18% 2 m urea gels. Aliquots of in vitro translated ppαf containing the signal peptide were loaded as controls. As shown in Fig. 2C, translocated deglycosylated protein (pαf) was signal-cleaved and therefore migrated faster than ppαf even after 5 min translocation into sbh1Δ sbh2Δ membranes. The slower mobility bands in sbh1Δ sbh2Δ membranes were removed by glycopeptidase F treatment and rendered pαf the same molecular weight as in the WT membranes. Collectively these results suggest that delayed signal peptide cleavage was not the cause for the higher molecular weight of 3gpαf in sbh1Δ sbh2Δ membranes.
Because the molecular weight of deglycosylated pαf from wild type and mutant membranes was identical, the higher molecular weight forms of 3gpαf in sbh1Δ sbh2Δ membranes are likely to represent differences in N-linked glycan processing. To analyze this wild type and mutant membranes were incubated with or without the glucosidase inhibitor castanospermine or castanospermine and the mannosidase inhibitor 1-deoxymannojirimycin prior to and during post-translational import of ppαf into the ER. These inhibitors rendered the migration of 3gpαf in sbh1Δ sbh2Δ and WT membranes identical, suggesting that sbh1Δ sbh2Δ cells have reduced N-glycan trimming by glucosidase and mannosidase (Fig. 2D). Reduced levels of mannosidase Mns1p in sbh1Δ sbh2Δ cells due to a constitutive translocation defect in these cells even at permissive temperature may explain this N-glycan trimming defect (supplementary Fig. S2).
Misfolded secretory protein export from the ER for ER-associated degradation is dependent on Sec61p for many substrates (10). We therefore asked if Sbh1p was required for ERAD. Incubation at 37 °C itself reduces ERAD in wild-type yeast cells and membranes in our hands, but conditional mutants in SEC61 display ERAD defects already at the permissive temperature for import into the ER (34). We therefore tested ERAD in WT and sbh1Δ sbh2Δ cells at 30 °C by first translocating a mutant form of α-factor precursor (pΔgpαf) that is subject to ERAD into wild type or sbh1Δ sbh2Δ microsomes. ERAD was initiated by adding yeast cytosol and ATP and an ATP-regenerating system. At the indicated time points, samples were precipitated with trichloroacetic acid and proteins analyzed by SDS-PAGE on 18% 2 m urea gels. As shown in Fig. 3 deletion of both SBH1 and SBH2 did not affect the kinetics of Δgpαf turnover. We also measured Δgpαf turnover in intact wild type or sbh1Δ sbh2Δ yeast in pulse-chase experiments and found no change in the mutant cells at 30 or 37 °C (data not shown). We conclude that Sbh1p and Sbh2p are dispensable for ERAD under these conditions.
FIGURE 3. Sbh1p and Sbh2p are dispensable for pΔgpαf ERAD.
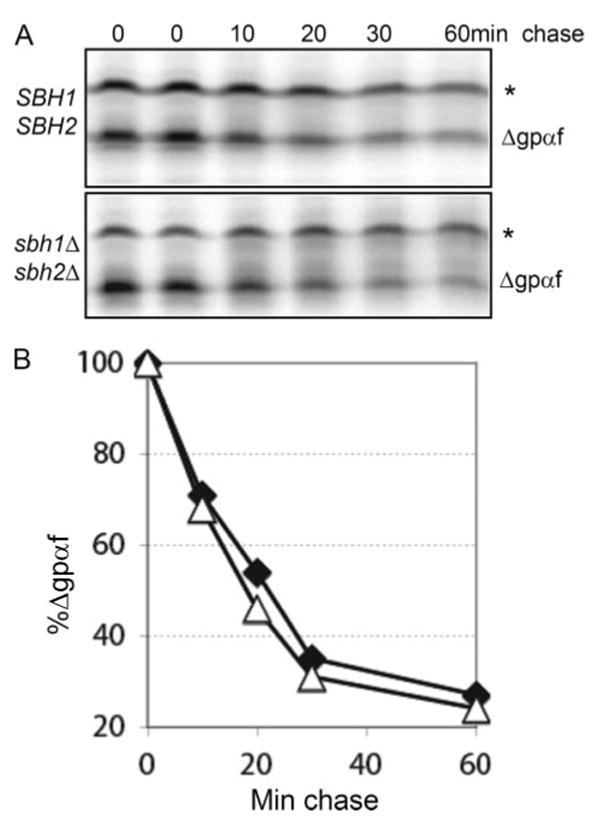
A, mutant α-factor precursor (pΔgpαf) was translocated into wild type or mutant microsomes, membranes were washed, and ERAD was initiated by adding yeast cytosol ATP and an ATP-regenerating system, and incubation at 30 °C. At the indicated time points, samples were precipitated with trichloroacetic acid and proteins analyzed by SDS-PAGE and phosphorimaging. The asterisk indicates untranslocated pΔgpαf associated with the cytosolic face of the microsomes. B, quantitation of Δgpαf on the gels shown in A. △, SBH1 SBH2; ♦, sbh1Δ sbh2Δ.
The Sbh1p Membrane Anchor Is Sufficient for Rescuing the Loss of Sbh1p and Sbh2p
To map functionally important domains in Sbh1p, we created a series of deletion mutants of SBH1 and assessed their ability to complement the temperature-sensitive growth defect of the sbh1Δ sbh2Δ cells. For this, sbh1Δ sbh2Δ cells were transformed with high copy plasmids expressing full-length SBH1 or its mutant versions or an empty vector (supplementary Table S2) and the growth of the trans-formants was monitored at 24 and 38 °C. Cells expressing the cytoplasmic and the TM domain, but lacking the luminal COOH-terminal part of Sbh1p rescued the temperature-sensitive growth (Fig. 4A). As shown before (17) the cytoplasmic part of Sbh1p was not able to rescue the growth (Fig. 4A, 1–54). Expression of the Sbh1p-TM domain alone rescued the growth phenotype as efficiently as the full-length Sbh1p (Fig. 4A, compare 1–82 with 50–75). Similar results were obtained when the different Sbh1p forms were expressed in the presence of methionine from the MET25 promoter in a low copy plasmid that results in close to endogenous level expression of Sbh1p (data not shown). Overexpression of different mutant forms of Sbh1p was not harmful to cell growth (Fig. 4A, 24 °C). To test the specificity of the growth rescue by the Sbh1p membrane anchor, we transformed sbh1Δ sbh2Δ cells with plasmids over-expressing the membrane anchor of Sbh2p or the γ subunit Sss1p. The Sss1p membrane anchor did not rescue this defect (Fig. 4B, compare Sss1-TM to Sbh1-TM), whereas the Sbh2p-TM domain was as efficient as Sbh1p in growth rescue of sbh1Δ sbh2Δ cells at the restrictive temperature.
FIGURE 4. The Sbh1p or Sbh2p-TM domains are sufficient to rescue loss of Sbh1p and Sbh2p.
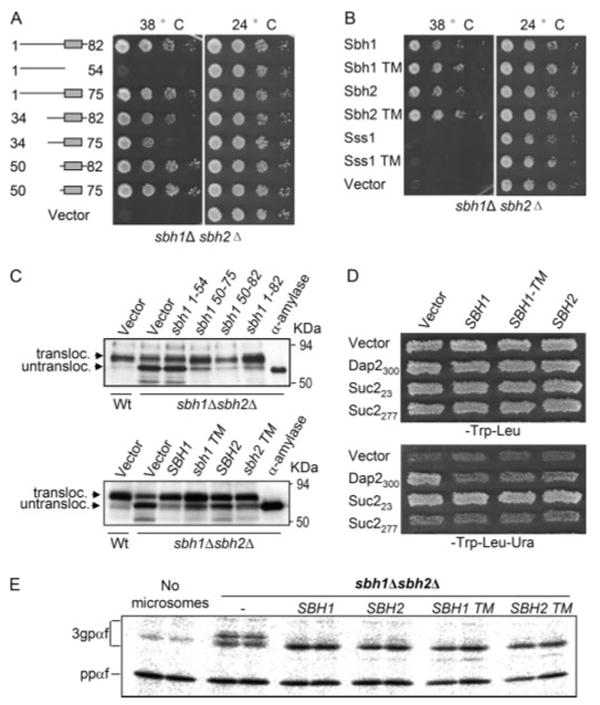
A, multicopy suppression of temperature sensitivity of sbh1Δ sbh2Δ cells by different mutant forms of SBH1. sbh1Δ sbh2Δ cells were transformed with SBH1 wild type or the indicated mutants expressed from the ADH1 promoter in the 2μ vector pVT102U and the growth of the cells was monitored at 38 and 24 °C. B, the Sbh1p-TM or Sbh2p-TM but not the Sss1p-TM domain rescues temperature sensitivity of sbh1Δ sbh2Δ cells. sbh1Δ sbh2Δ cells transformed with plasmids expressing either the full-length (FL) or only the TM domain (TM) of Sbh1p, Sbh2p, or Sss1p were replica plated to 38 and 24 °C and then rescued by the Sbh1p-or Sbh2p-TM domains. C, wild type cells (H3384) and sbh1Δ sbh2Δ (H3388) cells expressing bacterial α-amylase were transformed with plasmids YEpSBH1-(1–54), YEpSBH1-(50–75), YEpSBH1-(50–82), YEpSBH1-(1–82) or the empty vector (upper panel) or with plasmids YEpSBH1-(1–82), YEpSBH1-(50–75), YEpSBH2-(1–88), YEpSBH2-(57–82), or empty vector (lower panel), and the intracellular α-amylase was analyzed in cell lysates by Western blotting with antibodies to α-amylase. α-Amylase indicates the lane loaded with purified bacterial α-amylase. D, UTA translocation assay on sbh1Δ sbh2Δ cells. Strain H3543 was transformed with plasmids encoding reporter proteins Suc223, Sec2277, Dap2300, or the empty vector pRS314 and either the empty plasmid or plasmids encoding Sbh1p, Sbh1p-TM domain, or Sbh2p. The growth of patches of these transformants was tested on SCD-Trp-Leu or on SCD-Trp-Leu-Ura plates to score for translocation of the Ura3p containing reporters. E, the Sbh1p and Sbh2p-TM domains rescue the in vitro glycosylation defect in sbh1Δ sbh2Δ cells. Microsomes prepared from H3232 cells transformed with YEpSBH-(1–82), YEpSBH1 (50–75), YEpSBH2-(1– 88), YEpSBH2-(57– 82), or the empty vector pVT102U were assayed for in vitro translocation of ppαf and mobility of translocation products.
The pronounced translocation defect for α-amylase in sbh1Δ sbh2Δ cells provided us with a tool to study the functionality of the Sbh1p membrane anchor in protein translocation. We transformed α-amylase expressing sbh1Δ sbh2Δ cells (H3388) with plasmids YEpSBH1-(1–54), YEpSBH1-(50–75), YEpSBH1-(50–82), YEpSBH1-(1–82), or pVT102U. Liquid cultures of the cells were shifted to 37.5 °C for 1 h, lysed, and analyzed for different forms of α-amylase by Western blotting. When compared with Sbh1p-(1–82), the Sbh1p membrane anchor on its own was able to rescue the defect in α-amylase translocation nearly as efficiently (94 and 84%, respectively) as the wild type protein (Fig. 4C, upper panel). In sbh1Δ sbh2Δ cells containing only the empty vector or cells expressing the cytosolic fragment of Sbh1p, only 16% translocation of α-amylase was observed. The Sbh2p-TM domain also rescued the translocation defect in sbh1Δ sbh2Δ cells (Fig. 4C, lower panel).
In vitro translocation experiments showed that 3gpαf initially migrated as a group of bands at a higher molecular weight in sbh1Δ sbh2Δ membranes at all temperatures indicating a delay in glycan trimming (Fig. 2A). To assess whether also this phenotype could be rescued by Sbh1p or Sbh2p-TM domains, sbh1Δ sbh2Δ cells were transformed with a construct encoding full-length Sbh1p, Sbh2p, or their TM domains and membranes prepared from these cells were used for in vitro translocation assays. Expression of full-length Sbh1p or Sbh2p, or their TM domains rendered the glycosylation pattern of 3gpαf equal to that in WT cells (Fig. 4E).
To analyze in vivo the protein translocation defect of sbh1Δ sbh2Δ cells by more sensitive means we used the UTA assay (39, 40). The UTA assay measures coupling of translation to translocation by utilizing reporter proteins that contain a yeast signal sequence, a “spacer” peptide, ubiquitin, and finally Ura3p. When translation is stringently coupled to translocation, the reporter protein is translocated into the lumen of the ER before the ubiquitin moiety folds and is cleaved by cytosolic proteases. Such cells cannot survive without uracil in the growth medium as Ura3p needs to be in the cytosol to rescue lack of uracil in the growth medium. When there is a delay between translation and translocation, ubiquitin folds, is cleaved, and Ura3p is released into the cytosol. Strain H3543 (sbh1Δ sbh2Δ leu2–3,112 ura3–52 trp1Δ) was transformed with empty TRP1 plasmid without the Ura3 reporter or with reporter plasmids (TRP1 marker) encoding invertase or dipeptidyl aminopeptidase B (Dap2) signal sequence and 23 (Suc223), 277 (Suc2277), or 300 (Dap2300) amino acids of the proteins, respectively, before the ubiquitin moiety and Ura3p. The same cells were also transformed with plasmids encoding Sbh1p (YEpSBH1-(1–82)L), Sbh1p-TM domain (YEpSBH1-(50–75)L), Sbh2p (YEpSBH2-(1–88)L), or the empty vector. Suc2p uses the post-translational and Dap2p the co-translational translocation pathway (40). sbh1Δ sbh2Δ cells expressing Suc223 grew in all cases in the absence of uracil in the growth medium as the 23-amino acid linker enables rapid folding of the ubiqutin moiety, its cleavage, and release of the Ura3p in the cytosol (Fig. 4D, lower panel). sbh1Δ sbh2Δ cells expressing Suc2277 grew very weakly in the absence of uracil indicating efficient post-translational translocation of this reporter protein into the ER lumen in the absence of Sbh1 and Sbh2 proteins. However, the Dap2300 reporter expressing sbh1Δ sbh2Δ cells grew in the absence of uracil (Vector, Fig. 4D) indicating a defect in co-translational translocation. This defect was rescued by reintroduction of Sbh1p, Sbh1p-TM domain, or Sbh2p into these cells (Fig. 4D, lower panel). No growth was observed for cells containing the empty plasmid, lacking the Ura3 reporter (Fig. 4D, lower panel, top row). Collectively, these results show that the COOH-terminal membrane anchors of Sbh1p or Sbh2p are both necessary and sufficient for Sbh1p and Sbh2p function in protein translocation into the ER.
The Sbh1p-TM Domain Is Sufficient for Interaction with Sec61p and Sss1p
The observed ability of the Sbh1p-TM domain to rescue the loss of Sbh1p and Sbh2p prompted us to test whether the Sbh1p-TM domain can still interact with the Sec61 complex. As this TM domain is only 25 amino acids long and not recognized by the antibodies we have, for detection and pull-down experiments we made use of the BIO-tag derived from P. shermanii transcarboxylase that is biotinylated in vivo in yeast (29, 41). For this we generated gene fusions of full-length and the TM domain (amino acids 50–75) of Sbh1p placing the BIO-tag at the amino terminus under the control of ADH1 promoter in YEpBIO-SBH1-(1–82) or YEpBIO-sbh1-TM. The functionality of these fusion proteins was confirmed by transforming sbh1Δ sbh2Δ cells and scoring their ability to rescue the temperature sensitivity of this strain (data not shown). We then analyzed the interactions of the tagged Sbh1p and Sbh1p-TM domain (amino acids 50–75) with the Sec61 complex in sbh1Δ cells (H3227) transformed with YEpBIO-SBH1-(1–82), YEpBIO-sbh1-TM or just the empty vector pVT102U. Cells were lysed and a membrane-enriched fraction was prepared as previously described (4). BIO-tagged Sbh1p, Sbh1p-TM, and associated proteins were isolated with streptavidin-coated magnetic beads followed by washing and elution of bound proteins in SDS sample buffer, and analysis by SDS-PAGE and Western blotting with antibodies against Sec61p and Sss1p or with HRP-conjugated streptavidin to detect the BIO-tagged Sbh1p derivatives. Sec61p co-precipitated with both full-length BIO-Sbh1p and BIO-Sbh1-(50–75)p (7 and 2% of Sec61p in the input, respectively) (Fig. 5A). For Sss1p the corresponding co-precipitation efficiencies were 16 and 5%, whereas the efficiency of the streptavidin pulldown were 16% for BIO-Sbh1p and 24% for BIO-Sbh1p-(50–75) of these proteins present in the input. To exclude the possibility that BIO-Sbh1p co-precipitates proteins nonspecifically, the samples were analyzed for the presence of functionally non-related TM proteins Sso1p and Sso2p. No co-precipitation of Sso1 or Sso2 protein was observed (Fig. 5A, bottom panel). We conclude that the Sbh1p-TM domain can exist in a complex at least with Sec61p and Sss1p in the ER membrane.
FIGURE 5. The Sbh1p-TM domain is sufficient for co-precipitation with Sec61 translocation complex but not for functional interaction the exocyst.
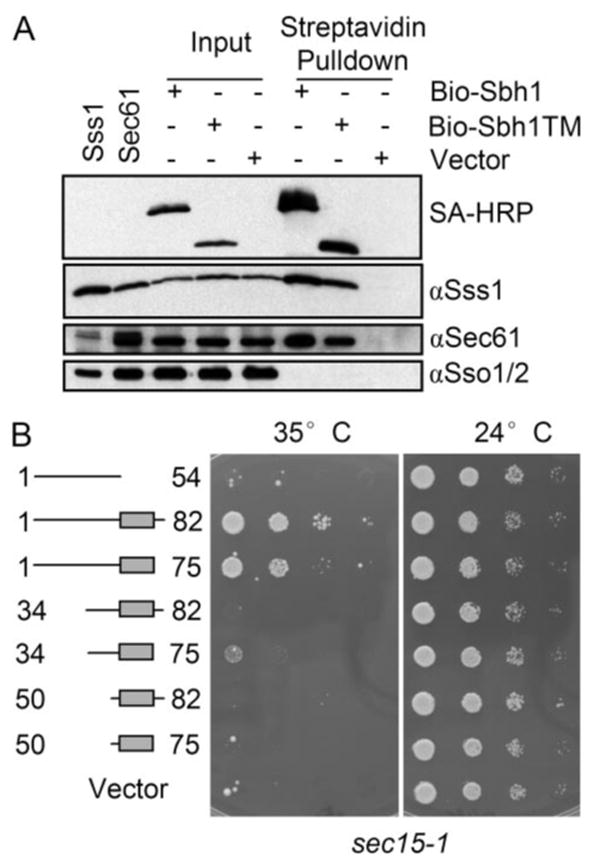
A, digitonin-solubilized membranes of SBH1 deleted cells (H3227) expressing N-terminal BIO-tagged full-length Sbh1p (amino acids 1–82), TM domain (amino acids 50–75), or the empty vector were subjected to pull-down with streptavidin-coated magnetic beads. Beads and input samples were resolved by SDS-PAGE, transferred to nitrocellulose, and analyzed by Western blotting with antibodies to Sec61p, Sss1p, or Sso1/2p, or with HRP-conjugated streptavidin to detect BIO-Sbh1p. The first two lanes are whole cell lysates overexpressing either Sss1p or Sec61p. B, the sec15–1 cells carrying YEpSBH1-(1–54), YEpSBH1-(34–75), YEpSBH1-(34–82), YEpSBH1-(50–75), YEpSBH1-(50–82), YEpSBH1-(1–82) or the empty vector pVT102U were grown on SCD-Ura plates at different temperatures. Sbh1p-TM or the cytosolic domain alone cannot suppress the temperature-sensitive growth phenotype of sec15-1 cells in contrast to full-length Sbh1p and the sbh1 mutant lacking the luminal part.
We have previously shown that overexpression of full-length Sbh1p, but not the cytosolic fragment alone, is able to rescue the temperature-sensitive growth phenotype of exocyst mutant sec15-1 (17). To address the role of the Sbh1p-TM domain is this process we transformed sec15-1 cells with plasmids YEpSBH1-(1–54), YEpSBH1-(34–75), YEpSBH1-(34–82), YEpSBH1-(50–75), YEpSBH1-(50–82), YEpSBH1-(1–82), or pVT102U. The growth of the transformants was then followed at the restrictive and permissive temperatures. Whereas full-length Sbh1p and a mutant lacking the luminal amino acids efficiently suppressed temperature sensitivity of sec15-1 cells, plasmids encoding the transmembrane domain, cytosolic domain, or the amino-terminal deleted Sbh1p-(34–75) were not able to do so (Fig. 5B). We observed the same when using another exocyst mutant sec8-9 (data not shown). This indicates that association of the Sbh1p cytosolic domain with the ER membrane is necessary and that the TM domain alone is not sufficient for the functional Sbh1-Exocyst interaction.
Rtn1p Is a Novel Sbh1p-interacting Protein
Recently, the ER-resident TM protein Rtn1p was shown to interact with the exocyst subunit Sec6p (42). We have previously demonstrated coimmunoprecipitation of Sbh1p with exocyst subunits Sec8p and Sec15p (17). This prompted us to test whether Rtn1p could be found in a protein complex with the Sec61 complex or with Sbh1p. For this we used strain H3431 in which Rtn1p is HA-tagged at its COOH terminus and is expressed from its own promoter. Cells were grown to A600 = 1, harvested, and microsomal membranes were prepared as described previously (4). Immunoprecipitations or pulldowns were carried out with anti-HA antibody coupled to magnetic beads. When Rtn1p-HA was immunoprecipitated from lysates prepared from cells containing endogenous Sbh1p, we observed reproducible coimmunoprecipitation of Sbh1p and Rtn1p-3HA (Fig. 6A). When the presence of other subunits of the Sec61 translocon was analyzed in these immunoprecipitates, no Sec61p and Sss1p were detected. This suggests that Rtn1p co-precipitates with Sbh1p that is not part of the Sec61 complex.
FIGURE 6. Sbh1p co-precipitation with Rtn1p.
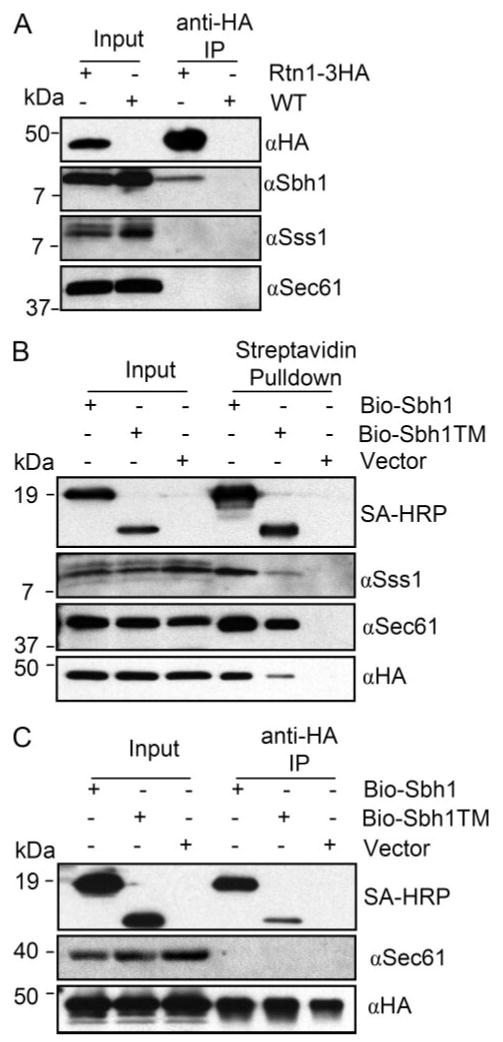
A, digitonin-solubilized membranes of cells expressing RTN1–3XHA (H3431) from its own promoter were subjected to immunoprecipitation with anti-HA antibody-coated magnetic beads. Input and immunoprecipitation samples were subjected to SDS-PAGE and Western blotting with anti-HA, Sbh1p, Sec61p, or Sss1p antibodies. Molecular weights are indicated on the left side of the blots. B, Rtn1p-HA copurifies both with BIO-Sbh1p and BIO-Sbh1p-TM. Cells (H3429) deleted for sbh1 and expressing RTN1–3XHA from its own promoter were transformed with YEpBIO-SBH1, YEpBIO-SBH1-(50–75), or an empty plasmid p426ADH. Lysates were prepared and subjected to pull-down with streptavidin-conjugated magnetic beads followed by SDS-PAGE and Western blotting. Proteins were detected with HRP-conjugated streptavidin and antibodies to Sec61p, Sss1p, and HA. Molecular weights are indicated on the left side of the blots. C, BIO-Sbh1p but not Sec61 copurifies with Rtn1p-HA. Lysates prepared from the same cells used in B were subjected to immunoprecipitations with anti-HA-coated magnetic beads and processed for SDS-PAGE and Western blotting with antibodies to Sec61p, HA, or with HRP-conjugated streptavidin to detect BIO-Sbh1p and BIO-Sbh1-TM.
To verify the Rtn1p-Sbh1p interaction, we constructed strain H3429 in which Rtn1p is HA-tagged and the SBH1 gene is deleted. This strain was transformed with YEpBIO-SBH1-(1–82), YEpBIO-sbh1-TM, or the empty vector. Lysates prepared of these cells were subjected to pulldowns with streptavidin-coated beads and the pulldowns were analyzed by SDS-PAGE and Western blotting for the presence of Sec61 translocon sub-units and Rtn1p. BIO-Sbh1p co-precipitated both Sec61p and Sss1p (Fig. 6B). In addition, Rtn1p-HA was detected in the precipitates (Fig. 6B, bottom panel). Similar albeit weaker co-precipitations were observed in lysates expressing only the BIO-tagged TM domain of Sbh1p. To further evaluate the possibility that Rtn1p interacts preferentially with Sbh1p that is not associated with the Sec61 complex, we performed anti-HA immunoprecipitations from cell lysates used in Fig. 6B. Immunoprecipitations were subjected to Western blotting and detection with antibodies to Sec61p and HA and with HRP-conjugated straptavidin. As shown for endogenous Sbh1p (Fig. 6A), BIO-Sbh1p also immunoprecipitated with Rtn1p-HA (Fig. 6C). In these immunoprecipitations no Sec61p was detected. Collectively, these results suggest that Sbh1p can exist complexed with the Sec61 translocon or with Rtn1p-HA. These complexes may define two different functional states of Sbh1p in the ER membrane.
DISCUSSION
The heterotrimeric Sec61/SecY complex performs an essential function in protein translocation conserved from bacteria to mammalian cells. Sec61p, the α subunit of this complex spans the membrane 10 times and forms the protein-conducting channel in the ER membrane (43). The functions of the smaller subunits β and γ are not well understood. In S. cerevisiae lack of the two homologous β subunits, Sbh1p and Sbh2p, results in temperature sensitivity of the cells (4, 7). Based on the recently revealed structure of the archaebacterial SecY complex, the γ subunit associates with a “hinge” region of Sec61p and may participate in the regulation of the translocon opening and closing dynamics (3). The α and γ subunits show significant sequence homology across the species (2), whereas the β subunits are conserved in eukaryotes and Archaea, but have minimal homology with the eubacterial SecG (3). Both α and γ are essential for cell viability, whereas β is non-essential at least in yeast and in eubacteria (2). This suggests that the β subunit may perform functions not tightly associated with protein translocation and this might have led to its diversification during evolution.
We set out to characterize the yeast Sec61 β subunits Sbh1p and Sbh2p as the data on their contribution to protein translocation is partially conflicting, no data existed on the contribution of these subunits on ERAD, and functionally important domains of these subunits had not been identified. We have previously shown that overexpression of Sbh1p results in enhanced secretion of the bacterial α-amylase (17). To further study the role of Sec61 β in this process, we analyzed how lack of Sbh1p and its homologue Sbh2p affects α-amylase secretion. Accumulation of an intracellular pool of α-amylase was observed in sbh1Δ sbh2Δ cells (Fig. 1A). The intracellular form of amylase retained in sbh1Δ sbh2Δ cells displayed faster mobility compared with WT or single deletant cells and therefore likely represented unglycosylated, cytosolic protein. This was confirmed by comparing the mobility of α-amylase in sbh1Δ sbh2Δ cells with its mobility in the known translocation mutants sec61-2, sec62-1, and sec63-1 under restrictive conditions (Fig. 1B). Because Sec62p has been implicated to be mainly active in post-translational translocation (6) the fact that α-amylase translocation was not severely compromised in sec62 mutant cells suggests that this protein makes use of the co-translational translocation pathway in yeast. That the fast migrating form of α-amylase represents a cytosolic, untranslocated form of the protein was supported by cell fractionation data: we found that in contrast to the slow migrating form in wild type cells, the fast migrating form was not associated with the microsomal pellet in sbh1Δ sbh2Δ cells but remained soluble (supplementary Fig. S1).
Our results on α-amylase translocation showed a more pronounced in vivo translocation defect than previous work where even at 38 °C sbh1Δ sbh2Δ cells displayed only a mild translocation defect for Kar2p and yeast α-factor precursor. However, in a heterologous translation-translocation system based on reticulocyte lysate and yeast microsomes, a strong defect was observed for ppαf and bacterial OmpA protein translocation at 37 °C (4). Using a homologous yeast translation-translocation assay, we observed relatively modest defects both for post- and co-translational translocation in vitro at the restrictive temperature. Differences in in vitro translocation could in part be due to the fact that in the earlier study combination of reticulocyte lysate and yeast membranes may have led to an exacerbation of the translocation defect (4). The apparently more severe translocation defect of the α-amylase may be due to intrinsically less efficient translocation of the bacterial protein that still has its original bacterial signal peptide (30). To obtain further insight into the role of Sbh1 and Sbh2 proteins in protein translocation, we used two S. cerevisiae reporter proteins with endogenous signal sequences in the UTA assay. No defect was observed for Suc2277-ubi-Ura3 reporter translocation into the ER in cells lacking both Sbh1p and Sbh2p. Suc2277 uses preferentially the post-translational translocation pathway (40). Analysis of the second reporter, Dap2300, showed a clear defect for signal recognition particle-dependent co-translational protein translocation in sbh1Δ sbh2Δ cells (Fig. 4D). The above and previous results suggest that Sbh1p and Sbh2p have an auxiliary function that is more important in co-translational than in post-translational protein translocation into the ER.
A glycan trimming defect was found in sbh1Δ sbh2Δ microsomes (Fig. 2). Activity of the trimming enzymes in sbh1Δ sbh2Δ ER could be disturbed indirectly or could be due to a reduced amount of the enzymes present in the ER due a translocation defect in these cells even at the permissive temperature. Compared with wild type cells, we noticed a 50% reduction in the amount of mannosidase 1 (Mns1p) present in sbh1Δ sbh2Δ membranes by quantitative immunoblotting (supplementary Fig. S2). This suggests that translocation of Mns1p into the ER is compromised in sbh1Δ sbh2Δ cells even under conditions permissive for growth and that this affects glycan trimming in the ER. In addition to its role in nascent polypeptide chain translocation into the ER, the Sec61 complex has been implicated in retrotranslocation in ERAD (10). However, we observed no defect for ERAD of a mutant non-glycosylated Δgpαf in sbh1Δ sbh2Δ cells or microsomes (not shown, and Fig. 2).
Sbh1p is a small 82-amino acid protein that contains a cytosolic amino terminus, a single TM domain at its carboxyl terminus, and seven amino acids that presumably face the lumen of the ER (7). To better understand the functional roles of Sbh1p we generated a set of mutants that mapped the functionality of these three domains of Sbh1p. Previously, the luminal part of the Sbh1p homologue in yeast Yarrowia lipolytica was shown to interact with calnexin (44). In S. cerevisiae the luminal domain is not essential for Sbh1p function as deletion of it did not affect the capability to rescue the sbh1Δ sbh2Δ temperature-sensitive phenotype or α-amylase translocation defect (Fig. 4A). Although the precise reason for the temperature sensitivity of sbh1Δ sbh2Δ cells is not known, one plausible explanation is that it is caused by compromised protein translocation into the ER. Interestingly, the TM domains (amino acids 50–75 of Sbh1p, amino acids 57–82 of Sbh2p) were as efficient as full-length Sbh1p in rescuing this phenotype (Fig. 4C). This effect was specific for Sbh1/2p as expression of the TM domain of the Sec61 γ subunit Sss1p could not rescue loss of Sbh1p and Sbh2p (Fig. 4B). The Sbh1p and Sbh2p transmembrane domain containing 25 amino acid peptides used here are 76% identical. This degree of identity allows the Sbh2p-TM domain to functionally replace the Sbh1p-TM domain in the Sec61 complex. It is possible that the more stringent binding determinant for formation of Sbh1p-Sec61p and Sbh2p-Ssh1p complexes is provided by the less homologous cytosolic domains of these proteins. The in vivo functionality of the Sbh1p-TM domain is further supported by the finding that reintroduction of Sbh1-(50–75) rescued the translocation defect of α-amylase and Dap2300 and also the glycan trimming defect in sbh1Δ sbh2Δ membranes (Fig. 4). In addition, the Sbh1p-TM domain was able to associate with Sec61p and Sss1p (Figs. 5 and 6). Previously a segment in the Sbh1p cytosolic domain was shown to display homology with the Sec7 domain responsible for guanine nucleotide exchange activity for ADP-ribosylation factor family GTPases and it was shown that Sbh1p can act as a guanine nucleotide exchange factor for signal recognition particle receptor β (14). The Sec61 β subunit was also shown to interact with the large ribosomal subunit and was proposed to mediate ribosome docking with the Sec61 complex (13). Our results suggest that the Sbh1p cytosolic domain is not essential for Sbh1p function in protein translocation and association with the Sec61 complex.
The non-essential nature, the putative peripheral association of Sec61 β with the Sec61 complex, and its low conservation in evolution compared with the highly conserved Sec61p raise the possibility that Sbh1p could have acquired functions independent of protein translocation into the ER or regulatory functions that coordinate translocation with other events. This notion is supported by the findings that Sbh1p in yeast was identified in a screen for suppressors of temperature-sensitive growth of the exocyst mutant sec15-1 (7) and that both in yeast and mammalian cells the β subunit can co-immunoprecipitate (16, 17) with subunits of the exocyst complex that is implicated in regulation of polarized secretion at the plasma membrane (18). The exocyst complex subunit Sec3 is required for efficient ER inheritance during cytokinesis, and overexpression of SEC3 results in enhanced localization of Sec61p in the emerging bud tip (45). More recently, Novick and colleagues (42) have shown that a resident ER TM protein Rtn1p localizes preferentially at the peripheral ER membranes and copurifies with the exocyst complex. As Sbh1p also copurified with the exocyst complex we tested whether Sbh1p can exist in a complex with Rtn1p. Interaction was observed both by immunoprecipitating Rtn1p-HA or by pulling down BIO-Sbh1p. Interestingly, Rtn1p-HA co-precipitated only Sbh1p, but not Sss1p or Sec61p (Fig. 6, A and C). This suggests that a fraction of Sbh1p can exist outside the Sec61 complex and interact directly or indirectly with Rtn1p. At the same time the TM or the cytosolic domains of Sbh1p were not able alone to suppress the temperature-sensitive growth phenotype of two exocyst mutants sec15-1 or sec8-9. This suggests that the cytosolic domain plays a role in the functional interaction between Sbh1p and the exocyst, and that association of the cytosolic domain with the ER membrane is needed to retain this link. To elucidate the contribution of the Sbh1p-Rtn1p interaction for ER inheritance and exocyst interactions requires a detailed biochemical and morphological analysis in the future.
Supplementary Material
Acknowledgments
We thank Jeremy Brown, Annette Herscovics, Nils Johnsson, Peter Kaiser, Michael Knop, Peter Novick, Randy Schekman, and Dieter Wolf, for strains, antibodies, and plasmids. Outi Könönen is acknowledged for skillful technical assistance, and Heike Kröger for help with preparing yeast microsomes.
Footnotes
The abbreviations used are: ER, endoplasmic reticulum; ERAD, endoplasmic reticulum-associated degradation; TM, transmembrane; HA, hemagglutinin; HRP, horseradish peroxidase; WT, wild type; UTA, ubiquitin-assisted translocation assay.
This work was supported in part by Academy of Finland Grants 52096 and 204373 (to S. K.), Academy Research Fellow Grant 211171 (to J. J.), and the Magnus Ehrnrooth Foundation (to J. J.), and was part of the research program “VTT Industrial Biotechnology” supported by the Academy of Finland, Finnish Center of Excellence Program, 2000–2005, Project 64330.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1 and S2 and Figs. S1 and S2.
References
- 1.Johnson AE, van Waes MA. Annu Rev Cell Dev Biol. 1999;15:799–842. doi: 10.1146/annurev.cellbio.15.1.799. [DOI] [PubMed] [Google Scholar]
- 2.Osborne AR, Rapoport TA, van den Berg B. Annu Rev Cell Dev Biol. 2005;21:529–550. doi: 10.1146/annurev.cellbio.21.012704.133214. [DOI] [PubMed] [Google Scholar]
- 3.van den Berg B, Clemons WM, Jr, Collinson I, Modis Y, Hartmann E, Harrison SC, Rapoport TA. Nature. 2004;427:36–44. doi: 10.1038/nature02218. [DOI] [PubMed] [Google Scholar]
- 4.Finke K, Plath K, Panzner S, Prehn S, Rapoport TA, Hartmann E, Sommer T. EMBO J. 1996;15:1482–1494. [PMC free article] [PubMed] [Google Scholar]
- 5.Hartmann E, Sommer T, Prehn S, Gorlich D, Jentsch S, Rapoport TA. Nature. 1994;367:654–657. doi: 10.1038/367654a0. [DOI] [PubMed] [Google Scholar]
- 6.Panzner S, Dreier L, Hartmann E, Kostka S, Rapoport TA. Cell. 1995;81:561–570. doi: 10.1016/0092-8674(95)90077-2. [DOI] [PubMed] [Google Scholar]
- 7.Toikkanen J, Gatti E, Takei K, Saloheimo M, Olkkonen VM, Soderlund H, De Camilli P, Keranen S. Yeast. 1996;12:425–438. doi: 10.1002/(SICI)1097-0061(199604)12:5%3C425::AID-YEA924%3E3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 8.Brodsky JL, Goeckeler J, Schekman R. Proc Natl Acad Sci U S A. 1995;92:9643–9646. doi: 10.1073/pnas.92.21.9643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Young BP, Craven RA, Reid PJ, Willer M, Stirling CJ. EMBO J. 2001;20:262–271. doi: 10.1093/emboj/20.1.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romisch K. Annu Rev Cell Dev Biol. 2005;21:435–456. doi: 10.1146/annurev.cellbio.21.012704.133250. [DOI] [PubMed] [Google Scholar]
- 11.Wilkinson BM, Tyson JR, Stirling CJ. Dev Cell. 2001;1:401–409. doi: 10.1016/s1534-5807(01)00043-0. [DOI] [PubMed] [Google Scholar]
- 12.Kalies KU, Rapoport TA, Hartmann E. J Cell Biol. 1998;141:887–894. doi: 10.1083/jcb.141.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levy R, Wiedmann M, Kreibich G. J Biol Chem. 2001;276:2340–2346. doi: 10.1074/jbc.M004867200. [DOI] [PubMed] [Google Scholar]
- 14.Helmers J, Schmidt D, Glavy JS, Blobel G, Schwartz T. J Biol Chem. 2003;278:23686–23690. doi: 10.1074/jbc.C300180200. [DOI] [PubMed] [Google Scholar]
- 15.Esnault Y, Feldheim D, Blondel MO, Schekman R, Kepes F. J Biol Chem. 1994;269:27478–27485. [PubMed] [Google Scholar]
- 16.Lipschutz JH, Lingappa VR, Mostov KE. J Biol Chem. 2003;278:20954–20960. doi: 10.1074/jbc.M213210200. [DOI] [PubMed] [Google Scholar]
- 17.Toikkanen JH, Miller KJ, Soderlund H, Jantti J, Keranen S. J Biol Chem. 2003;278:20946–20953. doi: 10.1074/jbc.M213111200. [DOI] [PubMed] [Google Scholar]
- 18.Guo W, Novick P. Trends Cell Biol. 2004;14:61–63. doi: 10.1016/j.tcb.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 19.Wiederkehr A, De Craene JO, Ferro-Novick S, Novick P. J Cell Biol. 2004;167:875–887. doi: 10.1083/jcb.200408001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Du Y, Ferro-Novick S, Novick P. J Cell Sci. 2004;117:2871–2878. doi: 10.1242/jcs.01286. [DOI] [PubMed] [Google Scholar]
- 21.Wach A, Brachat A, Pöhlmann R, Philippsen P. Yeast. 1994;10:1793–1808. doi: 10.1002/yea.320101310. [DOI] [PubMed] [Google Scholar]
- 22.Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, Maekawa H, Moreno-Borchart A, Doenges G, Schwob E, Schiebel E, Knop M. Yeast. 2004;21:947–962. doi: 10.1002/yea.1142. [DOI] [PubMed] [Google Scholar]
- 23.Sherman F. In: Guide to Yeast Genetics and Molecular Biology. Guthrie C, Fink GR, editors. Academic Press; New York: 1991. pp. 3–21. [Google Scholar]
- 24.Davydenko SG, Feng D, Jantti J, Keranen S. Yeast. 2005;22:993–1009. doi: 10.1002/yea.1286. [DOI] [PubMed] [Google Scholar]
- 25.Vernet T, Dignard D, Thomas DY. Gene (Amst) 1987;52:225–233. doi: 10.1016/0378-1119(87)90049-7. [DOI] [PubMed] [Google Scholar]
- 26.Mumberg D, Muller R, Funk M. Gene (Amst) 1995;156:119–122. doi: 10.1016/0378-1119(95)00037-7. [DOI] [PubMed] [Google Scholar]
- 27.Ruohonen L, Aalto MK, Keranen S. J Biotechnol. 1995;39:193–203. doi: 10.1016/0168-1656(95)00024-k. [DOI] [PubMed] [Google Scholar]
- 28.Sikorski RS, Hieter P. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tagwerker C, Zhang H, Wang X, Larsen LS, Lathrop RH, Hatfield GW, Auer B, Huang L, Kaiser P. Yeast. 2006;23:623–632. doi: 10.1002/yea.1380. [DOI] [PubMed] [Google Scholar]
- 30.Ruohonen L, Toikkanen J, Tieaho V, Outola M, Soderlund H, Keranen S. Yeast. 1997;13:337–351. doi: 10.1002/(SICI)1097-0061(19970330)13:4<337::AID-YEA98>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 31.Pilon M, Romisch K, Quach D, Schekman R. Mol Biol Cell. 1998;9:3455–3473. doi: 10.1091/mbc.9.12.3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jantti J, Keranen S, Toikkanen J, Kuismanen E, Ehnholm C, Soderlund H, Olkkonen VM. J Cell Sci. 1994;107:3623–3633. doi: 10.1242/jcs.107.12.3623. [DOI] [PubMed] [Google Scholar]
- 33.Kalies KU, Allan S, Sergeyenko T, Kroger H, Romisch K. EMBO J. 2005;24:2284–2293. doi: 10.1038/sj.emboj.7600731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pilon M, Schekman R, Romisch K. EMBO J. 1997;16:4540–4548. doi: 10.1093/emboj/16.15.4540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gillece P, Luz JM, Lennarz WJ, de La Cruz FJ, Romisch K. J Cell Biol. 1999;147:1443–1456. doi: 10.1083/jcb.147.7.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lyman SK, Schekman R. J Cell Biol. 1995;131:1163–1171. doi: 10.1083/jcb.131.5.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rexach MF, Schekman RW. J Cell Biol. 1991;114:219–229. doi: 10.1083/jcb.114.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hansen W, Garcia PD, Walter P. Cell. 1986;45:397–406. doi: 10.1016/0092-8674(86)90325-9. [DOI] [PubMed] [Google Scholar]
- 39.Johnsson N, Varshavsky A. EMBO J. 1994;13:2686–2698. doi: 10.1002/j.1460-2075.1994.tb06559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mason N, Ciufo LF, Brown JD. EMBO J. 2000;19:4164–4174. doi: 10.1093/emboj/19.15.4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cronan JE., Jr J Biol Chem. 1990;265:10327–10333. [PubMed] [Google Scholar]
- 42.De Craene JO, Coleman J, Estrada DM, Pypaert M, Anderson S, Yates JR, III, Ferro-Novick S, Novick P. Mol Biol Cell. 2006;17:3009–3020. doi: 10.1091/mbc.E06-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rapoport TA, Goder V, Heinrich SU, Matlack KE. Trends Cell Biol. 2004;14:568–575. doi: 10.1016/j.tcb.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 44.Boisrame A, Chasles M, Babour A, Beckerich JM, Gaillardin C. J Cell Sci. 2002;115:4947–4956. doi: 10.1242/jcs.00187. [DOI] [PubMed] [Google Scholar]
- 45.Wiederkehr A, Du Y, Pypaert M, Ferro-Novick S, Novick P. Mol Biol Cell. 2003;14:4770–4782. doi: 10.1091/mbc.E03-04-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hiller MM, Finger A, Schweiger M, Wolf DH. Science. 1996;273:1725–1728. doi: 10.1126/science.273.5282.1725. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.