

Abstract
Background
Short QT syndrome (SQT) is a primary electrical disease associated with a high risk of sudden cardiac death. A gain-of-function in IKr, due to a mutation in KCNH2, underlies SQT1.
Objective
To examine the cellular basis for arrhythmogenesis in an experimental model of SQT1 created using PD-118057, a novel IKr agonist.
Methods
Transmembrane action potentials were simultaneously recorded from epicardial, M and endocardial regions of arterially-perfused canine left ventricular (LV) wedge preparations, together with a pseudo-ECG.
Results
PD-118057 (10 µmol/L) abbreviated QT interval from 267±4 to 232±4 ms and increased transmural dispersion of repolarization (TDR) from 33.7±2.0 to 49.1±3.1 ms (p<0.001). T wave amplitude increased from 18.0±1.4 to 23.1±1.7% of R wave amplitude (p=0.027). Reversing the direction of activation of the LV wall (epicardial pacing) resulted in an increase in QT interval from 269±5 to 282±5 ms and increase in TDR from 34.1±2.0 to 57.6±3.3 ms (p<0.001) under baseline conditions. PD-118057 abbreviated the QT interval from 282±5 to 258±5 ms and produced a proportional decrease in effective refractory period (ERP). TDR increased from 57.6±3.3 to 77.6±4.3 ms (p<0.001). Polymorphic ventricular tachycardia (pVT) was induced in 10 out of 20 preparations with a single S2 applied to epicardium. Quinidine (10 µmol/L) increased the ERP and QT interval, did not significantly alter TDR, and prevented induction of pVT in 5/5 preparations.
Conclusion
Our results suggest that a combination of ERP abbreviation and TDR amplification underlie the development of pVT in SQT1 and that quinidine prevents pVT principally by prolonging ERP.
Keywords: arrhythmias, electrophysiology, antiarrhythmia drugs, inherited syndromes, sudden cardiac death, pharmacology
INTRODUCTION
Short QT syndrome (SQTS) is a recently recognized, inherited, primary electrical cardiac syndrome characterized by abnormally short QTc interval (<360 ms) and an increased propensity for development of atrial and ventricular tachyarrhythmias, including sudden cardiac arrest.1–5 It is a genetically heterogeneous disease and thus far mutations of 5 different genes encoding cardiac ion channels have been identified in familial or sporadic cases. Gain-of-function mutations in KCNH26, KCNQ17 and KCNJ28 genes encoding for the rapidly and slowly activating potassium channel currents (IKr and IKs) and the inward rectifier potassium channel current (IK1) give rise to the SQT1, SQT2 and SQT3 forms of SQTS. Our group recently described loss-of-function mutations in CACNA1c and CACNB2b genes encoding the α1 and β subunits of cardiac L-type calcium channels. This clinical phenotype, characterized by a new clinical entity consisting of a combined Brugada-like ST segment elevation and short QT intervals, has been designated SQT4 and SQT5.9
The ECG in SQTS is characterized by shorter than normal QT intervals (QTc<360 ms), short or absent ST segment, and in some cases, tall peaked T waves and augmented Tpeak-Tend intervals. The rate dependence of QT interval or QT/RR interval relationship has been reported to be less steep than normal.4, 5, 10 Electrophysiological testing in patients with SQTS shows marked abbreviation of atrial and ventricular refractory period. Polymorphic ventricular tachycardia (pVT) is readily inducible in a large fraction of affected individuals.3, 10
Due to the unavailability of IKr, IKs or IK1 agonists, the only previously developed models of short QT syndrome were ones in which the activator of the ATP-sensitive potassium current (IK-ATP), pinacidil, was used to augment outward current in canine left ventricular wedge preparations11 or Langendorff-perfused rabbit hearts.12 The present study involves the development and characterization of a specific model of SQT1 made possible by the recent availability of a selective IKr agonist, PD-118057.13 Available data indicate that PD-118057 binds to the IKr channel directly and increases its open channel probability without affecting the voltage-dependence and kinetics of gating parameters.
The SQT1 variant of SQTS is due to an N588K mutation in KCNH2, the gene that encodes the α subunit of the IKr channel. The mutation abolishes rectification of IKr current at positive voltages within the voltage range of the action potential, leading to a marked gain of function. We used the novel IKr agonist PD-118057 to mimic this gain-of-function and thus to examine the cellular basis for the electrocardiographic and arrhythmogenic manifestations of this model of SQT1. Our study also probes the mechanism underlying the antiarrhythmic effects of quinidine under SQT1 conditions.
METHODS
All experiments were performed in conformance with the guidelines of the Institutional Animal Care Committee. The left ventricular wedge was prepared as previously described.11, 14 Transmembrane action potentials were recorded from epicardial (Epi), subendocardial (M) and endocardial (Endo) regions of the wedge with the use of floating microelectrodes. A transmural pseudo-ECG was recorded using 2 AgCl half cells placed approximately 1 cm from the epicardial (+) and endocardial (−) surfaces of the preparation and along the same axis as the transmembrane recordings. QT interval was defined as the time interval between QRS onset and the point at which the line of maximal downslope of T wave crossed the isoelectric line. Tpeak-Tend interval was defined as the time interval between peak of the T wave and the point at which the line of maximal downslope of T wave crossed the isoelectric line. TDR was defined as the difference between the longest and the shortest repolarization times (activation time plus action potential duration (APD) measured at 90% repolarization (APD90)) of transmembrane action potentials recorded across the wall (typically M cell minus epicardial cell repolarization time).
The basic stimulus (S1) was delivered to endocardium at a BCL of 2000 ms. A premature stimulus (S2) was delivered to the epicardium at progressively briefer S1–S2 intervals until the refractory period was reached in an attempt to induce pVT. A similar protocol was repeated after switching the basic stimulus (S1) to epicardium as reversal of direction of activation of the left ventricle has been shown to amplify TDR and facilitate induction of pVT.15 The effective refractory period (ERP) was defined as the longest S1–S2 interval at which S2 failed to capture. Ventricular tachycardia (VT) inducibility was examined under each condition (control, PD-118057, + Quinidine).
Drugs
PD-118057, a kind gift of Pfizer Pharmaceuticals, was prepared as a 10mmol/L stock in 100% Dimethyl Sulfoxide (DMSO) and added to Tyrode’s solution to final concentration of 10 µmol/L. Quinidine was prepared as a 20mmol/L stock in Millipore water and added to Tyrode’s solution to achieve a final concentration of 10 µmol/L.
Statistical analysis
Statistical analysis was performed using a paired t-test or repeated measures analysis of variance (ANOVA) followed by Tukey’s test, as appropriate. All data are reported as mean±SEM.
RESULTS
Effect of PD-118057 on QT interval, TDR, Tpeak-Tend and APD90 (Endo Stimulation)
Figure 1 illustrates the effect of the IKr agonist PD-118057 to abbreviate the QT interval and amplify transmural dispersion of repolarization (TDR). Shown are action potentials simultaneously recorded from epicardial and M cells of a canine left ventricular wedge preparation, together with a pseudo-ECG. Figure 2 summarizes the effects of PD-118057 on APD90 as recorded in 24 wedge preparations. PD-118057 abbreviated the APD90 of epicardial and M cells leading to abbreviation of QT interval. QT interval abbreviated from 267.2±4.0 to 232.3±4.1 ms (n=20, p<0.001)(Fig. 3A). Preferential abbreviation of the epicardial action potential led to an increase of TDR from 33.7±2 to 49.1±3.1 and Tpeak-Tend from 37.7±1.2 to 52.6±2.7 (n=20, p<0.001, Figure 3A). The amplitude of the T wave increased from 18.0±1.4 to 23.1±1.7% of the R wave (n=20, p<0.05).
Figure 1.
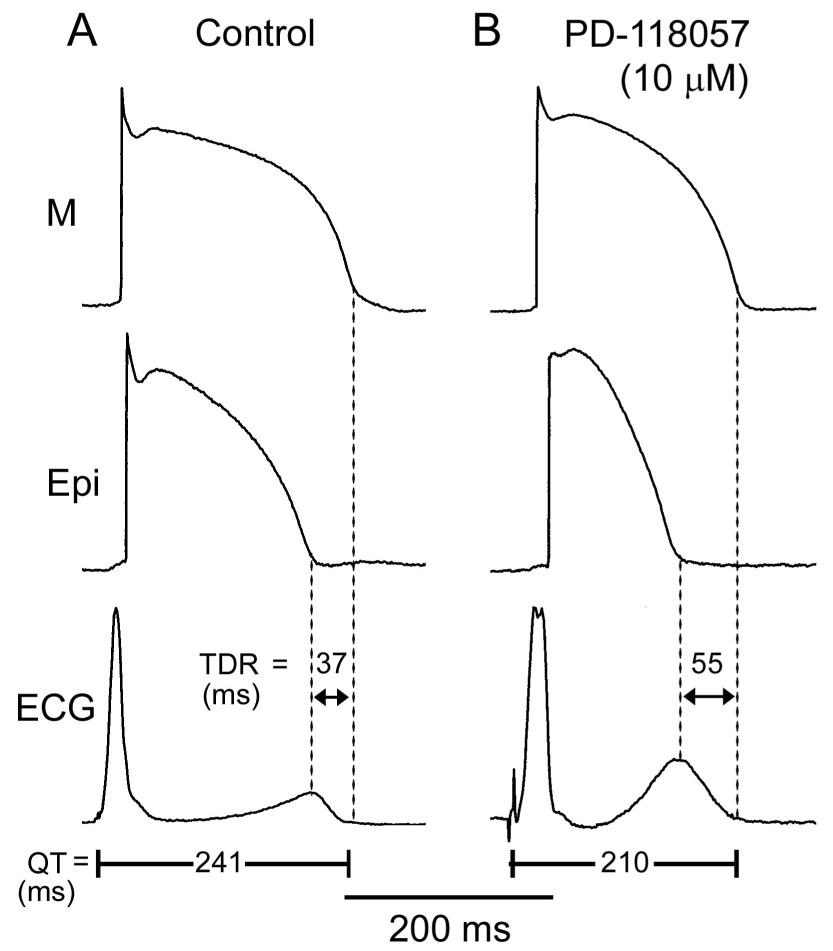
The effect of an IKr agonist to abbreviate action potential duration and QT interval and augment transmural dispersion of repolarization (TDR). A: Control. B: PD-118057 (10 µmol/L). Each panel shows transmembrane action potentials simultaneously recorded from an epicardial (Epi) and a deep subendocardial M-cell in an arterially perfused LV wedge preparation, together with a pseudo-ECG. Basic cycle length (BCL) =2000 ms.
Figure 2.
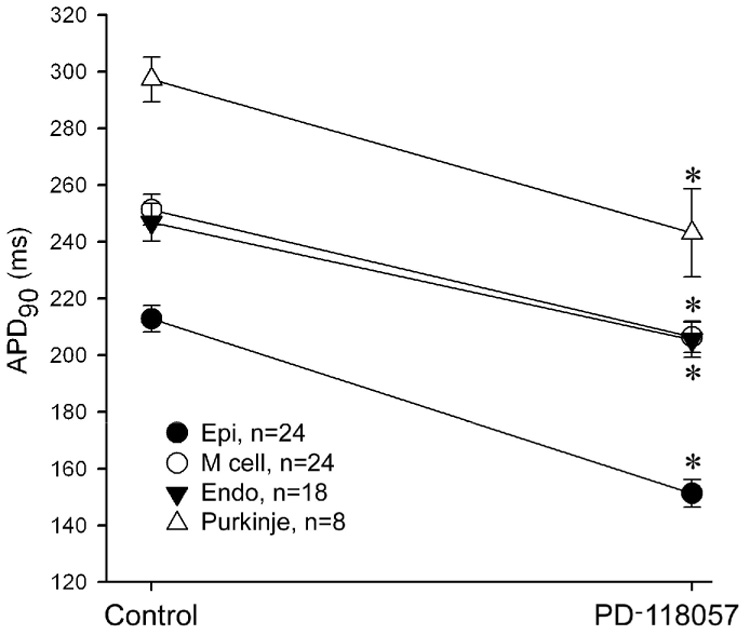
Effect of the IKr agonist PD-118057 10 µmol/L on APD90 of epicardial (Epi), M, endocardial (Endo) and Purkinje cell action potentials recorded from arterially-perfused LV wedge preparations. BCL=2000 ms. Endo Stimulation. * p<0.001, PD-110857 vs Control.
Figure 3.
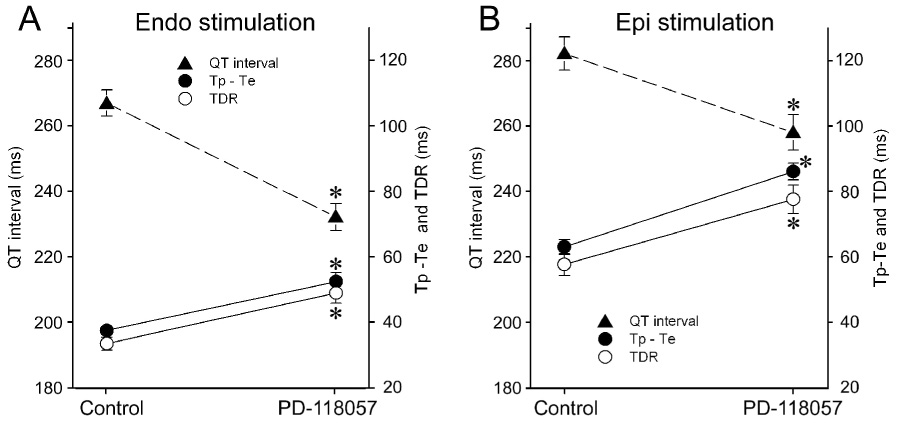
Composite data of the effects of PD-118057 10 µmol/L on QT, TDR, Tpeak-Tend interval when basic stimuli (S1) is delivered from endocardium (A. Endo stimulation, n= 20) or from epicardium (B. Epi stimulation, n=16). Shifting S1 from Endo to Epi increased QT, TDR and Tpeak-Tend. PD-118057 abbreviated QT interval and increased TDR and Tpeak-Tend in both cases. BCL= 2000 ms. * p <0.001, PD-118057 vs Control.
Effect of reversal of direction of activation of the left ventricular wall (Epi Stimulation)
Figure 4 illustrates the effect of reversing the direction of activation of the left ventricular wall. When basic stimulation (S1) was shifted from endocardium to epicardium, the epicardial cells activated earlier and repolarized earlier, whereas the M cells activated later and repolarized later. As a consequence, the QT interval prolonged from was 291 to 311 ms and TDR increased from 32 to 64 ms. Addition of PD-118057 (10 µmol/L) abbreviated the QT interval from 311 to 255 ms and increased TDR from 64 to 85 ms.
Figure 4.
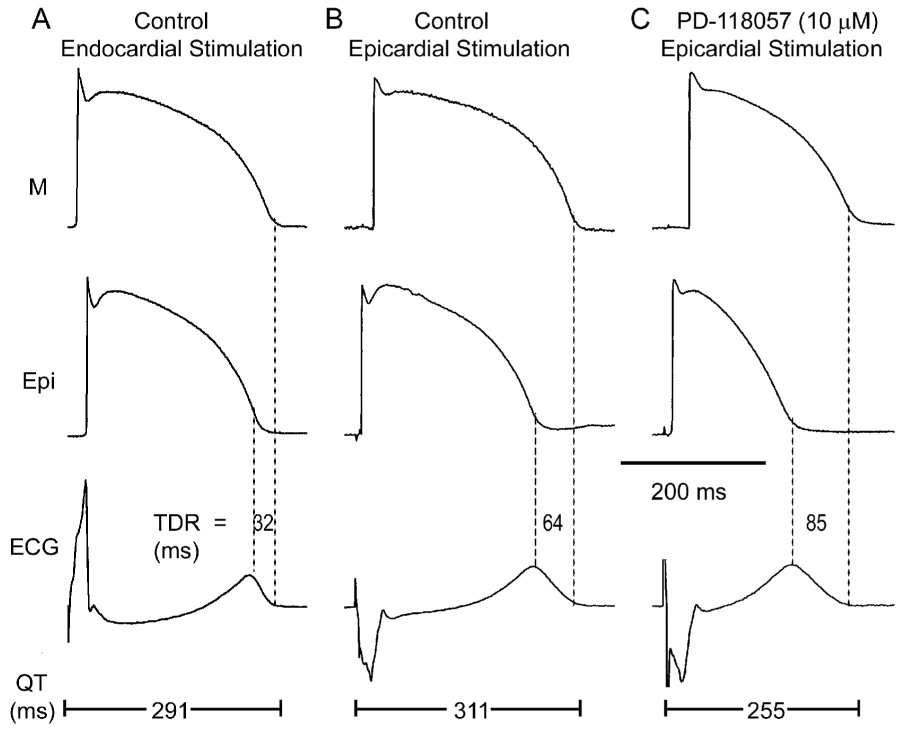
Effect of reversal of the direction of activation of the left ventricular wall on action potential morphology, QT interval and transmural dispersion of repolarization (TDR) in the absence and presence of the IKr agonist. A: Control (Endocardial stimulation). B: Control (Epicardial stimulation). C: Effect of PD-118057 (10µM; Epicardial stimulation). BCL = 2000 ms.
Composite data from 16 similar experiments are summarized in Table 1 and Figure 3B. Reversing the direction of activation across the ventricular wall by shifting basic stimulation (S1) from endocardium to epicardium caused an increase of the QT interval from 269.2±4.8 to 282.2±5.1, TDR from 34.1±2.0 to 57.7±3.3 and Tpeak-Tend from 37.3±1.4 to 63.1±2.3 under baseline conditions (BCL=2000, n=16, p<0.001). Addition of PD-118057(10µmol/L) abbreviated the QT interval from 282.2±5.1 to 258.1±5.4 and increased TDR and Tpeak-Tend from 57.7±3.3 to 77.6±4.3 and 63.1±2.3 to 86.1± 2.6, respectively (BCL=2000 ms, n=16, p<0.001). PD-118057 reduced ventricular ERP from 203.1±4.7 to 156.3±4.2 (BCL=2000 ms, n=16, p<0.001).
Table 1.
Effect of the IKr agonist PD-118057 on QT interval, TDR, pVT inducibility in the absence and presence of quinidine
| QT interval (ms) | TDR (ms) | pVT | ||||
|---|---|---|---|---|---|---|
| Endo Stimulation | Epi Stimulation | Endo Stimulation | Epi Stimulation | Endo Stimulation | Epi Stimulation | |
| Control (n=16–20) | 269.2±4.8 | 282.2±5.1* | 34.1±2.0 | 57.7±3.3* | 0/20 | 0/20 |
| PD-118057 (n=16–20) | 232.3±4.1† | 258.1±5.4*† | 46.9±3.7† | 77.6±4.3*† | 0/20 | 10/20 |
| Control (n=7) | 277.7±8.8 | 290.2±9.6* | 36.4±4.3 | 63.6±6.4* | 0/7 | 0/7 |
| PD-118057 (n=7) | 242.9±8.1† | 266.9±9.9*† | 55.1±5.2† | 89.0±6.4*† | 0/7 | 5/7 |
| PD-118057 + Quinidine (n=7) | 298±11.2‡ | 319.3±9.7*†‡ | 46.9±3.5 | 83.4±5.8*† | 0/7 | 0/7 |
pVT = polymorphic ventricular tachycardia. TDR = transmural dispersion of repolarization. BCL = 2000 ms.
p<0.05 vs. Endo Stimuli
p<0.05 vs. Control
p<0.05 vs. PD-118057.
IKr-agonist-induced arrhythmogenesis
With basic stimulation (S1) applied either to epicardium or endocardium, a premature stimulus (S2) was delivered to epicardium at progressively briefer S1–S2 intervals in an attempt to induce pVT. When S1 was applied to endocardium, programmed electrical stimulation failed to induce pVT in all 20 preparations, under control conditions and following PD-118057. With basic stimuli applied to epicardium, pVT could not be elicited under baseline conditions but was successfully induced in 10 out of 20 preparations following PD-118057 (Table 1).
Figure 5 illustrates an example of pVT induced with both S1 and S2 applied to epicardium in the presence but not absence of PD-118057. ERP abbreviated from 220 to 160 ms following exposure to PD-118057.
Figure 5.
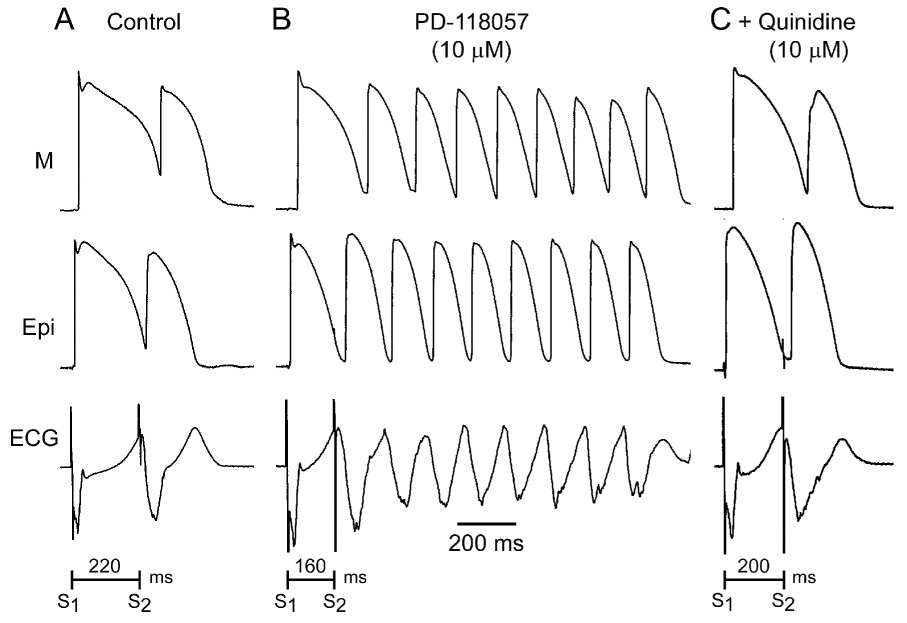
IKr agonist-induced short QT and polymorphic ventricular tachycardia (pVT). A: Control B: PD-118057 (10 µmol/L). C: PD-118057 (10 µM)+ quinidine (10 µM). Programmed electrical stimulation applied to the LV wedge preparation induced pVT when the QT interval and the effective refractory period were abbreviated with the IKr agonist, but not under baseline conditions or after the addition of quinidine. BCL = 2000 ms.
Effect of Quinidine
The effect of quinidine was evaluated in 5 preparations in which pVT could be induced and in 2 preparation in which pVT was not induced after PD-118057. Addition of quinidine(10µmol/L) to the coronary perfusate containing PD-118057 prolonged the QT interval from 266.9±9.9 to 319.3±9.7 (n=7, p<0.001), but TDR remained largely unchanged (89±6.4 vs. 83.4±5.8, n=7) (Figure 6, Table 1). Quinidine prolonged the ERP from 154.2±4.2 to 214.2±9.7 (n=7, p<0.001, Figure 6, Table 1). Programmed electrical stimulation failed to induce pVT in all 5 preparations in which pVT was elicited prior to quinidine (Figure 5, Table 1).
Figure 6.
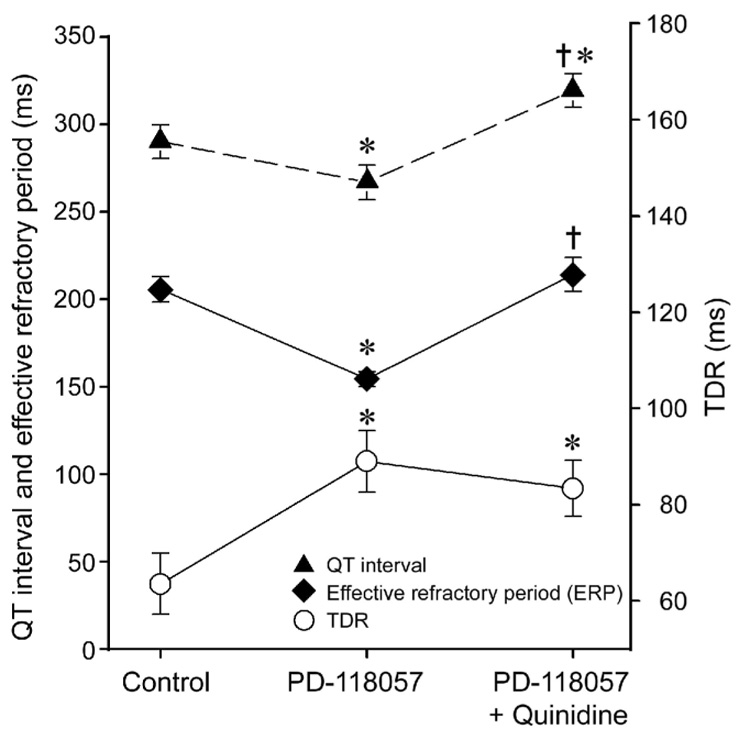
Quinidine (10 µmol/L) reverses the effects of PD-118057 to abbreviate QT interval and effective refractory period, but not its effects to augment transmural dispersion of repolarization (TDR). Basic cycle length = 2000 ms. Epicardial stimulation. * p<0.05 vs. Control. † p<0.001 vs. PD-118057.
DISCUSSION
To our knowledge this is the first experimental model of short QT syndrome in which the affected ion channel current is one previously shown to underlie a congenital form of the syndrome. The model recapitulates the electrocardiographic and arrhythmic manifestations of SQT1 and provides an understanding of the cellular basis for these changes utilizing the coronary-perfused LV wedge preparation. This preparation has been successfully used to reproduce the electrocardiographic and arrhythmic manifestations of long-QT syndrome, Brugada syndrome and catecholaminergic VT.16 A gain-of-function mutation in KCNH2, the gene that encodes the α-subunit of the IKr channel, has been shown to underlie SQT16 and we used PD-118057, a novel IKr agonist, to mimic this gain of function. PD-118057 at a concentration of 10 µmol/L has been shown to increase peak tail current of IKr by 111.1 % in human embryonic kidney cells and abbreviate QT interval by 25% in the isolated rabbit ventricle.13
Our principle findings are that: 1) PD-118057(10 µmol/L) abbreviates APD, QT interval, ERP and increases Tpeak-Tend interval and TDR; 2) Preferential abbreviation of epicardial APD is responsible for the increase in TDR and Tpeak-Tend interval; 3) Reversal of the direction of activation of the left ventricular wall by pacing from the epicardium instead of the endocardium increases TDR and exaggerates the effects of the IKr agonist to increase TDR, thus facilitating the induction of pVT; 4) Quinidine (10 µmol/L) prevents the induction of pVT by reversing the effect of PD-118057 on QT interval and ERP, but not on TDR. These results suggest that a gain of function of IKr predisposes to pVT via an abbreviation of ERP as well as an augmentation of TDR.
PD-118057 is thought to increase IKr by increasing open channel probability.13 The drug showed no major effect on INa, ICa-L, IK1, or IKs.13 The increase in net repolarizing current during phases 2 and 3 abbreviates action potential duration and hence QT interval. PD-118057 induces preferential abbreviation of epicardial APD as compared to M cell and endocardial APD, leading to increase in Tpeak-Tend interval and TDR. The ionic basis of preferential abbreviation of epicardial cell action potential is not clear but may be related to the fact that epicardium has the strongest repolarizing forces under baseline conditions. Interestingly, in rabbit left ventricular wedge preparations, Zhou at el 13 demonstrated preferential abbreviation of the endocardial APD in response to PD-118057.
Previous studies conjectured that a gain of function in IKr would have little effect on the Purkinje action potential because little IKr is expected to activate at the relatively negative plateau potentials encountered in action potentials recorded from isolated Purkinje myocytes.17 Our results clearly indicate that a gain of function of IKr produces an abbreviation of the Purkinje fiber action potential comparable to that observed in the ventricular myocardium.
Our data indicate that PD-118057 abbreviates ERP and increases TDR in the ventricular myocardium. The critical role of TDR as substrate for functional re-entry has been demonstrated in experimental models of long-QT syndrome, Brugada syndrome as well as in the short QT syndrome.16 Whereas increased TDR facilitates unidirectional block and the initiation of reentry, the abbreviated ERP reduces the wavelength (product of ERP and conduction velocity) and thus facilitates maintenance of reentry. When basic stimuli are applied to the endocardium, the increase in TDR was insufficient to permit induction of pVT. Shifting basic stimulation to epicardium increased TDR to 57.7±3.3 ms (71% increase) under baseline conditions and to 77.6±4.3 ms in the presence of the IKr agonist. The arrhythmogenic substrate created under these conditions permitted induction of pVT in 50% (10 out of 20) of preparations. The arrhythmogenic effect of reversing the direction of activation of the LV wall by pacing from epicardium instead of endocardium, has been demonstrated previously under a variety of conditions.14, 16, 18, 19
Quinidine (10 µmol/L) completely reversed the effect of PD-118057 on QT interval and ERP. In the presence of quinidine, the increase in TDR persisted but pVT could no longer be induced. Quinidine is known to block sodium channel current (INa), IKr, IKs, IK1, IK-ATP and the transient outward current (Ito). Through its action on K currents, it prolongs APD and ERP, thus reversing the effect of PD-118057 and leading to an increase in wavelength. Maintenance of reentrant wavefront requires that the wavelength be less than pathlength. Our data suggest that an increase in wavelength is a plausible explanation for the antiarrhythmic effect of quinidine in our model of SQT1. The inability of quinidine to alter TDR likely stems from the fact that quinidine, in the absence of PD-118057, causes an increase in TDR.20
Our results are consistent with a previously reported model of SQTS in which QT abbreviation was achieved by activation of IK-ATP using pinacidil. In left ventricular wedge preparation, pinacidil caused preferential abbreviation of M cell and endocardial APD leading to increase in TDR along with abbreviation of ERP and QT interval and inversion of the T wave.11 The threshold of TDR that permitted induction of pVT was 50 ms, significantly lower than in the present model.
Milberg et al.12 reported another experimental model of SQTS induced by pinacidil using a Langendorff-perfused rabbit heart. With simultaneous recording of eight monophasic action potentials (MAP), they demonstrated that pinacidil causes a concentration-dependent decrease in MAP and ERP and an increase in total and interventricular dispersion of repolarization, thereby increasing ventricular vulnerability to pVT. The effects of pinacidil were completely reversed by quinidine, but not sotalol or flecainide. In contrast to the present model, quinidine reversed the effect of pinacidil to increase dispersion of repolarization.
Clinical Implications
The present model is mechanistically similar but phenotypically different from the pinacidil models of SQTS in that it more closely mimics the gain-of-function mutation in KCNH2.11, 12 The ECG phenotype created by PD-118057 closely resembles the ECG observed in patients with SQT1. Along with abbreviation of QT interval, the model recapitulates the decrease in length of the ST segment, the increase in Tpeak-Tend interval and increase in amplitude of the T wave observed clinically. Data recorded from telemetry and implantable cardioverter defibrillators (ICDs) in patient with SQTS suggest that closely coupled premature ventricular complexes initiate episodes of pVT.21–23 Electrophysiological studies in these patients show marked abbreviation of ventricular ERP and programmed electrical stimulation inducibility of pVT in 61% of patients using two or three premature stimuli.3, 10 Consistent with these clinical findings, our experimental model displays reduced ventricular ERP and induction of pVT with closely coupled extrasystoles. Quinidine has been proposed as drug of choice for SQT1 as an adjunct to ICD. Treatment with quinidine in SQT1 patients increases the QT interval and ventricular effective refractory period and renders ventricular arrhythmia noninducible.10, 24 Proven efficacy of quinidine and failure of class IC and III antiarrhythmic drugs in SQT1 is primarily related to a secondary effect of the N588K mutation in KCNH2. The N588K missense mutation induces +90 mV shift in the voltage-dependence of inactivation of HERG channels. The inactivated state of the channel normally stabilizes the interaction of the channel with most IKr blockers.6, 17 Loss of inactivation accounts for the ineffectiveness of most IKr blockers in N588K KCNH2 channels. Quinidine is effective in SQT1 by the virtue of its interaction with the open state of the channel, whereas drugs like sotalol and dofetilide that have a higher affinity for the inactivated state of channel are largely ineffective.
IKr agonists have attracted the interest of the pharmaceutical industry because of their ability to suppress arrhythmogenesis encountered under long QT conditions. PD-118057 has been shown to be effective in reversing the long QT intervals and suppressing early afterdepolarization activity induced by IKr blockers.13 The present study highlights the proarrhythmic potential of IKr agonists and provides additional support for the arrhythmogenic potential QT abbreviation. Our data also extend the proarrhythmic potential of reversing the direction of activation of the ventricular wall to include the short QT syndrome. This finding has direct relevance to biventricular pacing and resynchronization therapy, commonly used in the treatment of congestive heart failure.
Limitations
As with any experimental study, extrapolation of our results to the understanding of the clinical syndrome must be approached with caution. While PD-118057 is effective in augmenting IKr, it does not precisely mimic the gain of function produced by the mutations in KCNH2 thus far uncovered. This notwithstanding, we take comfort in the fact that the model reproduces many of the characteristics of the clinical syndrome, as well as the response to drugs such as quinidine.
ACKNOWLEDGMENTS
We gratefully acknowledge the expert technical assistance of Judy Hefferon, Robert Goodrow, Jr., and Kathy Sullivan.
This study was supported by a grant HL-47687 from the NIH and the New York State and Florida Grand Lodges of Free and Accepted Masons.
Abbreviations
- ANOVA
analysis of variance
- APD
action potential duration
- APD90
APD measured at 90%
- BCL
basic cycle lengths
- DMSO
Dimethyl Sulfoxide
- Epi
epicardial
- Endo
endocardial
- ERP
effective refractory period
- ICDs
implantable cardioverter defibrillators
- IK1
inward rectifier potassium channel current
- IK-ATP
ATP-sensitive potassium current
- IKr
rapidly activating potassium channel currents
- IKs
slowly activating potassium channel currents
- Ito
transient outward current
- M
subendocardial
- MAP
monophasic action potentials
- pVT
Polymorphic ventricular tachycardia
- SQTS
Short QT syndrome
- TDR
transmural dispersion of repolarization
- VT
ventricular tachycardia
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Potential Conflicts of Interest: None
References
- 1.Gussak I, Brugada P, Brugada J, et al. Idiopathic short QT interval: a new clinical syndrome? Cardiology. 2000;94:99–102. doi: 10.1159/000047299. [DOI] [PubMed] [Google Scholar]
- 2.Gussak I, Brugada P, Brugada J, et al. ECG phenomenon of idiopathic and paradoxical short QT intervals. Cardiac Electrophysiol Rev. 2002;6:49–53. doi: 10.1023/a:1017931020747. [DOI] [PubMed] [Google Scholar]
- 3.Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: a familial cause of sudden death. Circulation. 2003;108:965–970. doi: 10.1161/01.CIR.0000085071.28695.C4. [DOI] [PubMed] [Google Scholar]
- 4.Bjerregaard P, Gussak I. Short QT syndrome: mechanisms, diagnosis and treatment. Nat Clin Pract Cardiovasc Med. 2005;2:84–87. doi: 10.1038/ncpcardio0097. [DOI] [PubMed] [Google Scholar]
- 5.Gussak I, Bjerregaard P. Short QT syndrome-5 years of progress. J Electrocardiol. 2005;38:375–377. doi: 10.1016/j.jelectrocard.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 6.Brugada R, Hong K, Dumaine R, et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation. 2004;109:30–35. doi: 10.1161/01.CIR.0000109482.92774.3A. [DOI] [PubMed] [Google Scholar]
- 7.Bellocq C, Van Ginneken AC, Bezzina CR, et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation. 2004;109:2394–2397. doi: 10.1161/01.CIR.0000130409.72142.FE. [DOI] [PubMed] [Google Scholar]
- 8.Priori SG, Pandit SV, Rivolta I, et al. A Novel Form of Short QT Syndrome (SQT3) Is Caused by a Mutation in the KCNJ2 Gene. Circ Res. 2005;96:800–807. doi: 10.1161/01.RES.0000162101.76263.8c. [DOI] [PubMed] [Google Scholar]
- 9.Antzelevitch C, Pollevick GD, Cordeiro JM, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115:442–449. doi: 10.1161/CIRCULATIONAHA.106.668392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giustetto C, Di Monte F, Wolpert C, et al. Short QT syndrome: clinical findings and diagnostic-therapeutic implications. Eur Heart J. 2006;27:2440–2447. doi: 10.1093/eurheartj/ehl185. [DOI] [PubMed] [Google Scholar]
- 11.Extramiana F, Antzelevitch C. Amplified transmural dispersion of repolarization as the basis for arrhythmogenesis in a canine ventricular-wedge model of short QT syndrome. Circulation. 2004;110:3661–3666. doi: 10.1161/01.CIR.0000143078.48699.0C. [DOI] [PubMed] [Google Scholar]
- 12.Milberg P, Tegelkamp R, Osada N, et al. Reduction of dispersion of repolarization and prolongation of postrepolarization refractoriness explain the antiarrhythmic effects of quinidine in a model of short QT syndrome. J Cardiovasc Electrophysiol. 2007;18:658–664. doi: 10.1111/j.1540-8167.2007.00813.x. [DOI] [PubMed] [Google Scholar]
- 13.Zhou J, Augelli-Szafran CE, Bradley JA, et al. Novel potent human ether-a-go-go-related gene (hERG) potassium channel enhancers and their in vitro antiarrhythmic activity. Mol Pharmacol. 2005;68:876–884. doi: 10.1124/mol.105.014035. [DOI] [PubMed] [Google Scholar]
- 14.Fish JM, Di Diego JM, Nesterenko VV, et al. Epicardial activation of left ventricular wall prolongs QT interval and transmural dispersion of repolarization: implications for biventricular pacing. Circulation. 2004;109:2136–2142. doi: 10.1161/01.CIR.0000127423.75608.A4. [DOI] [PubMed] [Google Scholar]
- 15.Nam G-B, Burashnikov A, Antzelevitch C. Cellular mechanisms underlying the development of catecholaminergic ventricular tachycardia. Circulation. 2005;111:2727–2733. doi: 10.1161/CIRCULATIONAHA.104.479295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Antzelevitch C. The role of spatial dispersion of repolarization in inherited and acquired sudden cardiac death syndromes. Am J Physiol Heart Circ Physiol. 2007;293:H2024–H2038. doi: 10.1152/ajpheart.00355.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cordeiro JM, Brugada R, Wu YS, et al. Modulation of IKr inactivation by mutation N588K in KCNH2: A link to arrhythmogenesis in short QT syndrome. Cardiovasc Res. 2005;67:498–509. doi: 10.1016/j.cardiores.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 18.Medina-Ravell VA, Lankipalli RS, Yan GX, et al. Effect of epicardial or biventricular pacing to prolong QT interval and increase transmural dispersion of repolarization. Does resynchronization therapy pose a risk for patients predisposed to long QT or torsade de pointes? Circulation. 2003;107:740–746. doi: 10.1161/01.cir.0000048126.07819.37. [DOI] [PubMed] [Google Scholar]
- 19.Antzelevitch C, Oliva A. Amplification of spatial dispersion of repolarization underlies sudden cardiac death associated with catecholaminergic polymorphic VT, long QT, short QT and Brugada syndromes. J Intern Med. 2006;259:48–58. doi: 10.1111/j.1365-2796.2005.01587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Antzelevitch C, Dumaine R. Electrical heterogeneity in the heart: physiological, pharmacological and clinical implications. In: Fozzard HA, Solaro RJ, editors. The Cardiovascular System. Volume 1, The Heart. New York: The American Physiological Society by Oxford University Press; 2002. pp. 654–692. [Google Scholar]
- 21.Schimpf R, Bauersfeld U, Gaita F, et al. Short QT syndrome: Successful prevention of sudden cardiac death in an adolescent by implantable cardioverter-defibrillator treatment for primary prophylaxis. Heart Rhythm. 2005;2:416–417. doi: 10.1016/j.hrthm.2004.11.026. [DOI] [PubMed] [Google Scholar]
- 22.Borggrefe M, Wolpert C, Antzelevitch C, et al. Short QT syndrome Genotype-phenotype correlations. J Electrocardiol. 2005;38(Suppl):75–80. doi: 10.1016/j.jelectrocard.2005.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu LX, Zhou W, Zhang X, et al. Short QT syndrome: a case report and review of literature. Resuscitation. 2006;71:115–121. doi: 10.1016/j.resuscitation.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 24.Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: pharmacological treatment. J Am Coll Cardiol. 2004;43:1494–1499. doi: 10.1016/j.jacc.2004.02.034. [DOI] [PubMed] [Google Scholar]