

Abstract
Snai2-deficient cells are radiosensitive to DNA damage. The function of Snai2 in response to DNA damage seems to be critical for its function in normal development and cancer. Here, we applied a functional genomics approach that combined gene-expression profiling and computational molecular network analysis to obtain global dissection of the Snai2-dependent transcriptional response to DNA damage in primary mouse embryonic fibroblasts (MEFs), which undergo p53-dependent growth arrest in response to DNA damage. Although examination of the response showed that overall expression of p53 target gene expression patterns was similarly altered in both control and Snai2-deficient cells, we have identified and validated candidate Snai2 target genes linked to Snai2 gene function in response to DNA damage. This work defines for the first time the effect of Snai2 on p53 target genes in cells undergoing growth arrest, elucidates the Snai2-dependent molecular network induced by DNA damage, points to novel putative Snai2 targets, and suggest a mechanistic model, which has implications for cancer management.
Keywords: Snai2, MEFs, microarray analysis, stem cells, DNA damage
Uncommitted progenitor cells express Snai2 and aberrant activation of Snai2 pathways is key in the development of cancers derived from many tissues (Inoue et al, 2002; Perez-Losada et al, 2002; Perez-Mancera et al, 2005). The implication of SNAI2 in human cancer seems to be wider than initially expected (Elloul et al, 2005; Gupta et al, 2005; Shih et al, 2005; Bermejo-Rodriguez et al, 2006; Come et al, 2006). Nevertheless, the molecular mechanisms by which SNAI2 participates in these biological processes are not yet clear. In vitro studies have shown that Snai2 confers resistance to cell death induced by the withdrawal of survival factors (Inoue et al, 2002; Perez-Losada et al, 2002; Perez-Mancera et al, 2005). To understand the relevance of SNAI2 to human cancer Snai2-expressing mice were generated (Perez-Mancera et al, 2005). The analysis of the Snai2-expressing mice identified that ‘uncontrolled’ Snai2 expression induces cancer in mice. These findings further indicated that overexpression of SNAI2 by human tumours could be of importance to both cell fate selection by genotoxic anticancer agents (Perez-Mancera et al, 2005) and clinical management of human cancer patients (Elloul et al, 2005; Shih et al, 2005; Bermejo-Rodriguez et al, 2006; Come et al, 2006). These results suggested that Snai2 expression was protecting cells from death by genetic alterations as a consequence of an inherent, basal level of genetic instability (Sanchez-Garcia, 1997; Pérez-Caro et al, 2005; Perez-Mancera et al, 2005). Consistent with this interpretation, in the haematopoietic system Snai2-deficient cells are radiosensitive to DNA damage (Inoue et al, 2002; Perez-Losada et al, 2003; Wu et al, 2005). In fact, SNAI2 expression seems to be associated with a patient's resistance to chemotherapeutic agents in human mesotheliomas (Catalano et al, 2004). Thus, constitutive activation of Snai2 could confer resistance properties to the tumour-target cells connecting DNA damage with the requirement of a critical level of Snai2 for cancer development. In agreement with these results, recently it has been shown Snai2 is a p53 target that antagonises p53-mediated apoptosis of haematopoietic progenitors (Wu et al, 2005). However, the effect of Snai2 on p53 target genes remains an open question (Haupt et al, 2006).
In this study, we have investigated the role of Snai2 in DNA damage. To explore the role of Snai2 in DNA damage response, we used primary mouse embryonic fibroblasts (MEFs), which undergo p53-dependent growth arrest in response to DNA damage. Snai2 wild-type and null MEFs were subjected to DNA damage and the global gene expression patterns were examined. Although overall the expression of the majority of the spotted genes was not altered in Snai2-deficient cells compared to control MEFs, we have identified candidate Snai2 target genes linked to Snai2 gene function in response to DNA damage.
MATERIALS AND METHODS
Preparation of MEF RNA populations
Wild-type, p53−/− and Snai2-deficient MEFs were isolated as described previously (Bermejo-Rodriguez et al, 2006). To prepare the RNA population for microarray studies, wild-type and Snai2-deficient MEFs (passage 4) were synchronised by growing to confluence in DMEM plus 10% FCS for 3 days. To stimulate re-entry into the cell cycle, the cells were reseeded into DMEM plus 10% FCS at 1.0 × 106 cells per 10-cm dish. After 6 h, 0.2 μg ml−1 doxorubicin (Sigma-Aldrich, Madrid, Spain) was added to induce G1 arrest. After 12 h treatment, cells were collected by trypsinisation and frozen as a pellet for subsequent RNA and/or protein preparation. Dishes of both untreated and treated wild-type and Snai2-deficient MEFs were collected after 16 h of doxorubicin treatment for FACS analysis to verify a p53-dependent G1 arrest as described previously (Attardi et al, 2000).
γ-Irradiation was performed using a 137Cs source. Asynchronously growing wild-type, p53−/− and Snai2-deficient MEFs were treated with 5 and 8 Gray (Gy) of γ-irradiation, which induces a well-characterised arrest in both G1 and G2 (Kastan et al, 1992). We chose to examine the effects at 18 h, a time point at which these arrest responses have been demonstrated previously.
RNA extraction
Total RNA was isolated in two steps using TRIzol (Life Technologies Inc., Grand Island, NY, USA) followed by Rneasy Mini-Kit (Qiagen Inc., Valencia, CA, USA) purification following the manufacturer's RNA Clean-up protocol with the optional on-column DNase treatment. The integrity and the quality of RNA was verified by electrophoresis and its concentration measured.
Microarray procedures
A measure of 30 μg of total RNA from each sample was directly labelled with cyanine 3-conjugated dUTP (Cy3), whereas 30 μg of RNA from the Universal Mouse Reference RNA (Stratagene, VWR International, Spain) was labelled with cyanine 5-conjugated dUTP (Cy5) as reference. For all of the microarray studies the CNIO MouseChip was used (Bermejo-Rodriguez et al, 2006). Hybridisations were performed as described (Bermejo-Rodriguez et al, 2006). After washing, the slides were scanned using a Scanarray 5000 XL (GSI Lumonics Kanata, Ontario, Canada) and images were analysed with the GenePix 4.0 program (Axon Instruments Inc., Union City, CA, USA). All experiments were repeated four times using cells from different embryos.
Data analysis
Data obtained from each hybridisation were stored in a database for analysis. The Cy3 : Cy5 ratios were normalised to the median ratio value of all of the spots in the array. After normalisation, spots with intensities for both channels (sum of medians) less than that of the local background were discarded. The ratios of the remaining spots were log transformed (base 2), and duplicated spots on the MouseChip were averaged to the median. To obtain the expression profile of wild-type and Snai2-deficient MEFs, we referred the ratios of control and Snai2-deficient DNA-damaged cells to the undamaged counterparts. The Ingenuity Pathways Analysis program (http://www.ingenuity.com/index.html) was used to further analyse the cellular functions and pathways that were significantly regulated by Snai2 in response to DNA damage.
Quantitative RT–PCR
Control and Snai2−/− MEF RNA (1 μg) was reverse-transcribed by using Advantage RT for PCR kit (BD Biosciences, Becton Dickinson, Madrid, Spain). SYBR Green PCR Master mix (Applied Biosystems, USA) was used for template amplification with the primers specific for each of the transcripts examined. PCR with RT sample were used as negative controls. Thermocycling for all targets were carried out in 30 μl reaction for 40 cycles in triplicate. Each cycle consisted of 94°C for 15 s, 56°C for 30 s and 72°C for 30 s. Incorporation of the SYBR Green dye into PCR products was monitored in real time with an ABI PRISM 7000 sequence detection system (Applied Biosystems). SDS system software was used to convert the fluorescent data into threshold cycle (Ct) at which exponential amplification of products begins. The differences in the Ct values (dCt) between the transcript of interest and endogenous control (GAPDH) were used to determine the relative expression of the gene in each sample and the ddCt method was used to calculate fold expression. To determine correlation between expression of two genes in the same set of samples dCt values were used to calculate regression coefficient.
Western blot analysis
Western blot analysis of control, p53−/−and Snai2−/− MEFs were carried out essentially as described (Castellanos et al, 1997). Extracts were normalised for protein content by Bradford analysis (Bio-Rad Laboratories Inc., Melville, NY, USA) and Coomassie blue gel staining. Lysates were run on a 10% SDS–PAGE gel and transferred on to a PVDF membrane. After blocking, the membrane was probed with the following primary antibodies: p53 (Ab3, Oncogene Research), Phospho-p53 (Ser15) (no. 9284, Cell Signaling Technology, IZASA, Barcelona, Spain), Puma/bbc3 (P4743, Sigma), Bid (sc-11423, Santa Cruz Biotechnology, Quimigen, Madrid, Spain), p27 (sc-1641, Santa Cruz Biotechnology), Atm (sc-1214, Santa Cruz) and actin (I-19) (sc-1616, Santa Cruz Biotechnology). Reactive bands were detected with an ECL system (Amersham, GE Healthcare, Madrid, Spain).
RESULTS
Normal G1 arrest in Snai2-deficient MEFs in response to doxorubicin treatment
Expression patterns were studied in wild-type MEFs and Snai2-deficient MEFs to search for Snai2-regulated species in response to DNA damage. We chose to use the DNA-damaging agent doxorubicin for this study as it effectively induced p53-dependent G1 arrest in MEFs (Attardi et al, 2000). To prepare RNA for screen wild-type MEFs and Snai2-deficient MEFs were treated with 0.2 μg ml−1 doxorubicin for 12 h. This time point was chosen because it had previously shown it to be a point of peak expression of known p53 target genes (Attardi et al, 2000). That G1 arrest had occurred was confirmed by FACS analysis, which showed a G1 arrest in both the wild-type and the Snai2-deficient cells (Figure 1).
Figure 1.

Doxorubicin induces a G1 arrest in Snai2-deficient mouse embryonic fibroblasts (MEFs). Dishes of both untreated (A) treated (B) wild-type and Snai2-deficient MEFs were collected after 16 h of doxorubicin treatment for FACS analysis to verify a G1 arrest. FACS profiles include G1-arrested without and with doxorubicin of both wild-type and Snai2-deficient MEFs.
Normal p53 activation in Snai2-deficient MEFs in response to doxorubicin treatment
It has been previously shown that Snai2-deficient bone marrow cells are radiosensitive to DNA damage induced by γ-irradiation (Inoue et al, 2002; Perez-Losada et al, 2003; Wu et al, 2005). p53 is centrally involved in the cellular response to DNA damage (Attardi et al, 2000). Exposure to DNA damage agents causes an increase in the intracellular levels of p53 (Attardi et al, 2000). Thus, we next explored if the DNA damage-protective potential of Snai2 was based on interference with p53 activation in MEFs in response to doxorubicin treatment. As shown in Figure 2, we measured the p53 protein levels in wild-type MEFs and Snai2-deficient MEFs after DNA damage induced by doxorubicin. The activation of p53 in both control and Snai2-deficient cells was similar (Figure 2), indicating that p53 regulation in response to DNA damage is not affected in Slug-deficient MEF.
Figure 2.

Levels of p53 protein in Snai2-deficient mouse embryonic fibroblast (MEF) cells in response to DNA damage treatment with doxorubicin. p53 protein was detected by western blotting in wild-type and Snai2-deficient MEFs. Actin was used as a loading control.
Identification of Snai2-target genes in response to DNA damage by mouse cDNA Microarray analysis in MEFs
Expression analyses were performing with a mouse cDNA microarray (Mousechip-CNIO) containing 15 000 clones (Bermejo-Rodriguez et al, 2006). Expression patterns were studied in wild-type MEFs and Snai2-deficient MEFs. Three independent mouse cDNA microarrays were used to search for Snai2-regulated species in MEFs in response to DNA damage. To obtain a global view of the number of genes regulated by Snai2 in response to DNA damage, we hybridised differentially labelled RNA from control MEFs versus the Snai2-deficient MEFs to a mouse cDNA microarray. This system allows us to identify physiologically relevant Snai2 targets in response to DNA damage. Overall the expression of the majority of the spotted genes was not altered in Snai2-deficient cells in response to doxorubicin treatment.
To confirm that this microarray analysis could be used to identify Snai2 target genes, we first examined whether known target genes were modulated in control MEFs after doxorubicin treatment. As expected, the main sensor of DNA damage p53 was upregulated close to genes involved in cell cycle arrest (Ku70), apoptosis (Casp3, Puma, Lrdd) and DNA repair (mdm2, Rad1) (Figure 3A). Puma is a Bcl-2 family member involved in p53-induced apoptosis (Jeffers et al, 2003) and Puma/bbc3 expression was upregulated in control MEFs in response to DNA damage. On the contrary, survival gene expression was downregulated in control MEFs after DNA doxorubicin treatment (Figure 3A). Analysis of the expression of the same set of genes revealed that overall expression was not altered in Snai2-deficient cells in response to doxorubicin treatment (Figure 3B). The expression of the previously implicated Snai2-target gene Puma/bbc3 (Wu et al, 2005; Bermejo-Rodriguez et al, 2006) was not significantly modulated in Snai2-deficient cells in response to DNA damage. To confirm this result, we next measured the Puma/bbc3 protein levels in wild-type MEFs and Snai2-deficient MEFs after DNA damage induced by doxorubicin. In agreement with our previous observations (Bermejo-Rodriguez et al, 2006), the Puma/bbc3 protein levels in Snai2-deficient MEFs were lower than in control MEFs (Figure 3C). However, the activation of Puma/bbc3 in both control and Snai2-deficient cells in response to doxorubicin treatment was similar (Figure 3C), indicating that Puma/bbc3 regulation in response to DNA damage is not affected in Snai2-deficient MEF. These results suggest that p53 does not require Snai2 for Puma/bbc3 regulation in response to DNA damage in MEFs.
Figure 3.
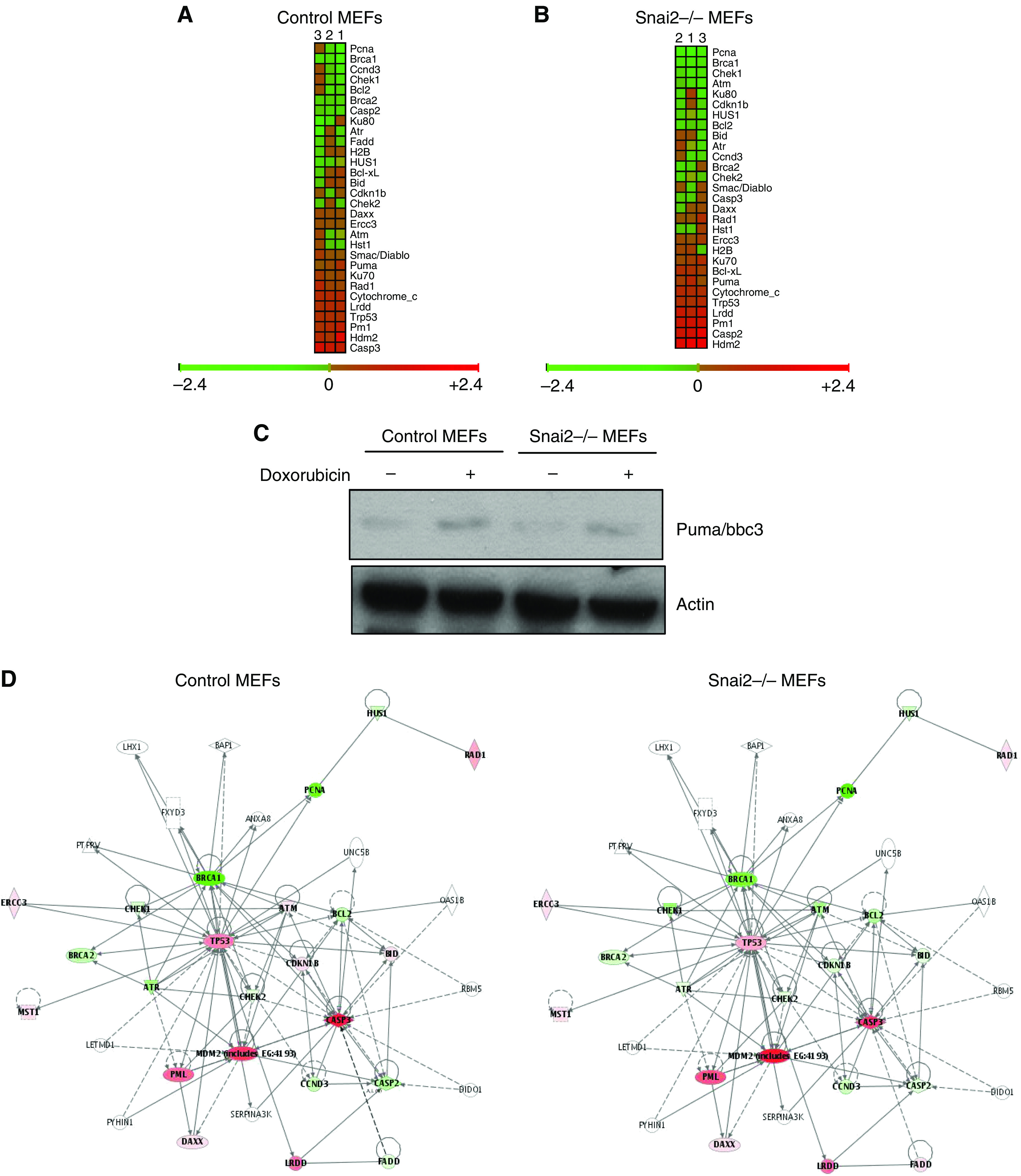
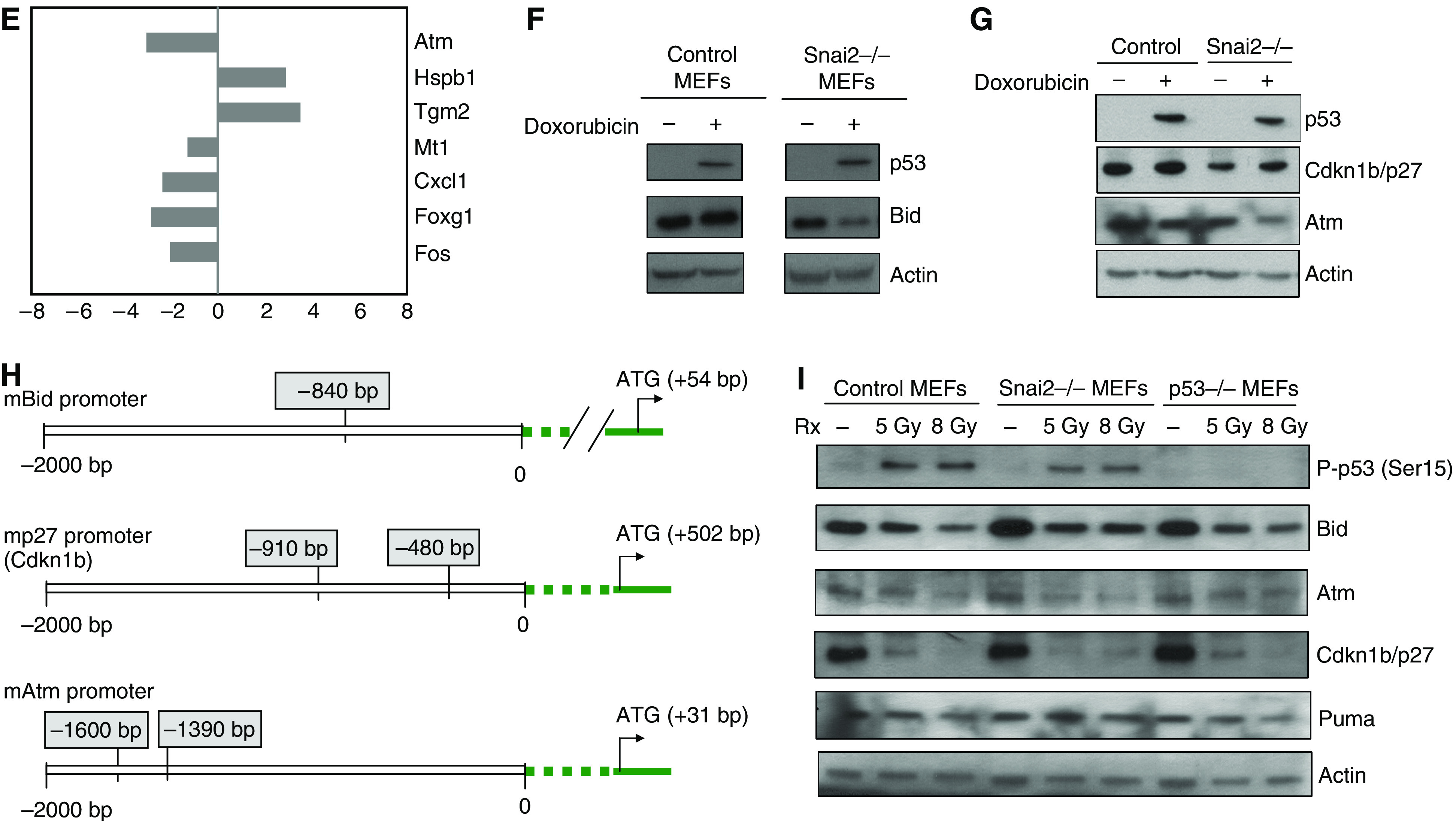
Graphical depiction of expression data for known target genes in response to DNA damage (cell cycle and apoptosis related genes) in wild-type mouse embryonic fibroblasts (MEFs) (A) and Snai2-deficient MEFs (B). Each gene (identified at right) represented by a single row of coloured boxes; each independent experiment is represented by one single column. (C) Puma expression in control MEFs and Snai2−/− MEFs in response to DNA damage treatment by doxorubicin. Puma protein analysis in control and Snai2−/− MEFs after DNA damage by western blot. Actin was used as a loading control. (D) Identification of Slug-dependent proteins using the Ingenuity database. Nodes are colour-coded in red (upregulated) or green (downregulated) according to their fold changes value. (E) Confirmation of the microarray results by qPCR performed as described in the Materials and Methods section. (F and G) Expression of Bid (F) and Atm and Cdkn1b/ p27 (G) in control MEFs and Snai2−/− MEFs in response to DNA damage treatment by doxorubicin. Bid, Atm and Cdkn1b/p27 protein analysis in control and Snai2−/− MEFs after DNA damage by western blot. Actin was used as a loading control. (H) Identification of Snai2 DNA-binding sites within promoter regions of murine Atm, Bid and p27 genes (bp, base pairs). (I) Expression of p53, Puma, Bid, Atm and Cdkn1b/ p27 (G) in control, Snai2−/− and p53−/− MEFs in response to DNA damage treatment by γ-irradiation. p53 protein was detected by western blot in wild-type and Snai2-deficient MEFs. Puma, Bid, Atm and Cdkn1b/p27 protein analysis by western blot in control, Snai2−/− and p53−/− MEFs after DNA damage treatment with 5 and 8 Gy of γ-irradiation. Actin was used as a loading control.
We further characterised our microarray data using the Ingenuity program (a software that identifies molecular networks by relating each gene entry with a database of known physical transcriptional or protein interactions). This study revealed the existence in the Snai2-dependent transcriptome of three proteins whose expression was altered in Snai2-deficient cells in response to DNA damage: Atm, Cdkn1b/p27 and Bid (Figure 3D). These results led us to demonstrate that these transcriptional factors were indeed downregulated in Snai2-deficient cells in response to DNA damage, by quantitative RT–PCR (qRT–PCR) in the case of Atm (Figure 3E) and by using specific antibodies to Bid, Atm and Cdkn1b/p27 (Figure 3F and G). Because Snai2 DNA-binding sites (Inukai et al, 1999) were present in promoter regions of Atm, and Cdkn1b/p27 and Bid (Figure 3H), Snai2 might be directly involved in the control of transcription-repression of these targets.
We next examined if Snai2 was regulating these three targets, Atm, Cdkn1b/p27 and Bid, in response to DNA damage by γ-irradiation. To prepare protein for analysis, wild-type MEFs, p53−/− and Snai2-deficient MEFs were treated with 5 and 8 Gy of γ-irradiation, which induces a well-characterised arrest in both G1 and G2 (Kastan et al, 1992). As shown in Figure 3I, these transcription factors were downregulated in Snai2-deficient MEFs in response to DNA damage by γ-irradiation. Thus, γ-irradiation is similar to doxorubicin in regulating the Snai2-dependent expression of these proteins. Moreover, the activation of Puma/bbc3 in both control and Snai2-deficient cells in response to γ-ray treatment was similar (Figure 3I), further indicating that Puma/bbc3 regulation in response to DNA damage is not affected in Snai2-deficient MEF.
Effect of Snai2 on p53 target genes in cells undergoing growth arrest in response to DNA damage
It is well established that there is a tissue specificity in the relative induction of several known p53 target genes in response to DNA damage (Burns et al, 2001; Burns and El-Deiry, 2003). Thus, we next examined how these genes were modulated in control and Snai2-deficient MEFs after doxorubicin treatment. As shown in Figures 4, 5 the overall expression of these p53 target gene expression patterns was similarly altered in both control and Snai2-deficient cells, indicating that p53 does not require Snai2 for regulation of majority of these genes in response to DNA damage in MEFs. However, the regulation of a significant number of p53 target genes in response to DNA damage in both control and Snai2-deficient MEFs differs from how they are regulated in the spleen and thymus in response to DNA damage (Burns et al, 2001; Burns and El-Deiry, 2003), confirming the striking cell specificity for p53-dependent transcription and repression (Burns et al, 2001; Burns and El-Deiry, 2003).
Figure 4.
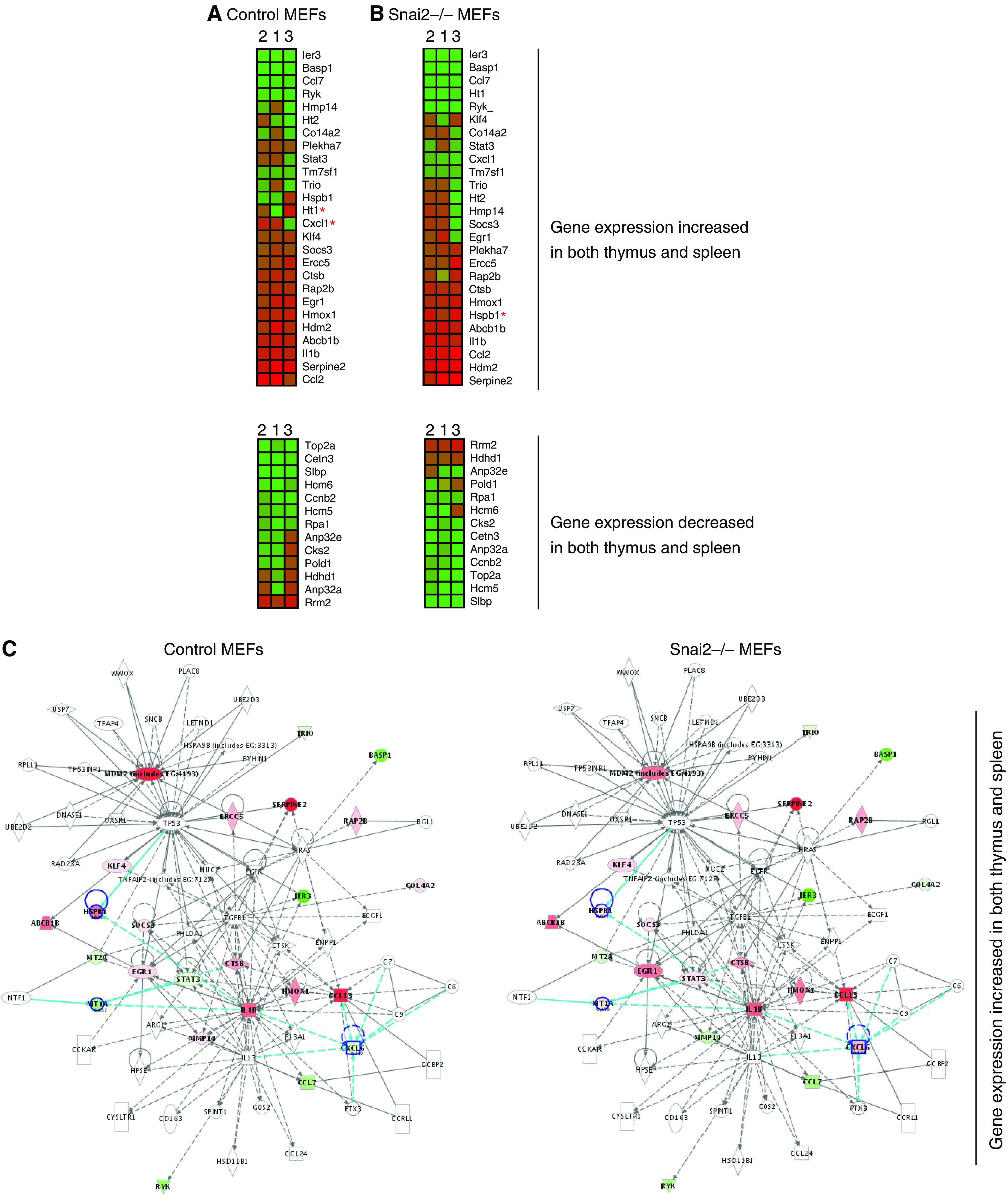
Graphical expression pattern in wild-type mouse embryonic fibroblasts (MEFs) (A) and Snai2-deficient MEFs (B) of genes upregulated and downregulated by p53 in response to DNA damage. Described genes upregulated and downregulated by p53 in the thymus and spleen (Burns and El-Deiry, 2003) in response to DNA damage were analysed. Each gene (identified at right) represented by a single row of coloured boxes; each independent experiment is represented by one single column. A red asterisk indicates major changes between control and Snai2-deficient MEFs. (C) Identification of Slug-dependent proteins using the Ingenuity database. Nodes are colour-coded in red (upregulated) or green (downregulated) according to their fold changes value.
Figure 5.


Graphical representation of the expression pattern in wild-type mouse embryonic fibroblasts (MEFs) (A) and Snai2-deficient MEFs (B) of genes known to be specifically increased by p53 in response to DNA damage. Genes known to be specifically increased by p53 in either thymus and spleen (Burns and El-Deiry, 2003) in response to DNA damage were studied. Each gene (identified at right) represented by a single row of coloured boxes; each independent experiment is represented by one single column. A red asterisk indicates the main expression differences between control and Snai2-deficient MEFs. (C) Identification of Slug-dependent proteins using the Ingenuity database. Nodes are colour-coded in red (upregulated) or green (downregulated) according to their fold changes value. (D) Identification of Snai2 DNA-binding sites within promoter regions of human and mouse of p53 target genes modulated by Snai2.
Analysis of the expression microarray data revealed six candidate p53 target genes (two genes whose expression was increased in Snai2-deficient MEFs compared to control MEFs in response to DNA damage: Hspb1 and Tgm2; and four genes whose expression was decreased in Snai2-deficient MEFs compared to control MEFs in response to DNA damage: Mt1, Cxcl1, Foxg1 and Fos). The Ingenuity program confirmed the existence in the Snai2-dependent transcriptome of six genes whose expression was altered in Snai2-deficient cells in response to DNA damage: Hspb1, Tgm2, Mt1, Cxcl1, Foxg1 and Fos (Figures 4C and 5C). Good concordance was found between array and qPCR data for the selected p53 target genes regulated by Snai2 (Figure 3E), suggesting an interesting link between these genes and Snai2. These p53 target genes modulated by Snai2 belong mainly to the following categories on the basis of the biological or pathological function: metastasis (Cxcl1); cell cycle/DNA/oxidative damage (Hspb1, Mt1, Fos); survival (Tgm2), and cell development (Foxg1b). These results indicate that Snai2 regulates a limited set of p53 target genes in MEFs in response to DNA damage. Moreover, the presence of Snai2 DNA-binding sites (Inukai et al, 1999) in the promoter regions of these p53 target genes modulated by Snai2 (Figure 5D) suggests Snai2 could be directly involved in the control of transcription-repression of these targets.
DISCUSSION
In this study, we have applied a functional genomics approach that combined gene expression profiling and computational molecular network analysis to obtain global dissection of the Snai2-dependent transcriptional response to DNA damage and to dissect the contribution of Snai2 on p53 target genes. As a model system, we used primary MEFs. Mouse embryonic fibroblasts represent an ideal cell system in which the activities of p53 can be studied. When treated with DNA-damaging agents, wild-type MEFs activate the cell cycle checkpoint by arresting in G1 (Attardi et al, 2000). This response is clearly p53 dependent as p53-null MEFs fail to undergo G1 arrest upon DNA damage treatment. Majority of approaches used to identify the role of p53-responsive genes such as p21 have typically entailed comparing gene-expression profiles of cell lines lacking p53 with cell lines overexpressing p53 (El-Deiry et al, 1993; Okamoto and Beach, 1994; Buckbinder et al, 1995; Polyak et al, 1996). Another advantage of the strategy used here is that it relies on the response of endogenous cellular p53 to DNA damage and is performed using primary cells, making it very physiological.
Before using this approach to identify Snai2 target genes linked to Snai2 gene function in response to DNA damage, we demonstrated that normal G1 arrest and normal p53 activation took place in Snai2-deficient MEFs in response to doxorubicin treatment. These observations indicated that p53 does not require Snai2 to activate the cell cycle checkpoint by arresting in G1. Moreover, these results indicate Snai2 does not require p53 for its DNA damage protective function in MEFs in agreement with previous data in haematopoietic precursors (Perez-Losada et al, 2002; Perez-Mancera et al, 2005; Wu et al, 2005). To confirm that our approach could be used to study the role of the p53-responsive gene Snai2 in DNA damage response, we next examined whether known target genes were modulated in control and Snai2-deficient MEFs after DNA doxorubicin treatment. Expression of these genes was modulated as expected in control MEFs and their expression was not altered in Snai2-deficient cells in response to doxorubicin treatment. These results further confirm the approach used is physiologically relevant.
In addition to the elucidation of the genetic program of Snai2 and the identification of transcriptionally regulated protein networks, one of the main results of this work is the observation that p53 triggers similar transcriptomal programs in Snai2-deficient cells in response to DNA damage. However, these results do not rule out that the functional specificity of Snai2 may be established outside the transcriptional program or, alternatively, that it may require additional regulatory components and/or specific cell backgrounds. Further work in this area will be required to address these possibilities.
Another important observation of this work is the identification of a limited set of protein network (Atm, Bid and p27) and p53-target genes (Hspb1, Tgm2, Mt1, Cxcl1, Foxg1 and Fos) whose expression is regulated by Snai2 in response to DNA damage. The presence of Snai2 DNA-binding sites (Inukai et al, 1999) in the promoter regions of these target genes modulated by Snai2 suggests Snai2 could be directly involved in the control of transcription-repression of these targets. Two of these targets (Atm and Bid) had already been identified as Snai2 targets describing the resistance to doxorubicin treatment of Snai2-expressing MCF7 cells (Kajita et al, 2004). In addition, Snai2 was shown in that study to bind Bid promoter (Kajita et al, 2004). The zinc-finger protein Snai2 is considered a transcriptional repressor. In agreement with this idea, expression of Hspb1 and Tgm2 was increased in Snai2-deficient MEFs compared to control MEFs in response to DNA damage. However, majority of Snai2-target genes (Atm, Bid, p27, Mt1, Cxcl1, Foxg1 and Fos) were downregulated in Snai2-deficient MEFs in response to DNA damage, supporting the view that Snai2 can also behave as a positive transcriptional regulator or act by repressing the transcription of a repressor (Bermejo-Rodriguez et al, 2006). All these novel Snai2 targets have been implicated in DNA damage and survival regulation (Wang et al, 1991; Beattie et al, 1998; Park et al, 2000; Nanda et al, 2001; Katoh and Katoh, 2004; Minn et al, 2005). Thus, the regulation of these genes by Snai2 in response to DNA damage could be important in preserving integrity of tumour target cells and supports the view that failure to control Snai2 expression can produce cancer and alterations in development (Perez-Mancera et al, 2005; Perez-Mancera et al, 2006). These Snai2-target genes could represent novel pharmacological targets and/or biomarkers in cancers linked to Snai2 (Perez-Mancera et al, 2005; Bermejo-Rodriguez et al, 2006; Perez-Caro and Sanchez-Garcia, 2006).
The expression of the previously implicated Snai2-target gene Puma/bbc3 (Wu et al, 2005; Bermejo-Rodriguez et al, 2006) was not significantly modulated in Snai2-deficient cells in response to DNA damage, indicating that Puma/bbc3 regulation in response to DNA damage is not affected in Snai2-deficient MEFs. These results suggest that p53 does not require Snai2 for Puma/bbc3 regulation in response to DNA damage in MEFs, suggesting cellular context is of great importance for interpretation of Snai2 function. This tissue specificity of Snai2 is also supported by the fact that many Snai2-expressing lineages showed no obvious phenotypes in Snai2 mutant mice (Jiang et al, 1998; Perez-Losada et al, 2002; Sanchez-Martin et al, 2002, 2004), suggesting that Snai2 function in these cell types either is not required or can be compensated through synergy with other Snail family members. Moreover, this observation is in agreement with the known tissue specificity in the relative induction of several known p53 target genes (Burns et al, 2001; Burns and El-Deiry, 2003). Our results clearly show that the regulation of a significant number of p53 target genes in response to DNA damage in both control and Snai2-deficient MEFs differ from how they are regulated in the spleen and thymus in response to DNA damage (Burns and El-Deiry, 2003), confirming the striking cell specificity for p53-dependent transcription and repression.
In summary, these results have provided a comprehensive picture of the transcriptional events regulated by Snai2 during the DNA-damage process. From this analysis, we have obtained information regarding the Snai2-dependent molecular routes induced by DNA damage, defined the effect of Snai2 on p53 target genes, pointed to novel putative Snai2 targets, and suggested a mechanistic model. The emerging model suggests that targeting the Snai2-mediated arm could effectively increase the radiosensitivity of Snai2-dependent cancers. Given that our work has been focused on MEF cells, it would be interesting to expand these studies to other cell types in the future to get an idea of the level of conservation of the transcriptional programs of Snai2 in different tissues. Future studies will define if cancer maintaining cells (or cancer stem cells) keep constitutively active part of the original genetic programme controlled by Snai2 in MEFs in response to DNA damage.
Acknowledgments
We thank all members of both lab 13 at IBMCC and Molecular Pathology at CNIO for their helpful comments and constructive discussions on this project. We are grateful to Dr Tyler Jacks for various useful reagents and to Dr T Gridley for the Slug mutant mice. Research in our group is supported partially by FEDER and by MEC (SAF2006-03726 and PETRI no. 95-0913.OP), Junta de Castilla y León (CSI03A05), FIS (PI050087), Fundación de Investigación MMA, Federación de Cajas de Ahorro Castilla y León (I Convocatoria de Ayudas para Proyectos de Investigación Biosanitaria con Células Madre), and CDTEAM (CENIT-Ingenio 2010) project.
References
- Attardi LD, Reczek EE, Cosmas C, Demicco EG, McCurrach ME, Lowe SW, Jacks T (2000) PERP, an apoptosis-associated target of p53, is a novel member of the PMP-22/gas3 family. Genes Dev 14(6): 704–718 [PMC free article] [PubMed] [Google Scholar]
- Beattie JH, Wood AM, Newman AM, Bremner I, Choo KH, Michalska AE, Duncan JS, Trayhurn P (1998) Obesity and hyperleptinemia in metallothionein (-I and -II) null mice. Proc Natl Acad Sci USA 95(1): 358–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermejo-Rodriguez C, Perez-Caro M, Perez-Mancera PA, Sanchez-Beato M, Piris MA, Sanchez-Garcia I (2006) Mouse cDNA microarray analysis uncovers Slug targets in mouse embryonic fibroblasts. Genomics 87(1): 113–118 [DOI] [PubMed] [Google Scholar]
- Buckbinder L, Talbott R, Velasco-Miguel S, Takenaka I, Faha B, Seizinger BR, Kley N (1995) Induction of the growth inhibitor IGF-binding protein 3 by p53. Nature 377(6550): 646–649 [DOI] [PubMed] [Google Scholar]
- Burns TF, Bernhard EJ, El-Deiry WS (2001) Tissue specific expression of p53 target genes suggests a key role for KILLER/DR5 in p53-dependent apoptosis in vivo. Oncogene 20(34): 4601–4612 [DOI] [PubMed] [Google Scholar]
- Burns TF, El-Deiry WS (2003) Microarray analysis of p53 target gene expression patterns in the spleen and thymus in response to ionizing radiation. Cancer Biol Ther 2(4): 431–443 [DOI] [PubMed] [Google Scholar]
- Castellanos A, Pintado B, Weruaga E, Arevalo R, Lopez A, Orfao A, Sanchez-Garcia I (1997) A BCR-ABL(p190) fusion gene made by homologous recombination causes B-cell acute lymphoblastic leukemias in chimeric mice with independence of the endogenous bcr product. Blood 90(6): 2168–2174 [PubMed] [Google Scholar]
- Catalano A, Rodilossi S, Rippo MR, Caprari P, Procopio A (2004) Induction of stem cell factor/c-Kit/slug signal transduction in multidrug-resistant malignant mesothelioma cells. J Biol Chem 279: 46706–46714 [DOI] [PubMed] [Google Scholar]
- Come C, Magnino F, Bibeau F, De Santa Barbara P, Becker KF, Theillet C, Savagner P (2006) Snail and slug play distinct roles during breast carcinoma progression. Clin Cancer Res 12(18): 5395–5402 [DOI] [PubMed] [Google Scholar]
- El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75(4): 817–825 [DOI] [PubMed] [Google Scholar]
- Elloul S, Elstrand MB, Nesland JM, Trope CG, Kvalheim G, Goldberg I, Reich R, Davidson B (2005) Snail, Slug, and Smad-interacting protein 1 as novel parameters of disease aggressiveness in metastatic ovarian and breast carcinoma. Cancer 103(8): 1631–1643 [DOI] [PubMed] [Google Scholar]
- Gupta PB, Kuperwasser C, Brunet JP, Ramaswamy S, Kuo WL, Gray JW, Naber SP, Weinberg RA (2005) The melanocyte differentiation program predisposes to metastasis after neoplastic transformation. Nat Genet 37(10): 1047–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt S, Alsheich-Bartok O, Haupt Y (2006) Clues from worms: a Slug at Puma promotes the survival of blood progenitors. Cell Death Differ 13(6): 913–9155 [DOI] [PubMed] [Google Scholar]
- Inoue A, Seidel MG, Wu W, Kamizono S, Ferrando AA, Bronson RT, Iwasaki H, Akashi K, Morimoto A, Hitzler JK, Pestina TI, Jackson CW, Tanaka R, Chong MJ, McKinnon PJ, Inukai T, Grosveld GC, Look AT (2002) Slug, a highly conserved zinc finger transcriptional repressor, protects hematopoietic progenitor cells from radiation-induced apoptosis in vivo. Cancer Cell 2: 279–288 [DOI] [PubMed] [Google Scholar]
- Inukai T, Inoue A, Kurosawa H, Goi K, Shinjyo T, Ozawa K, Mao M, Inaba T, Look AT (1999) SLUG, a ces-1-related zinc finger transcription factor gene with antiapoptotic activity, is a downstream target of the E2A-HLF oncoprotein. Mol Cell 4(3): 343–352 [DOI] [PubMed] [Google Scholar]
- Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, McKinnon PJ, Cleveland JL, Zambetti GP (2003) Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 4(4): 321–328 [DOI] [PubMed] [Google Scholar]
- Jiang R, Lan Y, Norton CR, Sundberg JP, Gridley T (1998) The Slug gene is not essential for mesoderm or neural crest development in mice. Dev Biol 198(2): 277–285 [PubMed] [Google Scholar]
- Kajita M, McClinic KN, Wade PA (2004) Aberrant expression of the transcription factors snail and slug alters the response to genotoxic stress. Mol Cell Biol 24(17): 7559–7566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastan MB, Zhan Q, el-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace Jr AJ (1992) A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 71(4): 587–597 [DOI] [PubMed] [Google Scholar]
- Katoh M, Katoh M (2004) Human FOX gene family. Int J Oncol 25(5): 1495–1500 [PubMed] [Google Scholar]
- Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massague J (2005) Genes that mediate breast cancer metastasis to lung. Nature 436(7050): 518–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanda N, Iismaa SE, Owens WA, Husain A, Mackay F, Graham RM (2001) Targeted inactivation of Gh/tissue transglutaminase II. J Biol Chem 276(23): 20673–20678 [DOI] [PubMed] [Google Scholar]
- Okamoto K, Beach D (1994) G is a transcriptional target of the p53 tumor suppressor protein. EMBO J 13(20): 4816–4822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SH, Cho HN, Lee SJ, Kim TH, Lee Y, Park YM, Lee YJ, Cho CK, Yoo SY, Lee YS (2000) Hsp25-induced radioresistance is associated with reduction of death by apoptosis: involvement of Bcl2 and the cell cycle. Radiat Res 154(4): 421–428 [DOI] [PubMed] [Google Scholar]
- Perez-Caro M, Sanchez-Garcia I (2006) Killing time for cancer stem cells (CSC): discovery and development of selective CSC inhibitors. Curr Med Chem 13(15): 1719–1725 [DOI] [PubMed] [Google Scholar]
- Pérez-Caro M, Pérez-Mancera P, Voces F, Sánchez-García I (2005) Of man in mouse: modelling human cancer genotype-phenotype correlations in mice. Curr Genomics 6: 81–88 [Google Scholar]
- Perez-Losada J, Sanchez-Martin M, Perez-Caro M, Perez-Mancera PA, Sanchez-Garcia I (2003) The radioresistance biological function of the SCF/kit signaling pathway is mediated by the zinc-finger transcription factor Slug. Oncogene 22: 4205–4211 [DOI] [PubMed] [Google Scholar]
- Perez-Losada J, Sanchez-Martin M, Rodriguez-Garcia A, Sanchez ML, Orfao A, Flores T, Sánchez-Garcia I (2002) Zincfinger transcription factor Slug contributes to the function of the stem cell factor c-kit signaling pathway. Blood 100: 1274–1286 [PubMed] [Google Scholar]
- Perez-Mancera PA, Gonzalez-Herrero I, Maclean K, Turner AM, Yip MY, Sanchez-Martin M, Garcia JL, Robledo C, Flores T, Gutierrez-Adan A, Pintado B, Sanchez-Garcia I (2006) SLUG (SNAI2) overexpression in embryonic development. Cytogenet Genome Res 114(1): 24–29 [DOI] [PubMed] [Google Scholar]
- Perez-Mancera PA, Gonzalez-Herrero I, Perez-Caro M, Gutierrez-Cianca N, Flores T, Gutierrez-Adan A, Pintado B, Sanchez-Martin M, Sanchez-Garcia I (2005) SLUG in cancer development. Oncogene 24(19): 3073–3082 [DOI] [PubMed] [Google Scholar]
- Polyak K, Waldman T, He TC, Kinzler KW, Vogelstein B (1996) Genetic determinants of p53-induced apoptosis and growth arrest. Genes Dev 10(15): 1945–1952 [DOI] [PubMed] [Google Scholar]
- Sanchez-Garcia I 1997. Consequences of chromosomal abnormalities in tumor development. Annu Rev Genet 31: 429–453 [DOI] [PubMed] [Google Scholar]
- Sanchez-Martin M, Gonzalez-Herrero I, Sanchez-Garcia I (2004) SNAI2 (SNAIL HOMOLOG 2). Atlas Genet Cytogenet Oncol Haematol April 2004. URL: http://atlasgeneticsoncology.org/Genes/SNAI2ID453.html
- Sanchez-Martin M, Rodriguez-Garcia A, Perez-Losada J, Sagrera A, Read AP, Sanchez-Garcia I (2002) SLUG (SNAI2) deletions in patients with Waardenburg disease. Hum Mol Genet 11(25): 3231–3236 [DOI] [PubMed] [Google Scholar]
- Shih JY, Tsai MF, Chang TH, Chang YL, Yuan A, Yu CJ, Lin SB, Liou GY, Lee ML, Chen JJ, Hong TM, Yang SC, Su JL, Lee YC, Yang PC (2005) Transcription repressor slug promotes carcinoma invasion and predicts outcome of patients with lung adenocarcinoma. Clin Cancer Res 11(22): 8070–8078 [DOI] [PubMed] [Google Scholar]
- Wang H, Wang JY, Johnson LR, Scott RE (1991) Selective induction of c-jun and jun-B but not c-fos or c-myc during mitogenesis in SV40-transformed cells at the predifferentiation growth arrest state. Cell Growth Differ 2(12): 645–652 [PubMed] [Google Scholar]
- Wu WS, Heinrichs S, Xu D, Garrison SP, Zambetti GP, Adams JM, Look AT (2005) Slug antagonizes p53-mediated apoptosis of hematopoietic progenitors by repressing puma. Cell 123(4): 641–653 [DOI] [PubMed] [Google Scholar]