

Abstract
Cell division involves a cyclical biochemical process composed of several step-wise reactions that have to occur once per cell cycle. Dysregulation of cell division is a hallmark of all cancers. Genetic and epigenetic mechanisms frequently result in deranged expression and/or activity of cell-cycle proteins including the cyclins, cyclin-dependent kinases (Cdks), Cdk inhibitors and checkpoint control proteins. The critical nature of these proteins in cell cycling raises hope that targeting them may result in selective cytotoxicity and valuable anticancer activity.
Keywords: cell cycle, targeted therapy
STRATEGIES FOR TARGETING THE CELL CYCLE
Dysregulation of the cell cycle is a hallmark of malignancy (Table 1). Many different strategies for targeting the cell cycle have been described. Attention has focused primarily on targeting mitosis (by targeting tubulin and more recently mitotic kinases including KSP/Eg5, Polo-like kinase 1 and Aurora kinases) as well as chemical inhibitors of cyclin-dependent kinase (Cdk) catalytic activity. Other potential strategies include therapeutics that can inhibit the interaction between cyclins and Cdks; decrease cyclin expression; promote the degradation of cyclins by increasing their phosphorylation; and restore endogenous Cdk inhibitor function. Other possibilities include the inhibition of the ‘noncycling’ cyclin-activating kinase complex of Cdk7/cyclin H, and the indirect targeting of the late G2 to M checkpoint by inhibiting CDC25 or by activating WEE1 or MYT1. This review will, however, focus on the chemical Cdk inhibitors evaluated in clinical trials to date and will highlight the main trial results reported to date.
Table 1. (a) Deregulated cyclins and Cdk's and associated tumours and (b) deregulated endogenous Cdk inhibitors and associated tumours.
| Target | Oncogenic changes | Associated tumours |
|---|---|---|
| (a) | ||
| Cyclin D1 | Gene amplification | 40–80% Breast carcinoma |
| Overexpression | 70% Familial polyposis | |
| Translocation | 50% B-cell lymphoma | |
| 50% NSCLC | ||
| 35% Head and neck carcinoma | ||
| 25–50% Oesophageal carcinoma | ||
| 25% Bladder carcinoma | ||
| Cyclin E | Gene amplification | 90% Colorectal carcinoma |
| Overexpression | 30–80% Breast carcinoma | |
| 70% Prostate carcinoma | ||
| 18% Ovarian carcinoma | ||
| Gastric carcinoma | ||
| Cervical carcinoma | ||
| Cyclin E2 | Overexpression | Breast carcinoma |
| Small-cell lung carcinoma | ||
| Cervical carcinoma | ||
| Cyclin B1 | Overexpression | 90% Colorectal carcinoma |
| Cyclin A | Amplification or overexpression | Hepatocellular carcinoma |
| CDK2 | Overexpression | Colorectal carcinoma |
| CDK4 | Amplification | Sarcomas, gliomas |
| (b) | ||
| p16INK4a | Mutation (5% of human cancers) | Pancreatic cancer |
| Deletion (14% of human cancers) | Melanoma | |
| Epigenetic (19% of human cancers) | Gliomas | |
| Bladder cancer | ||
| Head and neck cancer | ||
| NSCLC | ||
| Lymphoma/leukaemia | ||
| p21cip−1/waf−1 | Mutation/deletion rare | Oral (rare mutations) |
| Downregulation rare | Oesophageal (rare mutations) | |
| Intracellular mislocalis ation? | Breast (rare mutations) | |
| p27kip−1 | Mutations/deletions rare | Breasta |
| Downregulation (increased degradation)a | Colona | |
| Prostatea | ||
NSCLC=non-small-cell lung cancer.
Increased degradation.
PHARMACOLOGIC CDK INHIBITORS
Considerable progress has been made in the identification of pharmacologic agents targeting the Cdks (Senderowicz, 2003). The first generation of Cdk inhibitors lacked specificity, with flavopiridol, staurosporine and its analogue UCN-01 and E7070 being nonselective inhibitors of not only Cdks but many other targets. Second-generation inhibitors are more selective, with many of these compounds specifically developed to target selected Cdks (Table 2).
Table 2. Chemical Cdk inhibitors and their targets.
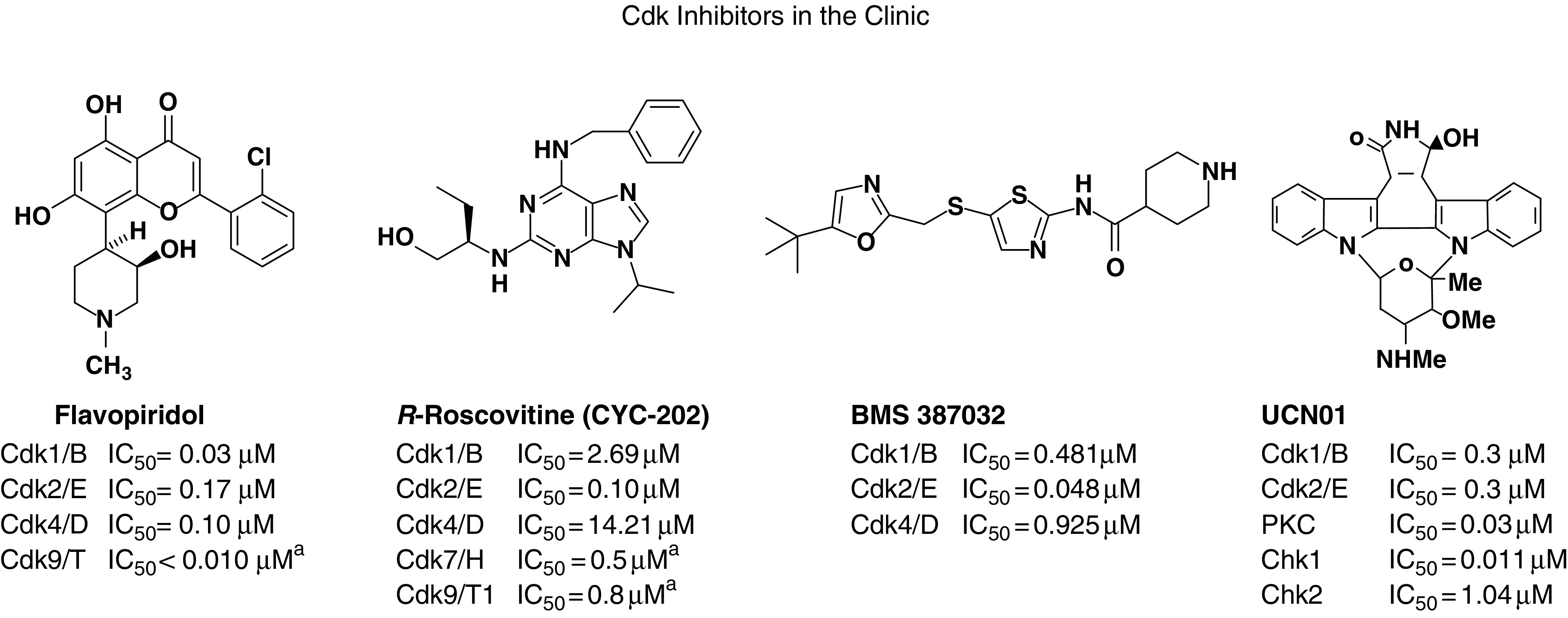
|
aIncreased degradation downregulate transcription, for example, ↓cyclin D1 expression.
Flavopiridol
Flavopiridol (Alvocidib™) has several mechanisms of anticancer activity (Senderowicz and Sausville, 2000) and is a broad-spectrum Cdk inhibitor targeting Cdks 1, 2, 4, 6 and 7, interacting with the adenosine triphosphate (ATP) binding site. Flavopiridol also inhibits the Cdk9/cyclin T complex, broadly repressing transcription and decreasing cyclin D1 mRNA expression (Carlson et al, 1999). It induces cell-cycle arrest in G1 and is cytotoxic to cells undergoing DNA synthesis. It also inhibits other kinases including PKC and PKA at higher concentrations, inducing apoptosis, (Patel et al, 1998) and is active in many xenograft models.
The first Phase I trial of flavopiridol utilised a 72-h continuous infusion every 2 weeks, a schedule supported by preclinical models (Senderowicz et al, 1998). The maximum-tolerated dose (MTD) achieved in this clinical trial was 50 mg m2 day−1, the initial dose-limiting toxicity being secretory diarrhoea. The MTD with antidiarrhoeal prophylaxis was 78 mg m2 day−1 (Kahn et al, 2001; Messmann et al, 2003) The achieved plasma levels were sufficient for Cdk inhibition (200–400 nM), with one partial response in renal cell cancer and minor responses in renal, colorectal carcinomas and non-Hodgkin's lymphoma.
This 72-h schedule was subsequently investigated in further Phase I and Phase II trials in patients with mantle cell lymphoma, renal cancer, melanoma, gastric cancer, non-small-cell lung cancer (NSCLC) and colorectal cancers (Bennett et al, 1999; Stadler et al, 2000; Schwartz et al, 2001; Shapiro et al, 2001; Lin et al, 2002; Thomas et al, 2002). An unusually high incidence of arterial and venous thromboembolic events was documented in the majority of these trials, including deep venous thrombosis, pulmonary thromboembolism, myocardial infarction and transient neurological ischaemic events. This serious drug-related toxicity was not appreciated in the initial studies but was reported in more than 30% of patients in later trials and is of unknown aetiology.
A study of flavopiridol administered as a 1-h infusion was then pursued supported by data indicating that higher plasma concentrations than those achieved in the 72-h infusion studies were required to induce apoptosis (Arguello et al, 1998; Tan et al, 2002). Initially, a 1-h infusion administered for 5 days every 3 weeks was investigated. The recommended Phase II dose for this schedule was 37.5 mg m2 day−1, with grade 4 neutropenia and grade 3 fatigue being dose limiting. The three- and one-day schedules administered every 3 weeks were also evaluated with the aim of further increasing peak serum levels of drug. Grade 4 neutropenia was also dose limiting in these studies, with the recommended Phase II dose of these schedules being 50 mg m2 day−1 and 62.5 mg m−2, respectively. The peak plasma concentrations, however, remained lower than the concentrations required in preclinical studies to induce apoptosis (5–7 μmol l−1), with no objective tumour responses being observed. Phase II studies of flavopiridol administered by 1-h infusion daily for 3 days every 3 weeks have been reported. No responses were observed in advanced melanoma (Burdette-Radoux et al, 2002); however, in patients with advanced mantle cell lymphoma, three responses out of 28 evaluable patients (11%) were reported, with 20 patients having stable disease (71%) for a median duration of 3.4 months (Kouroukis et al, 2003).
An alternative administration schedule has been recently evaluated for flavopiridol in patients with chronic lymphatic leukaemia. Previous Phase I/II studies had shown that, while flavopiridol induces apoptosis in CLL cells in a p53-independent manner in vitro, the drug is inactive using a 24- to 72-h CIVI schedule. Based on pharmacokinetic modelling data demonstrating high drug binding to human plasma proteins, an optimised dosing schedule of 30-min i.v. bolus (IVB) followed by 4-h CIVI has been pursued. This alternative schedule has demonstrated promising activity including flavopiridol-induced tumour lysis syndrome, suggesting that this agent may warrant further evaluation using this novel schedule (Lin et al, 2004).
The use of flavopiridol in combination studies has also been pursued (Bible and Kaufmann, 1997; Motwani et al, 2001). Treatment scheduling appears to be important in ensuring that flavopiridol augments the effects of other agents (Schwartz et al, 2002; Wittmann et al, 2003).
UCN-01
The second chemical Cdk inhibitor to be evaluated was UCN-01 (7-hydroxystaurosporine) (Senderowicz and Sausville, 2000), which also has several mechanisms of action including Cdk1 (cdc2) and Cdk2 inhibition (IC50 of 300–600 nM). UCN-01 causes cell-cycle arrest and apoptosis at concentrations above 100nM (Akiyama et al, 1997), abrogating the G2 checkpoint in response to DNA damage and promoting the induction of p53-independent apoptosis by inhibiting Chk1 and Chk2 (IC50 for both approximately 10–30 nM). (Wang et al, 1996; Busby et al, 2000; Yu et al, 2002). UCN-01 also abrogates S-phase arrest in CHO cells treated with cisplatin, promoting the induction of apoptosis (Bunch and Eastman, 1997), and may target Akt signalling by inhibiting PDK1 (IC50=33 nM) (Sato et al, 2002). Frequent dosing is required to optimise the antitumour activity of UCN-01 (Sausville et al, 1998), and 72 h of drug exposure is required to achieve growth inhibition. A Phase I trial of UCN-01 administered as a 72-h continuous infusion every 2 weeks to patients with advanced malignancy revealed long elimination half-lives due to high-affinity binding of UCN-01 to α1-acid glycoprotein in human plasma (Fuse et al, 1998; Sausville et al, 2001). This schedule was therefore changed to a 72-h continuous infusion administered every 4 weeks, with a recommended dose of 42.5 mg m2 day−1. Dose-limiting toxicities at 53 mg m−2 included hyperglycaemia, pulmonary toxicity with hypoxaemia, emesis and hypotension. The basis for the pulmonary toxicity was unclear but did not involve altered cardiac motility or pulmonary thromboembolism, being associated with small, transient, pleural effusions. Hyperglycaemia occurred at all dose levels. Analysis of immunoreactive C peptide levels suggested that this was related to peripheral tissue insulin resistance rather than a decrease in insulin secretion. Pharmacokinetic analysis revealed a terminal half-life of 588 h, with mean drug plasma concentrations in the micromolar range. Analysis of free salivary levels of UCN-01 showed drug levels of approximately 100 nm, which is sufficient to affect cell-cycle parameters in vitro. A partial response of 6 months duration was observed in patient with melanoma, and a complete response, sustained for over 38 months in a patient with anaplastic large-cell lymphoma.
E7070
E7070 is a chloroindolyl sulphonamide that induces G1/S cell cycle arrest at low nanomolar concentrations, inhibiting Cdk2/cyclin E, downregulating cyclin H, upregulating p53 and p21, and inducing apoptosis (Ozawa et al, 2001). Its potency is enhanced by longer drug exposures (Ozawa et al, 2001). E7070 has a broad spectrum of in vitro and in vivo preclinical antitumour activity, inducing regression of established tumours. This efficacy is schedule dependent, with a daily-for-8-days schedule being more efficacious than 4- and 1-day schedules.
A number of Phase I trials of this agent have been performed investigating different dosing regimens. When given daily by intravenous infusion for 5 days every 3 weeks, the DLTs consisted of neutropenia, thrombocytopenia, diarrhoea and stomatitis, with a recommended Phase II dose of 130 mg m2 day−1. One partial response was seen in a patient with heavily pretreated breast cancer (Punt et al, 2001). A second trial investigated an alternative schedule – a 1-h infusion administered every 3 weeks (Raymond et al, 2002). Toxicities were similar to the 5-day regimen and comprised myelosuppression, acne-like skin rash, alopecia, mucositis, conjunctivitis, hypoglycaemia, nausea and fatigue. Recommended doses were 700 mg m−2 for the heavily pretreated group and 800 mg m−2 for the lightly pretreated group. No partial responses were observed. A weekly 1-h infusion schedule for 4 consecutive weeks and a continuous 120-h infusion have also been evaluated with similar toxicities being observed. The recommended doses for the weekly and continuous infusion schedules were 400 and 96 mg m−2, respectively. Pharmacokinetic studies indicate that the clearance and volume of distribution at steady state of E7070 decrease with increasing dose.
Phase II single-agent studies are under way in a number of different tumour types investigating the once-every-3-weeks and daily-for-5-days schedules. In patients with fluorouracil refractory colorectal cancer, two of 21 patients receiving the once-every-4-weeks schedule, and two of 23 patients receiving the daily-for-5-days schedule, had objective responses, with 10 and 13% of patients, respectively, having stable disease at 6 months (Mainwaring et al, 2002). In a Phase II study of patients with NSCLC, who had previously received one chemotherapy regimen, only one of 44 patients had an objective response (Talbot et al, 2002). No objective responses were observed in patients with metastatic melanoma receiving a dose of 700 mg m−2 for over 1 h every 3 weeks (Aamdal et al, 2002).
R-Roscovitine (CYC202)
The purine analogue R-roscovitine (CYC202) is a highly selective, orally bioavailable, small molecule inhibitor of several Cdks competing with their ATP binding sites: it is a relatively potent inhibitor of human Cdk2/cyclin E, Cdk7/cyclin H, Cdk9/cyclin T1 with IC50 of 0.1, 0.5 and 0.8 μM, respectively (Meijer et al, 1997), but inhibits Cdk4/cyclin D1 with an IC50 of 14.2 μM. It has an average IC50 against the NCI cell-line panel of 16 μM. It also blocks the degradation of p53 through the inhibition of MDM2 expression (Lu et al, 2001). CYC202 induces G1 and G2/M arrest and cell death from all compartments of the cell cycle (Schutte et al, 1997; McClue et al, 2002). The antitumour efficacy of CYC202 has also been tested in a panel of human tumour xenografts (McClue et al, 2002). Continuous exposure to CYC202 at dosages ranging from 0.3 to 100 μM demonstrated dose-dependent antitumour activity.
Recent molecular pharmacology studies have shown that treatment of colorectal cancer cells with CYC202 results in a decrease in pRb phosphorylation (serines 780, 608, 807, 811 and Thr-821), indicative of direct Cdk2 inhibition (Whittaker et al, 2004). In addition, CYC202 causes a downregulation of various cyclins, including cyclin D1; this is likely to lead to a secondary inhibition of various cyclins, which would explain the reduced phosphorylation at multiple sites on RB that is seen at later time points.
CYC202 has been evaluated in Phase I clinical trials. Two such trials have recently been reported. In the first trial, CYC202 was administered orally, twice daily for 7 days out of every 21. In all, 19 patients were treated for a total of 36 cycles of CYC202. At 800 mg BD, DLTs comprising grade 3 skin rash and grade 4 hypokalaemia were observed. Other toxicities seen included reversible renal impairment, mild reversible transaminitis and emesis. MAG-3 studies indicated that the renal impairment was related to altered renal blood flow. No evidence of renal tubular damage was detected. The aetiology of these renal changes is unknown but could be related to the effects of CYC202 on adenosine receptors. The pharmacokinetics of the compound were dose proportional, with CYC202 being widely distributed (720.8 l, 95% CI 384.9–1056.8) and rapidly cleared (142.6 l/h, 95% CI 80.5–204.9) with a mean terminal elimination half-life of 4.02 h (95% CI 2.8–5.2). This trial is still recruiting patients, but no objective responses have yet been seen (Benson et al, 2003; White et al, 2004).
In the second trial, a twice-daily-for-5-days schedule, administered every 3 weeks was evaluated. The maximum twice-daily dose achieved was 1600 mg BD with a recommended dose of 1250 mg BD. Toxicities reported have included grade 4 emesis, grade 3 asthenia and skin rash. No objective responses were seen but stable disease was recorded in three out of 29 patients treated. (Pierga et al, 2003) Exploration of a 10-day schedule is now under way.
BMS-387032
High-throughput screening followed by lead optimisation has resulted in the identification of the 2-aminothiazole BMS-387032 as a potent, selective and competitive small molecule inhibitor of the Cdk2/cyclin E complex, with an IC50 of 48 nM. The 2-aminothiazoles have been reported to be 10- and 30-fold more potent against Cdk2 than Cdk1 and Cdk4, and three to five orders of magnitude less potent against all other tested non-Cdk kinases. The X-ray crystal structure of this compound with Cdk2 has been reported, and has revealed the mechanisms by which this compound interacts with the Cdk2 ATP binding site (Misra et al, 2004). In vitro, BMS-387032 inhibits Cdk2 phosphorylation in the A2780 ovarian carcinoma cell line, inhibiting the phosphorylation of downstream targets of Cdk2 including pRb, histone H1 and DNA polymerase-α.
The compound displays potent in vitro cytotoxicity against the A2780 cell line with an IC50 of 50 nM, and is active against a broad array of cell lines. In vivo studies have confirmed oral bioavailability and activity against a variety of cell lines, including P388 murine leukaemia, A2780 ovarian and A431 human squamous cell carcinoma. Combination studies indicate that BMS-387032 is synergistic with cisplatin in SV-1 colon carcinoma cells, this synergy being dependent on the drug sequence (Lane et al, 2003). Phase I clinical trials with BMS-387032 are ongoing utilising different schedules (Jones et al, 2003; McCormick et al, 2003; Shapiro et al, 2003).
PHARMACODYNAMIC STUDIES
As with other molecularly targeted therapeutics under investigation, the clinical development of the Cdk inhibitors in early trials requires the study of their biological effects in tumour cells acquired through serial tumour biopsies. These studies are required to select optimal biological dosing and schedule, and identify the patient population most likely to benefit from these agents. Potential pharmacodynamic parameters under investigation include the inhibition of pRb phosphorylation; the depletion of cyclins and Cdks; increases in Cdk inhibitor protein expression; and the suppression of mdm2 expression and induction of p53 expression. Pharmacodynamic studies have been reported for flavopiridol (Lam et al, 2001) and CYC202 (Whittaker et al, 2004). More recent studies indicate that 18F-labelled 3′-deoxy-3′fluorothymidine may be a useful imaging modality for the selective Cdk2 inhibitor BMS-387032 (Fischman et al, 2004). These translational studies are critically important in the optimal development of these agents.
CONCLUDING REMARKS
Significant progress has been made in the clinical targeting of the Cdks. Newer and more specific Cdk inhibitors are envisioned to result in decreased toxicity and more selective cytotoxicity. Increased specificity may not, however, spare noncycling cells since recent data have implicated the Cdks 5, 7, 8 and 9 in cellular functions that do not involve the cell cycle. Cdks 7, 8 and 9 have been reported to regulate RNA transcription through the phosphorylation of RNA polymerase II, while Cdk5 has been shown to be involved in regulating insulin secretion, synaptic vesicle recycling, neuronal survival and tau (microtubule associated protein) phosphorylation and aggregation (Sausville, 2002). These findings may explain why hyperglycaemia has been observed with many chemical Cdk inhibitors. The RNA polymerase II regulatory activity of Cdk 7, 8 and 9 may, however, enhance the antitumour effect of these agents, and it has indeed been suggested that the anticancer activity of flavopiridol may be in part related to the inhibition of RNA transcription. It remains to date unclear whether the inhibition of Cdk 7, 8 and 9 activity is desirable for an anticancer agent, or whether the toxicity associated with inhibiting these noncycling Cdks will substantially decrease cytotoxic selectivity and the therapeutic index.
Overall, these data suggest that highly selective small molecule inhibitors of specific Cdks may be preferable in order to decrease toxicity. Generating this specificity, however, not only remains a significant challenge to chemists but may also decrease anticancer efficacy in view of the inherent functional redundancy of this family of kinases. A broader-spectrum inhibitor that can, for example, selectively inhibit Cdk1, Cdk2, Cdk4 and Cdk6 at low nanomolar IC50 concentrations may therefore be preferable. The recent observations that selectively inhibiting Cdk2 in certain cell lines is not sufficient for antitumour activity would support this view (Tetsu and McCormick, 2003), as would the demonstration that the Cdk2 knockout mouse shows no major abnormalities and in particular no effects on proliferation (Ortega et al, 2003). The high sequence homology of the ATP binding sites of these and the noncycling kinases Cdk 5, 7, 8 and 9, as well as that of glycogen synthase kinase-3β and that of the adenosine receptors makes this a difficult task. However, much has already been achieved and while many questions need to be answered we are moving closer to the Holy Grail: the development of compounds selectively cytotoxic to tumour cells, yet sparing normal cells.
References
- Aamdal S, Smyth J, Awada A, Dittrich C, Caponigro F, Djurasinovic N, Marchal B, Yule M (2002) Phase II study of E7070 in patients with metastatic melanoma (stage IV). Eur J Cancer 38: S50. [DOI] [PubMed] [Google Scholar]
- Akiyama T, Yoshida T, Tsujita T, Shimizu M, Mizukami T, Okabe M, Akinaga S (1997) G1 phase accumulation induced by UCN-01 is associated with dephosphorylation of Rb and CDK2 proteins as well as induction of CDK inhibitor p21/Cip1/WAF1/Sdi1 in p53-mutated human epidermoid carcinoma A431 cells. Cancer Res 57: 1495–1501 [PubMed] [Google Scholar]
- Arguello F, Alexander M, Sterry JA, Tudor G, Smith EM, Kalavar NT, Greene Jr JF, Koss W, Morgan CD, Stinson SF, Siford TJ, Alvord WG, Klabansky RL, Sausville EA (1998) Flavopiridol induces apoptosis of normal lymphoid cells, causes immunosuppression, and has potent antitumor activity in vivo against human leukemia and lymphoma xenografts. Blood 91: 2482–2490 [PubMed] [Google Scholar]
- Bennett P, Mani S, O'Reilly S, Wright J, Schilsky RL, Vokes EE, Grochow L (1999) Phase II trial of flavopiridol in metastatic colorectal cancer: preliminary results. Proc Am Soc Clin Oncol 18: 277 [Google Scholar]
- Benson C, White J, Twelves C, O'Donnell A, Cruickshank C, Tan S, Gianella-Borradori A, Judson I (2003) A phase I trial of the oral cyclin dependent kinase inhibitor CYC202 in patients with advanced malignancy. Proc Am Soc Clin Oncol 22: 209 [Google Scholar]
- Bible KC, Kaufmann SH (1997) Cytotoxic synergy between flavopiridol (NSC 649890, L86-8275) and various antineoplastic agents: the importance of sequence of administration. Cancer Res 57: 3375–3380 [PubMed] [Google Scholar]
- Bunch RT, Eastman A (1997) 7-Hydroxystaurosporine (UCN-01) causes redistribution of proliferating cell nuclear antigen and abrogates cisplatin-induced S-phase arrest in Chinese hamster ovary cells. Cell Growth Differ 8: 779–788 [PubMed] [Google Scholar]
- Burdette-Radoux S, Tozer R, Lohmann R, Quirt I, Ernst D, Walsh W, Wainman N, Colevas D, Eisenhauer E (2002) A. NCIC CTG phase II study of flavopiridol in patients with previously untreated metastatic malignant melanoma. Proc Am Soc Clin Oncol 21: 346 [Google Scholar]
- Busby EC, Leistritz DF, Abraham RT, Karnitz LM, Sarkaria JN (2000) The radiosensitizing agent 7-hydroxystaurosporine (UCN-01) inhibits the DNA damage checkpoint kinase hChk1. Cancer Res 60: 2108–2118 [PubMed] [Google Scholar]
- Carlson B, Lahusen T, Singh S, Loaiza-Perez A, Worland PJ, Pestell R, Albanese C, Sausville EA, Senderowicz AM (1999) Down-regulation of cyclin D1 by transcriptional repression in MCF-7 human breast carcinoma cells induced by flavopiridol. Cancer Res 59: 4634–4641 [PubMed] [Google Scholar]
- Fischman A, Letrent S, Bonab A, Livni E, Carter E, Rubin R, Mauro D, Tarby C, Galbraith S, Griffin T (2004) PET as a biomarker of the antitumor effects of the CDK2 inhibitor BMS-387032. Proc Am Soc Clin Onc 23: 135 [Google Scholar]
- Fuse E, Tanii H, Kurata N, Kobayashi H, Shimada Y, Tamura T, Sasaki Y, Tanigawara Y, Lush RD, Headlee D, Figg WD, Arbuck SG, Senderowicz AM, Sausville EA, Akinaga S, Kuwabara T, Kobayashi S (1998) Unpredicted clinical pharmacology of UCN-01 caused by specific binding to human alpha1-acid glycoprotein. Cancer Res 58: 3248–3253 [PubMed] [Google Scholar]
- Jones S, Burris H, Kies M, Willcutt N, Degen P, Woo M, Letrent S, Youssoufian H, DeCiliis A, Papadimtrakopoulou V (2003) A Phase I study to determine the safety and pharmacokinetics (PK) of BMS-387032 given intravenously every three weeks in patients with metastatic refractory solid tumors. Proc Am Soc Clin Onco 22: 199 [Google Scholar]
- Kahn ME, Senderowicz A, Sausville EA, Barrett KE (2001) Possible mechanisms of diarrheal side effects associated with the use of a novel chemotherapeutic agent, flavopiridol. Clin Cancer Res 7: 343–349 [PubMed] [Google Scholar]
- Kouroukis CT, Belch A, Crump M, Eisenhauer EA, Gascoyne RD, Meyer R, Lohmann R, Lopez P, Powers J, Turner R, Connors JM (2003) Flavopiridol in untreated or relapsed mantle-cell lymphoma: results of a phase II study of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 21: 1740–1745 [DOI] [PubMed] [Google Scholar]
- Lam LT, Pickeral OK, Peng AC, Rosenwald A, Hurt EM, Giltnane JM, Averett LM, Zhao H, Davis RE, Sathyamoorthy M, Wahl LM, Harris ED, Mikovits JA, Monks AP, Hollingshead MG, Sausville EA, Staudt LM (2001) Genomic-scale measurement of mRNA turnover and the mechanisms of action of the anti-cancer drug flavopiridol. Genome Biol 2(10): 41.1–41.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane M, Temple K, Yu B, Nguyen H, Wadler S (2003) Combined treatment with cisplatin and a novel cdk2 specific inhibitor (BMS 387032), results in cytotoxic synergy in human colon carcinoma cells that is dependent on dose, sequence and cyclin D1 expression.a. Proc Am Assoc Can Res 44: 161 [Google Scholar]
- Lin TS, Dalton JT, Wu D, Fischer B, Moran M, Lucas D, Cunningham K, Colevas AD, Grever MR, Byrd JC (2004) Flavopiridol given as a 30-min intravenous (IV) bolus followed by 4-h continuous IV infusion (CIVI) results in clinical activity and tumor lysis in refractory chronic lymphocytic leukemia (CLL). J Clin Oncol ASCO Annual Meeting Proceedings (Post-Meeting Edition). Vol 22, No 14S (July 15 Supplement), 2004: 6564
- Lin TS, Howard OM, Neuberg DS, Kim HH, Shipp MA (2002) Seventy-two hour continuous infusion flavopiridol in relapsed and refractory mantle cell lymphoma. Leukemia Lymphoma 43: 793–797 [DOI] [PubMed] [Google Scholar]
- Lu W, Chen L, Peng Y, Chen J (2001) Activation of p53 by roscovitine-mediated suppression of MDM2 expression. Oncogene 20: 3206–3216 [DOI] [PubMed] [Google Scholar]
- Mainwaring P, Van Cutsem E, Van Laetham J-L, Propper D, Twelves C, Bridgewater J, Audhuy B, Carmichael J, Punt C, Cassidy J, Stuart N, Ravic M (2002) A multicentre randomised phase II study of E7070 in patients with colorectal cancer who have failed 5-fluorouracil-based chemotherapy. Proc Am Soc Clin Oncol 21: 153 [Google Scholar]
- McClue SJ, Blake D, Clarke R, Cowan A, Cummings L, Fischer PM, MacKenzie M, Melville J, Stewart K, Wang S, Zhelev N, Zheleva D, Lane DP (2002) In vitro and in vivo antitumor properties of the cyclin dependent kinase inhibitor CYC202 (R-roscovitine). Int J Cancer 102: 463–468 [DOI] [PubMed] [Google Scholar]
- McCormick J, Gadgeel S, Helmke W, Chaplen R, van Leeuwen B, Woo M, Youssoufian H, DeCiliis A, Letrent S, LoRusso P (2003) Phase I study of BMS-387032, a cyclin dependent kinase (CDK) 2 inhibitor. Proc Am Soc Clin Oncol 22: 208 [Google Scholar]
- Meijer L, Borgne A, Mulner O, Chong JP, Blow JJ, Inagaki N, Inagaki M, Delcros JG, Moulinoux JP (1997) Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur J Biochem 243: 527–536 [DOI] [PubMed] [Google Scholar]
- Messmann RA, Ullmann CD, Lahusen T, Kalehua A, Wasfy J, Melillo G, Ding I, Headlee D, Figg WD, Sausville EA, Senderowicz AM (2003) Flavopiridol-related proinflammatory syndrome is associated with induction of interleukin-6. Clin Cancer Res 9: 562–570 [PubMed] [Google Scholar]
- Misra RN, Xiao HY, Kim KS, Lu S, Han WC, Barbosa SA, Hunt JT, Rawlins DB, Shan W, Ahmed SZ, Qian L, Chen BC, Zhao R, Bednarz MS, Kellar KA, Mulheron JG, Batorsky R, Roongta U, Kamath A, Marathe P, Ranadive SA, Sack JS, Tokarski JS, Pavletich NP, Lee FY, Webster KR, Kimball SD (2004) N-(cycloalkylamino)acyl-2-aminothiazole inhibitors of cyclin-dependent kinase 2. N-[5-[[[5-(1,1-dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4-piperidinecarboxamide (BMS-387032), a highly efficacious and selective antitumor agent. J Med Chem 47(7): 1719–1728 [DOI] [PubMed] [Google Scholar]
- Motwani M, Jung C, Sirotnak FM, She Y, Shah MA, Gonen M, Schwartz GK (2001) Augmentation of apoptosis and tumor regression by flavopiridol in the presence of CPT-11 in Hct116 colon cancer monolayers and xenografts. Clin Cancer Res 7: 4209–4219 [PubMed] [Google Scholar]
- Ortega S, Prieto I, Odajima J, Martin A, Dubus P, Sotillo R, Barbero JL, Malumbres M, Barbacid M (2003) Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat Genet 35(1): 25–31 [DOI] [PubMed] [Google Scholar]
- Ozawa Y, Sugi NH, Nagasu T, Owa T, Watanabe T, Koyanagi N, Yoshino H, Kitoh K, Yoshimatsu K (2001) E7070, a novel sulphonamide agent with potent antitumour activity in vitro and in vivo. Eur J Cancer 37: 2275–2282 [DOI] [PubMed] [Google Scholar]
- Patel V, Senderowicz AM, Pinto Jr D, Igishi T, Raffeld M, Quintanilla-Martinez L, Ensley JF, Sausville EA, Gutkind JS (1998) Flavopiridol, a novel cyclin-dependent kinase inhibitor, suppresses the growth of head and neck squamous cell carcinomas by inducing apoptosis. J Clin Invest 102: 1674–1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierga J, Faivre S, Vera K, Laurence V, Delbaldo C, Bekradda M, Armand JP, Gianella-Borradori A, Dieras V, Raymond E (2003) A phase I and pharmacokinetic (PK) trial of CYC202, a novel oral cyclin-dependent kinase (CDK) inhibitor, in patients (pts) with advanced solid tumors. Proc Am Soc Clin Oncol 22: 210 [Google Scholar]
- Punt CJ, Fumoleau P, van de WB, Faber MN, Ravic M, Campone M (2001) Phase I and pharmacokinetic study of E7070, a novel sulfonamide, given at a daily times five schedule in patients with solid tumors. A study by the EORTC-Early Clinical Studies Group (ECSG). Ann Oncol 12: 1289–1293 [DOI] [PubMed] [Google Scholar]
- Raymond E, Bokkel Huinink WW, Taieb J, Beijnen JH, Faivre S, Wanders J, Ravic M, Fumoleau P, Armand JP, Schellens JH (2002) Phase I and pharmacokinetic study of E7070, a novel chloroindolyl sulfonamide cell-cycle inhibitor, administered as a one-hour infusion every three weeks in patients with advanced cancer. J Clin Oncol 20: 3508–3521 [DOI] [PubMed] [Google Scholar]
- Sato S, Fujita N, Tsuruo T (2002) Interference with PDK1-Akt survival signaling pathway by UCN-01 (7-hydroxystaurospine). Oncogene 7(21): 1727–1738 [DOI] [PubMed] [Google Scholar]
- Sausville EA (2002) Complexities in the development of cyclin-dependent kinase inhibitor drugs. Trends Mol Med 8(4 Suppl): S32–S37, Review [DOI] [PubMed] [Google Scholar]
- Sausville EA, Arbuck SG, Messmann R, Headlee D, Bauer KS, Lush RM, Murgo A, Figg WD, Lahusen T, Jaken S, Jing X, Roberge M, Fuse E, Kuwabara T, Senderowicz AM (2001) Phase I trial of 72-h continuous infusion UCN-01 in patients with refractory neoplasms. J Clin Oncol 19: 2319–2333 [DOI] [PubMed] [Google Scholar]
- Sausville EA, Lush RD, Headlee D, Smith AC, Figg WD, Arbuck SG, Senderowicz AM, Fuse E, Tanii H, Kuwabara T, Kobayashi S (1998) Clinical pharmacology of UCN-01: initial observations and comparison to preclinical models. Cancer Chemother Pharmacol 42(Suppl): S54–S59 [DOI] [PubMed] [Google Scholar]
- Schutte B, Nieland L, van Engeland M, Henfling ME, Meijer L, Ramaekers FC (1997) The effect of the cyclin-dependent kinase inhibitor olomoucine on cell cycle kinetics. Exp Cell Res 236: 4–15 [DOI] [PubMed] [Google Scholar]
- Schwartz GK, Ilson D, Saltz L, O'Reilly E, Tong W, Maslak P, Werner J, Perkins P, Stoltz M, Kelsen D (2001) Phase II study of the cyclin-dependent kinase inhibitor flavopiridol administered to patients with advanced gastric carcinoma. J Clin Oncol 19: 1985–1992 [DOI] [PubMed] [Google Scholar]
- Schwartz GK, O'Reilly E, Ilson D, Saltz L, Sharma S, Tong W, Maslak P, Stoltz M, Eden L, Perkins P, Endres S, Barazzoul J, Spriggs D, Kelsen D (2002) Phase I study of the cyclin-dependent kinase inhibitor flavopiridol in combination with paclitaxel in patients with advanced solid tumors. J Clin Oncol 20: 2157–2170 [DOI] [PubMed] [Google Scholar]
- Senderowicz AM (2003) Small-molecule cyclin-dependent kinase modulators. Oncogene 22: 6609–6620 [DOI] [PubMed] [Google Scholar]
- Senderowicz AM, Headlee D, Stinson SF, Lush RM, Kalil N, Villalba L, Hill K, Steinberg SM, Figg WD, Tompkins A, Arbuck SG, Sausville EA (1998) Phase I trial of continuous infusion flavopiridol, a novel cyclin-dependent kinase inhibitor, in patients with refractory neoplasms. J Clin Oncol 16: 2986–2999 [DOI] [PubMed] [Google Scholar]
- Senderowicz AM, Sausville EA (2000) Preclinical and clinical development of cyclin-dependent kinase modulators. J Natl Cancer Inst 92: 376–387 [DOI] [PubMed] [Google Scholar]
- Shapiro G, Lewis N, van Leeuwen B, Letrent S, Woo M, Youssoufian H, DeCiliis A, Cohen R (2003) A Phase I study to determine the safety and pharmacokinetics (PK0 of BMS-387032 with a 24-h infusion given every three weeks in patients with metastatic refractory solid tumors. Proc Am Soc Clin Oncol 22: 199 [Google Scholar]
- Shapiro GI, Supko JG, Patterson A, Lynch C, Lucca J, Zacarola PF, Muzikansky A, Wright JJ, Lynch Jr TJ, Rollins BJ (2001) A phase II trial of the cyclin-dependent kinase inhibitor flavopiridol in patients with previously untreated stage IV non-small cell lung cancer. Clin Cancer Res 7: 1590–1599 [PubMed] [Google Scholar]
- Stadler WM, Vogelzang NJ, Amato R, Sosman J, Taber D, Liebowitz D, Vokes EE (2000) Flavopiridol, a novel cyclin-dependent kinase inhibitor, in metastatic renal cancer: a University of Chicago Phase II Consortium Study. J Clin Oncol 18: 371–375 [DOI] [PubMed] [Google Scholar]
- Talbot D, Norbury C, Slade M, von Pawel J, Bosquee L, Gatzemeier U, Ravic M (2002) A phase II and pharmacodynamic study of E7070 in patients with non-small cell lung cancer (NSCLC) who have failed platinum-based chemotherapy. Proc Am Soc Clin Oncol 21: 327 [Google Scholar]
- Tan AR, Headlee D, Messmann R, Sausville EA, Arbuck SG, Murgo AJ, Melillo G, Zhai S, Figg WD, Swain SM, Senderowicz AM (2002) Phase I clinical and pharmacokinetic study of flavopiridol administered as a daily 1-h infusion in patients with advanced neoplasms. J Clin Oncol 20: 4074–4082 [DOI] [PubMed] [Google Scholar]
- Tetsu O, McCormick F (2003) Proliferation of cancer cells despite CDK2 inhibition. Cancer Cell 3(3): 233–245 [DOI] [PubMed] [Google Scholar]
- Thomas JP, Tutsch KD, Cleary JF, Bailey HH, Arzoomanian R, Alberti D, Simon K, Feierabend C, Binger K, Marnocha R, Dresen A, Wilding G (2002) Phase I clinical and pharmacokinetic trial of the cyclin-dependent kinase inhibitor flavopiridol. Cancer Chemother Pharmacol 50: 465–472 [DOI] [PubMed] [Google Scholar]
- Wang Q, Fan S, Eastman A, Worland PJ, Sausville EA, O'Connor PM (1996) UCN-01: a potent abrogator of G2 checkpoint function in cancer cells with disrupted p53. J Natl Cancer Inst 88: 956–965 [DOI] [PubMed] [Google Scholar]
- White JD, Cassidy J, Tweleves C, Benson C, Pacey S, Judson I, McGrath H, Rose F, Frenz L (2004) Phase I trial of the oral cyclin dependent kinase inhibitor CYC202 in patients with advanced malignancy. Proc Am Soc Clin Oncol 23: 205 [Google Scholar]
- Whittaker SR, Walton MI, Garrett MD, Workman P (2004) The cyclin-dependent kinase inhibitor CYC202 (R-roscovitine) inhibits RB phosphorylation, causes loss of cyclin D1 and activates the mitogen activated protein kinase pathway. Cancer Res 1(64): 262–272 [DOI] [PubMed] [Google Scholar]
- Wittmann S, Bali P, Donapaty S, Nimmanapalli R, Guo F, Yamaguchi H, Huang M, Jove R, Wang HG, Bhalla K (2003) Flavopiridol down-regulates antiapoptotic proteins and sensitizes human breast cancer cells to epothilone B-induced apoptosis. Cancer Res 63: 93–99 [PubMed] [Google Scholar]
- Yu Q, La Rose J, Zhang H, Takemura H, Kohn KW, Pommier Y (2002) UCN-01 inhibits p53 up-regulation and abrogates gamma-radiation-induced G (2)-M checkpoint independently of p53 by targeting both of the checkpoint kinases, Chk2 and Chk1. Cancer Res 62(20): 5743–5748 [PubMed] [Google Scholar]