

Abstract
Systemic virus dissemination is a potential problem during local gene delivery in solid tumours. However, the kinetics and pathways of the dissemination have not been well characterised during the first 24 h after the infusion is started. To this end, we infused adenoviral vectors for luciferase or enhanced green fluorescence protein into three different tumour models in mice. During and/or after the infusion, we determined the amount of adenoviruses in the tumour, blood, and liver, and examined the transgene expression in the liver, lung, blood, and tumour. In addition, we intravenously injected tumour cells expressing luciferase and examined the biodistribution of these cells in the body. We observed transgene expression in the liver and tumour at 24 h after the infusion, but could not detect transgene expression in the blood and lung. The peak concentration of viral vectors in the plasma occurred during the intratumoral infusion. At 10 min after the infusion, few viral vectors remained in the blood and the ratio of copy numbers of adenoviruses between liver and tumour was >2 in 80% and ⩾10 in 40% of the mice. Most tumour cells injected intravenously accumulated in the lung within the first 24 h. Taken together, these data indicated that systemic virus dissemination occurred mainly during the first 10 min after the intratumoral infusion was started, and that the dissemination was due to infusion-induced convective transport of viral vectors into leaky tumour microvessels.
Keywords: gene delivery, virus dissemination, intratumoral infusion
Gene therapy represents a new paradigm to treat cancer at the genetic level (Ledley, 1996; Anderson, 1998). Its success relies on the ability to deliver sufficient genetic materials into target cells. The most widely used vectors for gene delivery are genetically engineered viruses, because they, in the long-term evolution of nature, are equipped with specific machineries that facilitate DNA transport into the nucleus of cells (Baranowski et al, 2001). However, a main drawback of viral vectors is the toxicity caused by undesirable acute immune response to viral proteins (Brown and Lillicrap, 2002; Bowers et al, 2003; Chen et al, 2003; Jooss and Chirmule, 2003; Lowenstein, 2003). Moreover, proteins from transgene expression may also exert adverse effects on normal cells either directly or indirectly through conversion of prodrugs to cytotoxic agents (Freeman et al, 1993; Rosenfeld et al, 1995; Alvarez and Curiel, 1997; Pope et al, 1997; Chada et al, 2003). Both toxicity and efficacy of gene therapy increase with the dose of treatment. Therefore, the goal of research in gene delivery is to improve the efficacy/toxicity ratio (Crystal, 1995; Alemany et al, 2000a).
The improvement can be achieved through increasing the specificity of either vector delivery or transgene expression in tumour cells. One way to increase the delivery specificity is to physicochemically or genetically modify viral surface proteins so that the vectors will target specific biomarkers in tumours (Douglas et al, 1996; Hall et al, 1997). However, this approach has its limitations because such biomarkers may exist in normal tissues and distribute heterogeneously in tumours. To reduce transgene expression in normal tissues, unique promoters have been integrated into viral vectors. These promoters can be activated only by specific stimuli in tumours that are either endogenous or applied externally (Vaupel et al, 1989; Gossen and Bujard, 1992; Garver et al, 1994; Harris et al, 1994; Hallahan et al, 1995; Kumagai et al, 1996; Jaggar et al, 1997; Jenster et al, 1997; Blackburn et al, 1998; Chung et al, 1999; Abdul-Ghani et al, 2000; Huang et al, 2000; Koshikawa et al, 2000; Pollock et al, 2000; Spitz et al, 2000). However, the control of transgene expression will not reduce the acute toxicity in normal tissues caused by viral proteins.
Intratumoral infusion is another strategy for improving the efficacy/toxicity ratio in viral gene therapy. It has been widely used in cancer clinical trials. Researchers often assume that viral vectors and transgene expression are confined to solid tumours after the infusion, thereby causing minimal toxicity in normal tissues (Lu et al, 1999; Paielli et al, 2000). On the other hand, researchers in some studies have observed that transgene expression in normal tissues can be as strong as that in tumours (Toloza et al, 1996; Bramson et al, 1997; Nasu et al, 1999; Tjuvajev et al, 1999; Lohr et al, 2001; Wang et al, 2003), that adenoviral vectors are present in systemic circulation of patients in clinical trials (DeWeese et al, 2001; Nemunaitis et al, 2001), and that some animals die within a short period after intratumoral infusion (Toloza et al, 1996; Tjuvajev et al, 1999). These observations indicate that viral vectors can escape from tumours during and/or after intratumoral infusion. Thus, the improvement in efficacy/toxicity ratio via the intratumoral infusion is likely to be far from optimal.
Although virus dissemination has been observed in preclinical and clinical studies, its kinetics and pathways are still controversial. Therefore, we characterised the biodistributions of adenoviral vectors and transgene expression in the tumour, liver, blood, and lung at different time points after the infusion was started. We observed that the systemic virus dissemination occurred mainly within the first 10 min. The amount of viruses disseminated into the liver could be one order of magnitude higher than that retained in the tumour, and the mechanism of dissemination was the infusion-induced convective transport of viral vectors into leaky tumour microvessels.
MATERIALS AND METHODS
Adenoviral vectors
The Ad5-based recombinant system was used to produce the nonreplicating adenoviruses: AdCMVEGFP and AdCMVLuc encoding enhanced green fluorescence protein (EGFP) and luciferase, respectively. The cDNA for EGFP or luciferase was inserted into the E1 region of the adenovirus and the transgene expression was driven by the cytomegalovirus (CMV) promoter. Adenoviruses were propagated in 293 cells (ATCC, Manassas, VA, USA), harvested at 48 h after infection, and purified by the method of cesium chloride gradient centrifugation according to a standard protocol (Graham and Prevec, 1995). The viral vectors were stored in 10% glycerol at −82°C.
Tumour cell lines
Three tumour cell lines (4T1, B16.F10, and IMR-23) were used in the study. 4T1 is a murine mammary carcinoma; B16.F10 is a metastatic subline of B16 murine melanoma; IMR-23 is a human neuroblastoma. All tumour cells were cultured in DMEM supplemented with 10% newborn bovine serum (Hyclone, Logan, UT, USA) and 1% penicillin/streptomycin (Gibco/Life Technologies, Grand Island, NY, USA) at 37°C with 5% CO2 and 95% air. For tumour implantation in mice, the cells were removed from culture flasks with 0.25% trypsin/EDTA and rinsed twice in PBS.
Animals
Female Balb/c and C57BL/6 mice, 4–6 weeks old, were ordered from Charles River Laboratory (Wilmington, MA, USA) for the implantation of 4T1 and B16.F10 tumours, respectively. Female Balb/c nude mice at the same age as Balb/c mice were ordered from the National Cancer Institute (Bethesda, MD, USA) for the implantation of IMR-23 tumour. For tumour cell implantation, mice were first anesthetised with an intraperitoneal injection (0.15 ml) of a mixture of ketamine and xylazine (80 mg ketamine and 10 mg xylazine per kg body weight). Then, one million tumour cells in 50 μl PBS were injected subcutaneously into the right hind limb of mice. After implantation, the mice were placed on a warm pad until they woke up. All subcutaneous tumours were ready for experiments when they reached 5–8 mm in diameter. After experiments, fully anaesthetised mice were killed with cervical dislocation. The use of animals and all experimental procedures involving animals in this study had been approved by the Duke University Institutional Animal Care and Use Committee. These procedures were also consistent with the United Kingdom Co-ordinating Committee on Cancer Research (UKCCCR) Guidelines (UKCCCR, 1998).
Intratumoral infusion
Mice were anesthetised with an intraperitoneal injection (0.15 ml) of a mixture of ketamine and xylazine (80 mg ketamine and 10 mg xylazine per kg body weight). The tip of a 30-gauge needle was carefully placed near the centre of tumours by controlling the depth of needle insertion relative to the tumour size measured by a caliper. Virus suspension was infused into tumours via the needle mounted on a syringe pump (model 22, Harvard Apparatus Co., Cambridge, MA, USA). The volume of infusion was fixed at 50 μl although the dose of viruses varied with experiments as indicated below. The infusion rate was chosen to be 1 μl s−1 based on a preliminary study in which we infused 50 μl of a solution of Evans blue dye or a suspension of an adenoviral vector for LacZ, AdCMVLacZ, into 4T1 tumours and observed that the distribution volume of Evans blue or β-galactosidase in tumours was the highest when the infusion rate was 1 μl s−1.
Luciferase and EGFP expression in different organs/tissues
At 24 h after intratumoral infusion of AdCMVLuc or AdCMVEGFP (2.0 × 108 plaque forming units (pfu)), mice were anesthetised again with the same method as described above and transgene expression in tumours was examined. For luciferase analysis, 50 μl of aqueous D-luciferin solution was injected intraperitoneally into the mice placed on a warm pad at 20 min before the examination of bioluminescence in the body. The bioluminescence was recorded by the In Vivo Imaging System (IVIS) (Xenogen Corp., Alameda, CA, USA), with an exposure time of 30 s (Wang et al, 2003) and the bioluminescence intensity was represented by pseudo-colours as indicated by a colour bar. The final images were generated by superimposing the pseudo-colour bioluminescence images on the reference images. For EGFP analysis, tissue samples from the blood, lung, liver, and tumour were harvested. Except the blood, tissue samples were sectioned into thin slices (300 μm thick), using a Vibratome (Model 3000; Technical Products International, St Louis, MO, USA). Enhanced green fluorescence protein expression in the blood and sliced tissues was examined under a confocal laser-scanning microscope (LSM 510, Carl Zeiss, Thornwood, NY, USA).
In a separate experiment, we injected 106 luciferase-expressing 4T1 cells in a 100-μl suspension into the tail vein of Balb/c mice anesthetised with the same method as described above. At both 10 min and 24 h after the cell injection, luciferase expression in the body was examined using the same method as described above.
Qualitative analysis of adenoviruses in the blood
At predetermined time points (25 s, 50 s, 5 min, 10 min, 2, and 24 h) after starting the infusion of AdCMVEGFP (3.0 × 108 pfu) into 4T1 tumours, blood samples (approximately 20 μl in each sample) from anesthetised mice were collected through the orbital sinus with a heparinised capillary, stored at room temperature for 10 min, and centrifuged at 10 000 g for 3 min to separate the plasma from blood cells. The plasma and cell suspension were then exposed to four cycles of thawing (37°C) and freezing (−196°C in liquid nitrogen) treatment. In all, 1 μl of plasma solution or suspension of lysed blood cells was added into 96-well plates with 70–80% confluent 293 cells. After 24 h, EGFP expression in these cells was examined under a fluorescence microscope (Axiovert 100, Carl Zeiss).
Quantitative analysis of adenovirus dissemination in liver and tumours
At 10 min after the infusion of AdCMVEGFP (3.0 × 108 pfu) into 4T1 tumors, mice were killed with cervical dislocation and the liver and tumours were immediately harvested, frozen in liquid nitrogen, and stored at −82°C. For viral DNA analysis, the tissue samples were immersed in liquid nitrogen and ground into fine tissue powder in a mortar. Viral DNA was isolated from the tissue powder using a DNeasy Tissue kit (Qiagen, Clarita, CA, USA). The DNeasy mini column was eluted twice with 200 μl of Tris buffer (10 mM, pH=8.5) to obtain viral DNA solution, in which Ad E4 copy number was measured with the Smart Cycler System (Cepheid, Sunnyvale, CA, USA). In the real-time PCR analysis, the primers and probes were purchased from Sigma-Genosis (Woodlands, TX USA). The forward primer sequence for Ad E4 region was: 5′-TGACACGCATACTCGGAGCTA-3′: 34 885–34 905; the reverse primer sequence was: 5′-TTTGAGCAGCACCTTGCATT-3′: 34 977–34 958; the fluorogenic probe sequence was: [DFAM]CGCCGCCCATGCAACAAGCTT[DBH1]: 34 930–34 951 (Adachi et al, 2001). Each PCR reaction was performed in duplicate in a 25-μl reaction mixture containing 0.15 μl of AmpliTaq Gold DNA polymerase (5 U μl−1) (Applied Biosystems, Foster City, CA USA), 300 nM each primer, and 200 nM fluorogenic probe. The amplification consisted of an initial heating at 95°C for 10 min, followed by 40 cycles at 95°C for 15 s and 60°C for 45 s. The standard curve was generated by using serial dilutions of pFG140 plasmid DNA (Microbix Biosystems Inc., Ontario, Canada) in the genomic liver DNA solution.
RESULTS
Systemic AdCMVLuc dissemination from tumours
To demonstrate the systemic dissemination of viral vectors, AdCMVLuc suspension was infused into two murine tumour models, 4T1 and B16.F10, respectively, and one human tumour xenograft, IMR-23. Bleeding was observed in many tumours immediately after the removal of infusion needle but the amount of bleeding was much less than the infused volume (i.e. 50 μl). At 24 h after infusion, strong bioluminescence, an indication of luciferase expression, was observed in the liver and tumour in all animal models (see Figure 1). The bioluminescence from the liver indicated that AdCMVLuc had escaped from tumours in local gene delivery, because luciferase is an exogenous and nonsecretable protein. It should be retained in transfected, viable cells.
Figure 1.

Typical images of luciferase distribution at 24 h after AdCMVLuc infusion into different tumours: (A) 4T1 in a Balb/c mouse, (B) B16.F10 in a C57BL/6 mouse, and (C) IMR-23 in a Balb/c nude mouse. The dose of infusion was 2.0 × 108 pfu tumour−1.
Kinetics of adenovirus dissemination
The plasma from blood samples at different time points, both during and after the intratumoral infusion of AdCMVEGFP, contained the viral vectors that could transfect 293 cells in vitro (see Figure 2A). Qualitatively, the amount of AdCMVEGFP in the plasma reached the peak at the end of intratumoral infusion and then decreased quickly with time. It was significantly lower than the peak value at 10 min and nearly undetectable at 2 h after the infusion. Cells from blood samples contained only a few AdCMVEGFP that could transfect 293 cells in vitro (see Figure 2B), indicating that the virus dissemination was not mediated by transfected tumour or blood-borne cells. To our best knowledge, this was the first direct evidence that demonstrated the adenovirus dissemination during intratumoral infusion.
Figure 2.
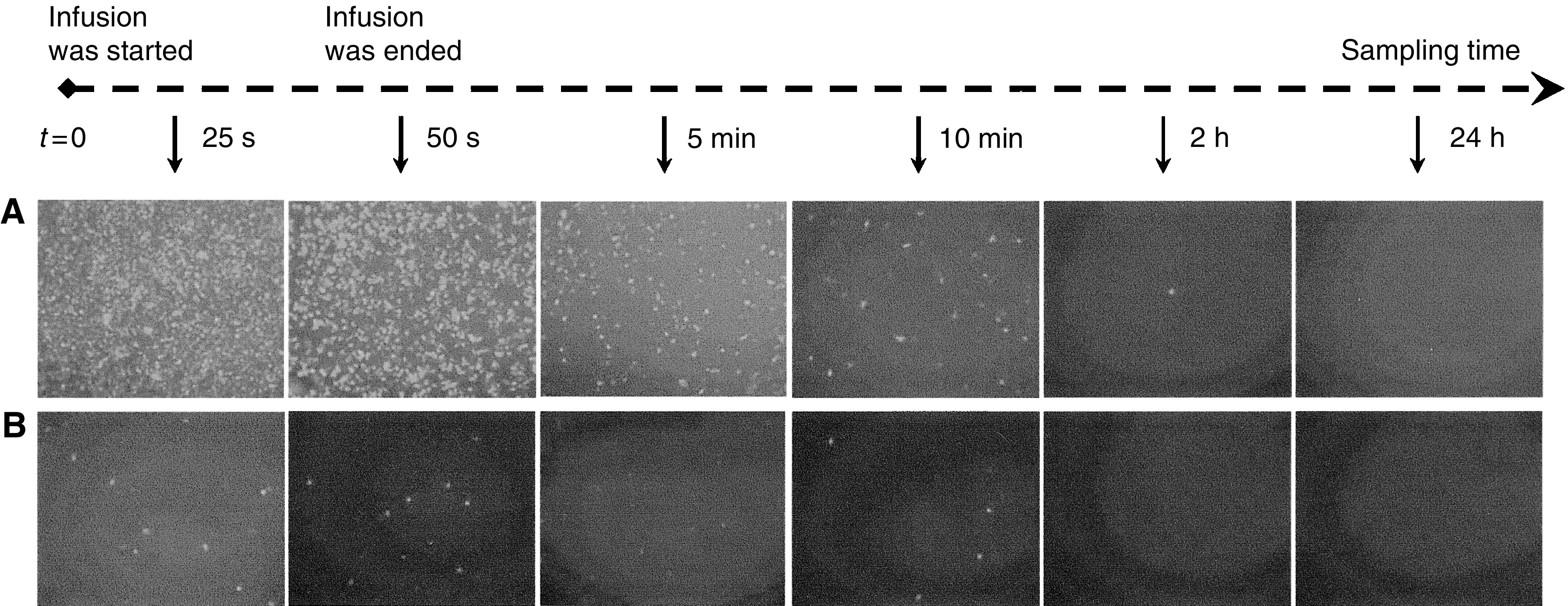
EGFP expression in 293 cells transfected by AdCMVEGFP that was isolated from (A) the plasma and (B) the lysed blood cells in mice. The blood samples were collected at different time points after the intratumoral infusion of AdCMVEGFP was started. The dose of infusion was 3.0 × 108 pfu tumour−1. The infusion was performed at 1 μl s−1 over a 50-s period.
Role of tumour or blood-borne cells in virus dissemination
To further investigate the role of tumour or blood-borne cells in virus dissemination, we performed two additional experiments. Firstly, we infused luciferase-expressing cells into the tail vein of mice, and observed that nearly all infused cells accumulated in the lung (Figure 3). Secondly, we infused AdCMVEGFP vectors into 4T1 tumours and examined EGFP expression in the liver, tumour, lung, and blood at 24 h after the infusion. We observed EGFP expression in liver and tumour tissues but could not detect EGFP-expressing cells in the lung and blood (Figure 4). These results confirmed it again that the virus dissemination was not mediated by transfected tumour or blood-borne cells.
Figure 3.

Typical images of luciferase distribution in three mice at (A) 10 min and (B) 24 h after intravenous injection of 4T1 cells expressing luciferase. The dose of injection was 106 cells per mouse.
Figure 4.

Typical images of EGFP distribution in different tissues at 24 h after the infusion of AdCMVEGFP into 4T1 tumours. Bar=100 μm. The dose of infusion was 3.0 × 108 pfu tumour−1.
Quantitative measurement of adenoviruses in liver and tumour tissues
To quantitatively understand the significance of systemic adenovirus dissemination at early time points, we quantified the copy numbers of adenoviruses in liver and tumour tissues at 10 min after the intratumoral infusion of AdCMVEGFP. We chose this time point for three reasons. Firstly, data in the literature show that adenoviruses have a plasma half-life of ∼2 min (Worgall et al, 1997; Alemany et al, 2000b) (see also Figure 2A in this study). Thus, approximately 97% of disseminated viruses have been cleared from the blood at 10 min. Secondly, the clearance in mice is mainly through the liver (Herz and Gerard, 1993; Hackett et al, 2000). Finally, the amount of active adenoviral vectors in the liver decreases with time (Worgall et al, 1997). As a result, the copy numbers of adenoviruses in the liver at 10 min after the infusion provided the best estimate of the total amount of adenoviruses disseminated from the tumour.
The ratio of the copy numbers of adenoviruses between liver and tumour is shown in Figure 5. The minimal value was 0.2; eight out of 10 mice had a ratio greater than 2; and four mice had a ratio close to or greater than 10. These data demonstrated that a significant amount of adenoviruses had disseminated from tumours within 10 min after the intratumoral infusion.
Figure 5.

Ratio of virus copy numbers between liver and tumour at 10 min after the infusion of AdCMVEGFP into 4T1 tumours implanted in 10 different mice.
DISCUSSION
We observed the systemic virus dissemination in all three animal models after intratumoral infusion of adenoviral vectors. In 4T1 tumours, the dissemination occurred mainly during the first 10 min after the infusion was started. The amount of viruses disseminated into the liver could be one order of magnitude higher than that retained in the tumour. The blood and lung in mice contained few transfected cells after the intratumoral infusion. However, tumour cells injected intravenously into the mice accumulated mainly in the lung. These observations in combination with data in the literature allowed us to determine the mechanism of systemic virus dissemination during the first 24 h after the intratumoral infusion was started.
Potentially, the mechanisms of dissemination include cell-mediated transport and diffusion and convection of viral vectors from the interstitial space into the lumen of tumour microvessels. We have showed previously that the diffusion process is too slow to cause the virus dissemination after intratumoral infusion (Wang et al, 2003). Based on diffusion, the maximum amount of disseminated viruses would be several orders of magnitude less than that retained in the tumour. This was inconsistent with the observations shown in Figures 1, 2, 4 and 5 and data in the literature (DeWeese et al, 2001; Nemunaitis et al, 2001; Sauthoff et al, 2003). Therefore, diffusion is negligible during the virus dissemination.
The cell-mediated transport was likely to be insignificant as well. The reasons are as follows. First, the amount of adenoviral vectors associated with cells in blood samples shown in Figure 2B was extremely low compared with that in the plasma. Even the cell-associated vectors could be originally in the plasma and then nonspecifically bind to erythrocytes (Cichon et al, 2003) or be co-precipitated with blood cells during the centrifuging process. Second, most tumour cells injected intravenously accumulated in the lung (Figure 3), which was consistent with data in the literature that lung is the primary target of metastatic tumour cells in mice (Lee, 1983). Third, few transfected cells existed in the blood and lung at 24 h after the intratumoral infusion (Figure 4). Therefore, the observations shown in Figure 2B indicated that blood-borne cells played an insignificant role in the virus dissemination, while the observations shown in Figures 3 and 4 indicated that the virus dissemination was not mediated by transfected tumour cells either.
Convection is in general negligible in the centre of solid tumours, due to uniformly elevated interstitial fluid pressure and intrinsic leakiness of tumour microvessels (Jain, 1997). Convection is important only during the intratumoral infusion and the first few minutes after the intratumoral infusion when a fluid pressure gradient is created around the tip of the infusion needle. After the infusion, the pressure gradient should decrease rapidly with time since (a) the equilibrium time constant between the microvascular pressure and the interstitial fluid pressure is approximately 10 s (Netti et al, 1995) and (b) the microvascular pressure in the tumour should return to the undisturbed level within a few minutes after the infusion, due to blood circulation. Therefore, convection can only cause virus dissemination at early time points but becomes insignificant at late time points.
Taken together, the virus dissemination occurred after the adenoviral vectors entered the system circulation via infusion-induced convective transport. The entrances were the leaky microvessels that were either intrinsically hyperpermeable, with pores in the microvessel wall being larger than adenoviruses (Hobbs et al, 1998; Yuan, 1998) or damaged by the infusion procedures. In the blood, adenoviruses were rapidly taken up by Kupffer cells and hepatocytes in the liver, with a plasma half-life of ∼2 min (Alemany et al, 2000b). The dissemination of the adenoviral vectors was significant only during the first few minutes (<10 min) after the intratumoral infusion.
The ratio of copy numbers of adenoviruses between liver and tumour shown in Figure 5 had a large variation among different animals. This result was qualitatively consistent with the ratio of bioluminescence intensities between liver and tumour after intratumoral infusion of AdCMVLuc (data not shown). Such variations were unlikely to be caused by the experimental procedures because the tip of the needle had been carefully placed near the centre of all tumours and the infusion protocol had been standardised by using a syringe pump. The variation could be due to the heterogeneous distribution of microvessels, a hallmark of solid tumours. As a result, the number of leaky microvessels involved in virus dissemination would depend on vascularisation and location of the needle tip in tumours.
Systemic virus dissemination after intratumoral infusion has been investigated in previous studies (DeWeese et al, 2001; Nemunaitis et al, 2001; Sauthoff et al, 2003). Sauthoff et al (2003) have detected a self-replicating adenoviral vector in mouse blood at 10 min after intratumoral infusion, based on the assay of virus titration on 293 cells. The number of viral DNA copies in the blood is one order of magnitude lower than that retained in the tumour and no adenoviruses can be detected in the liver during the first week after the intratumoral infusion. The liver data are inconsistent with those in the literature, which have demonstrated that nearly 99% of adenoviruses in the systemic circulation will eventually accumulate in the liver of mice (Herz and Gerard, 1993; Hackett et al, 2000). This discrepancy is likely to be caused by the inappropriate use of virus titration assay. It is well known that most adenoviruses in the liver are internalised by Kupffer cells or hepatocytes. The internalisation and the subsequent intracellular trafficking of adenoviruses may make these vectors lose their ability to transfect 293 cells. Therefore, a better way to determine the virus accumulation in the liver is to quantify the copy number of unique viral DNA sequences as shown in this study. Sauthoff et al (2003) have also shown that the adenoviral vectors can be detected in the blood at 2–8 weeks after the intratumoral infusion, presumably due to the replication of vectors in tumours. We used nonreplicating vectors so that we did not observe virus dissemination at late time points (Wang et al, 2003).
In a clinical study, the amount of adenoviral DNA in the blood at 30 min after the intratumoral infusion has varied between 0.007 and 0.129% of that infused into tumours (DeWeese et al, 2001). The amount of adenoviruses disseminated into the liver or other normal tissues is not determined in patients. Based on the blood data alone, one cannot estimate the amount of disseminated adenoviruses because it is likely that most adenoviruses disseminated during the intratumoral infusion have already been taken up by the liver at 30 min (Alemany et al, 2000b) (see also Figures 2 and 5).
In summary, we demonstrated that the virus dissemination occurred during the first few minutes after the intratumoral infusion was started. It was due to infusion-induced convective transport of viral vectors into leaky tumour microvessels. In future studies, we will investigate effects of infusion rate and pressure as well as structures of tumour tissues on virus dissemination. The findings in this study suggest that it is desirable to develop polymeric delivery systems that can increase the concentration of adenoviruses in solid tumours and simultaneously reduce the amount of disseminated adenoviruses in normal tissues, particularly in the liver, thereby improving the efficacy/toxicity ratio in cancer gene therapy.
Acknowledgments
We thank Drs David Zaharoff and Sheng Tong for the scientific discussion, and Drs Jun Chen and Lori Setton for the help in PCR experiment. This work is supported in part by grants from the National Science Foundation (BES-9984062) and the National Institutes of Health (CA81512).
References
- Abdul-Ghani R, Ohana P, Matouk I, Ayesh S, Ayesh B, Laster M, Bibi O, Giladi H, Molnar-Kimber K, Sughayer MA, de Groot N, Hochberg A (2000) Use of transcriptional regulatory sequences of telomerase (hTER and hTERT) for selective killing of cancer cells. Mol Ther 2: 539–544 [DOI] [PubMed] [Google Scholar]
- Adachi Y, Reynolds PN, Yamamoto M, Wang M, Takayama K, Matsubara S, Muramatsu T, Curiel DT (2001) A midkine promoter-based conditionally replicative adenovirus for treatment of pediatric solid tumors and bone marrow tumor purging. Cancer Res 61: 7882–7888 [PubMed] [Google Scholar]
- Alemany R, Balague C, Curiel DT (2000a) Replicative adenoviruses for cancer therapy. Nat Biotechnol 18: 723–727 [DOI] [PubMed] [Google Scholar]
- Alemany R, Suzuki K, Curiel DT (2000b) Blood clearance rates of adenovirus type 5 in mice. J Gen Virol 81: 2605–2609 [DOI] [PubMed] [Google Scholar]
- Alvarez RD, Curiel DT (1997) A phase I study of recombinant adenovirus vector-mediated intraperitoneal delivery of herpes simplex virus thymidine kinase (HSV-TK) gene and intravenous ganciclovir for previously treated ovarian and extraovarian cancer patients. Hum Gene Ther 8: 597–613 [DOI] [PubMed] [Google Scholar]
- Anderson WF (1998) Human gene therapy. Nature 392: 25–30 [DOI] [PubMed] [Google Scholar]
- Baranowski E, Ruiz-Jarabo CM, Domingo E (2001) Evolution of cell recognition by viruses. Science 292: 1102–1105 [DOI] [PubMed] [Google Scholar]
- Blackburn RV, Galoforo SS, Corry PM, Lee YJ (1998) Adenoviral-mediated transfer of a heat-inducible double suicide gene into prostate carcinoma cells. Cancer Res 58: 1358–1362 [PubMed] [Google Scholar]
- Bowers WJ, Olschowka JA, Federoff HJ (2003) Immune responses to replication-defective HSV-1 type vectors within the CNS: implications for gene therapy. Gene Ther 10: 941–945 [DOI] [PubMed] [Google Scholar]
- Bramson JL, Hitt M, Gauldie J, Graham FL (1997) Pre-existing immunity to adenovirus does not prevent tumor regression following intratumoral administration of a vector expressing IL-12 but inhibits virus dissemination. Gene Ther 4: 1069–1076 [DOI] [PubMed] [Google Scholar]
- Brown BD, Lillicrap D (2002) Dangerous liaisons: the role of ‘danger’ signals in the immune response to gene therapy. Blood 100: 1133–1140 [DOI] [PubMed] [Google Scholar]
- Chada S, Ramesh R, Mhashilkar AM (2003) Cytokine- and chemokine-based gene therapy for cancer. Curr Opin Mol Ther 5: 463–474 [PubMed] [Google Scholar]
- Chen D, Murphy B, Sung R, Bromberg JS (2003) Adaptive and innate immune responses to gene transfer vectors: role of cytokines and chemokines in vector function. Gene Ther 10: 991–998 [DOI] [PubMed] [Google Scholar]
- Chung I, Schwartz PE, Crystal RG, Pizzorno G, Leavitt J, Deisseroth AB (1999) Use of L-plastin promoter to develop an adenoviral system that confers transgene expression in ovarian cancer cells but not in normal mesothelial cells. Cancer Gene Ther 6: 99–106 [DOI] [PubMed] [Google Scholar]
- Cichon G, Boeckh-Herwig S, Kuemin D, Hoffmann C, Schmidt HH, Wehnes E, Haensch W, Schneider U, Eckhardt U, Burger R, Pring-Akerblom P (2003) Titer determination of Ad5 in blood: a cautionary note. Gene Ther 10: 1012–1017 [DOI] [PubMed] [Google Scholar]
- Crystal RG (1995) Transfer of genes to humans: early lessons and obstacles to success. Science 270: 404–410 [DOI] [PubMed] [Google Scholar]
- DeWeese TL, van der Poel H, Li S, Mikhak B, Drew R, Goemann M, Hamper U, DeJong R, Detorie N, Rodriguez R, Haulk T, DeMarzo AM, Piantadosi S, Yu DC, Chen Y, Henderson DR, Carducci MA, Nelson WG, Simons JW (2001) A phase I trial of CV706, a replication-competent, PSA selective oncolytic adenovirus, for the treatment of locally recurrent prostate cancer following radiation therapy. Cancer Res 61: 7464–7472 [PubMed] [Google Scholar]
- Douglas JT, Rogers BE, Rosenfeld ME, Michael SI, Feng M, Curiel DT (1996) Targeted gene delivery by tropism-modified adenoviral vectors. Nat Biotechnol 14: 1574–1578 [DOI] [PubMed] [Google Scholar]
- Freeman SM, Abboud CN, Whartenby KA, Packman CH, Koeplin DS, Moolten FL, Abraham GN (1993) The ‘bystander effect’: tumor regression when a fraction of the tumor mass is genetically modified. Cancer Res 53: 5274–5283 [PubMed] [Google Scholar]
- Garver Jr RI, Goldsmith KT, Rodu B, Hu PC, Sorscher EJ, Curiel DT (1994) Strategy for achieving selective killing of carcinomas. Gene Ther 1: 46–50 [PubMed] [Google Scholar]
- Gossen M, Bujard H (1992) Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA 89: 5547–5551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham FL, Prevec L (1995) Methods for construction of adenovirus vectors. Mol Biotechnol 3: 207–220 [DOI] [PubMed] [Google Scholar]
- Hackett NR, El Sawy T, Lee LY, Silva I, O'Leary J, Rosengart TK, Crystal RG (2000) Use of quantitative TaqMan real-time PCR to track the time-dependent distribution of gene transfer vectors in vivo. Mol Ther 2: 649–656 [DOI] [PubMed] [Google Scholar]
- Hall FL, Gordon EM, Wu L, Zhu NL, Skotzko MJ, Starnes VA, Anderson WF (1997) Targeting retroviral vectors to vascular lesions by genetic engineering of the MoMLV gp70 envelope protein. Hum Gene Ther 8: 2183–2192 [DOI] [PubMed] [Google Scholar]
- Hallahan DE, Mauceri HJ, Seung LP, Dunphy EJ, Wayne JD, Hanna NN, Toledano A, Hellman S, Kufe DW, Weichselbaum RR (1995) Spatial and temporal control of gene therapy using ionizing radiation. Nat Med 1: 786–791 [DOI] [PubMed] [Google Scholar]
- Harris JD, Gutierrez AA, Hurst HC, Sikora K, Lemoine NR (1994) Gene therapy for cancer using tumour-specific prodrug activation. Gene Ther 1: 170–175 [PubMed] [Google Scholar]
- Herz J, Gerard RD (1993) Adenovirus-mediated transfer of low density lipoprotein receptor gene acutely accelerates cholesterol clearance in normal mice. Proc Natl Acad Sci USA 90: 2812–2816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs SK, Monsky W, Yuan F, Roberts WG, Griffith L, Torchillin VP, Jain RK (1998) Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Nat Acad Sci USA 95: 4607–4612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q, Hu JK, Lohr F, Zhang L, Braun R, Lanzen J, Little JB, Dewhirst MW, Li CY (2000) Heat-induced gene expression as a novel targeted cancer gene therapy strategy. Cancer Res 60: 3435–3439 [PubMed] [Google Scholar]
- Jaggar RT, Chan HY, Harris AL, Bicknell R (1997) Endothelial cell-specific expression of tumor necrosis factor-alpha from the KDR or E-selectin promoters following retroviral delivery. Hum Gene Ther 8: 2239–2247 [DOI] [PubMed] [Google Scholar]
- Jain RK (1997) Delivery of molecular and cellular medicine to solid tumors. Microcirculation 4: 1–23 [DOI] [PubMed] [Google Scholar]
- Jenster G, Spencer TE, Burcin MM, Tsai SY, Tsai MJ, O'Malley BW (1997) Steroid receptor induction of gene transcription: a two-step model. Proc Natl Acad Sci USA 94: 7879–7884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jooss K, Chirmule N (2003) Immunity to adenovirus and adeno-associated viral vectors: implications for gene therapy. Gene Ther 10: 955–963 [DOI] [PubMed] [Google Scholar]
- Koshikawa N, Takenaga K, Tagawa M, Sakiyama S (2000) Therapeutic efficacy of the suicide gene driven by the promoter of vascular endothelial growth factor gene against hypoxic tumor cells. Cancer Res 60: 2936–2941 [PubMed] [Google Scholar]
- Kumagai T, Tanio Y, Osaki T, Hosoe S, Tachibana I, Ueno K, Kijima T, Horai T, Kishimoto T (1996) Eradication of Myc-overexpressing small cell lung cancer cells transfected with herpes simplex virus thymidine kinase gene containing Myc-Max response elements. Cancer Res 56: 354–358 [PubMed] [Google Scholar]
- Ledley FD (1996) Pharmaceutical approach to somatic gene therapy. Pharm Res 13: 1595–1614 [DOI] [PubMed] [Google Scholar]
- Lee YT (1983) Breast carcinoma: pattern of metastasis at autopsy. J Surg Oncol 23: 175–180 [DOI] [PubMed] [Google Scholar]
- Lohr F, Huang Q, Hu K, Dewhirst MW, Li CY (2001) Systemic vector leakage and transgene expression by intratumorally injected recombinant adenovirus vectors. Clin Cancer Res 7: 3625–3628 [PubMed] [Google Scholar]
- Lowenstein PR (2003) Virology and immunology of gene therapy, or virology and immunology of high MOI infection with defective viruses. Gene Ther 10: 933–934 [DOI] [PubMed] [Google Scholar]
- Lu Y, Carraher J, Zhang Y, Armstrong J, Lerner J, Rogers WP, Steiner MS (1999) Delivery of adenoviral vectors to the prostate for gene therapy. Cancer Gene Ther 6: 64–72 [DOI] [PubMed] [Google Scholar]
- Nasu Y, Bangma CH, Hull GW, Lee HM, Hu J, Wang J, McCurdy MA, Shimura S, Yang G, Timme TL, Thompson TC (1999) Adenovirus-mediated interleukin-12 gene therapy for prostate cancer: suppression of orthotopic tumor growth and pre-established lung metastases in an orthotopic model. Gene Ther 6: 338–349 [DOI] [PubMed] [Google Scholar]
- Nemunaitis J, Khuri F, Ganly I, Arseneau J, Posner M, Vokes E, Kuhn J, McCarty T, Landers S, Blackburn A, Romel L, Randlev B, Kaye S, Kirn D (2001) Phase II trial of intratumoral administration of ONYX-015, a replication-selective adenovirus, in patients with refractory head and neck cancer. J Clin Oncol 19: 289–298 [DOI] [PubMed] [Google Scholar]
- Netti PA, Baxter LT, Boucher Y, Skalak R, Jain RK (1995) Time-dependent behavior of interstitial fluid pressure in solid tumors: implications for drug delivery. Cancer Res 55: 5451–5458 [PubMed] [Google Scholar]
- Paielli DL, Wing MS, Rogulski KR, Gilbert JD, Kolozsvary A, Kim JH, Hughes J, Schnell M, Thompson T, Freytag SO (2000) Evaluation of the biodistribution, persistence, toxicity, and potential of germ-line transmission of a replication-competent human adenovirus following intraprostatic administration in the mouse. Mol Ther 1: 263–274 [DOI] [PubMed] [Google Scholar]
- Pollock R, Issner R, Zoller K, Natesan S, Rivera VM, Clackson T (2000) Delivery of a stringent dimerizer-regulated gene expression system in a single retroviral vector. Proc Natl Acad Sci USA 97: 13221–13226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope IM, Poston GJ, Kinsella AR (1997) The role of the bystander effect in suicide gene therapy. Eur J Cancer 33: 1005–1016 [DOI] [PubMed] [Google Scholar]
- Rosenfeld ME, Feng M, Michael SI, Siegal GP, Alvarez RD, Curiel DT (1995) Adenoviral-mediated delivery of the herpes simplex virus thymidine kinase gene selectively sensitizes human ovarian carcinoma cells to ganciclovir. Clin Cancer Res 1: 1571–1580 [PubMed] [Google Scholar]
- Sauthoff H, Hu J, Maca C, Goldman M, Heitner S, Yee H, Pipiya T, Rom WN, Hay JG (2003) Intratumoral spread of wild-type adenovirus is limited after local injection of human xenograft tumors: virus persists and spreads systemically at late time points. Hum Gene Ther 14: 425–433 [DOI] [PubMed] [Google Scholar]
- Spitz DR, Sim JE, Ridnour LA, Galoforo SS, Lee YJ (2000) Glucose deprivation-induced oxidative stress in human tumor cells. A fundamental defect in metabolism? Ann N Y Acad Sci 899: 349–362 [DOI] [PubMed] [Google Scholar]
- Tjuvajev JG, Chen SH, Joshi A, Joshi R, Guo ZS, Balatoni J, Ballon D, Koutcher J, Finn R, Woo SL, Blasberg RG (1999) Imaging adenoviral-mediated herpes virus thymidine kinase gene transfer and expression in vivo. Cancer Res 59: 5186–5193 [PubMed] [Google Scholar]
- Toloza EM, Hunt K, Swisher S, McBride W, Lau R, Pang S, Rhoades K, Drake T, Belldegrun A, Glaspy J, Economou JS (1996) In vivo cancer gene therapy with a recombinant interleukin-2 adenovirus vector. Cancer Gene Ther 3: 11–17 [PubMed] [Google Scholar]
- UKCCCR (1998) United Kingdom Co-ordinating Committee on Cancer Research (UKCCCR) guidelines for the welfare of animals in experimental neoplasia (second edition). Br J Cancer 77: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaupel P, Kallinowski F, Okunieff P (1989) Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res 49: 6449–6465 [PubMed] [Google Scholar]
- Wang Y, Hu JK, Krol A, Li YP, Li CY, Yuan F (2003) Systemic dissemination of viral vectors during intratumoral injection. Mol Cancer Ther 2: 1233–1242 [PubMed] [Google Scholar]
- Worgall S, Wolff G, Falck-Pedersen E, Crystal RG (1997) Innate immune mechanisms dominate elimination of adenoviral vectors following in vivo administration. Hum Gene Ther 8: 37–44 [DOI] [PubMed] [Google Scholar]
- Yuan F (1998) Transvascular drug delivery in solid tumors. Semin Radiat Oncol 8: 164–175 [DOI] [PubMed] [Google Scholar]