

Abstract
Microarray experiments designed to identify genes differentially expressed in the E11.5 lung and trachea showed that melanoma inhibitory activity (Mia1) was expressed only in the lung. Mia1 was abundantly expressed during early lung development, but was virtually absent by the end of gestation. Distal embryonic lung epithelium showed high levels of Mia1 expression, which was suppressed by treatment with either retinoic acid or the FGF signaling antagonist SU5402. Late-gestation fetuses in which lung epithelial hyperplasia was induced by misexpression of FGF7 or FGF10 showed continued expression of Mia1 in areas of aberrant morphogenesis. Mia1 expression was also significantly increased in urethane-induced lung adenomas. Treatment of E18.5 lung explants with exogenous MIA caused significant reductions in the expression of the lung differentiation markers Sftpa, Sftpb, Sftpc, and Abca3. Bitransgenic mice expressing MIA under the control of the SFTPC promoter after E16.5, the age when Mia1 is normally silenced, died from respiratory failure at birth with morphologically immature lungs associated with reduced levels of saturated phosphatidylcholine and mature SP-B. Microarray analysis showed significant reductions expression of Sftpa, Sftpb, Abca3, Aqp5, Lzp-s, Scd2, and Aytl2 in lungs misexpressing MIA. These results suggest that the silencing of Mia1 that occurs in late gestation may be required for maturation of the surfactant system.
Keywords: lung development, gene expression, melanoma inhibitory activity, surfactant
Introduction
The embryonic mouse lung begins as paired evaginations from the floor of the foregut endoderm on day E9.5 of gestation. A series of dichotomous and lateral branchings gives rise to the pulmonary tree, which is followed by a period of alveolization that continues postnatally. Concomitant with the processes of branching morphogenesis and alveolization is the differentiation of specialized epithelial cell types. Experiments using techniques in both classical embryology and molecular biology have shown that lung branching morphogenesis and epithelial differentiation are induced by diffusible cues produced by the associated pulmonary mesenchyme (Rudnick, 1933; Shannon et al., 1998; Spooner and Wessells, 1970). Identification of the molecules regulating lung morphogenesis and differentiation has been a subject of intense interest. These include a large number of diffusible signaling molecules as well as components of the extracellular matrix (ECM) (for reviews see (Cardoso and Lu, 2006; Shannon and Hyatt, 2004; Warburton et al., 2003). All of these factors must be expressed in the correct place and at the proper time for normal lung development to occur.
In experiments using tissue recombinants (Shannon, 1994; Shannon et al., 1998) and mesenchyme-free culture (Deterding and Shannon, 1995; Hyatt et al., 2004), we have demonstrated that the entire embryonic respiratory tract epithelium, from the trachea to the distal lung tips, exhibits substantial plasticity in its eventual phenotype that is dependent on the inductive cues it receives from its associated mesenchyme. This observation led us to speculate that the differences between embryonic trachea and lung are defined by limited subset of genes, and that the molecular comparison of these two tissues might provide new information on genes that are important for both lung and trachea development. In the present study we used microarray analysis to identify genes differentially expressed in the embryonic lung and trachea. We provide detailed analysis of one of these differentially expressed genes, melanoma inhibitory activity (Mia1), also known as cartilage-derived retinoic acid-sensitive protein (CD-RAP). Our results demonstrate that Mia1 is a developmentally regulated, FGF-sensitive ECM protein whose expression is associated with areas of morphogenetic expansion in the normal lung, in lungs with FGF-induced hyperplastic growth, and in carcinogen-induced lung adenomas. We also present in vivo and in vitro data suggesting that MIA can suppress distal lung epithelial maturation when misexpressed during late gestation.
Materials and methods
Microarray Analysis
Lung vs Trachea
Lung/trachea complexes were removed from day E11.5 FVB/N mouse embryos, and then further dissected to produce distinct lung and trachea populations. Lung tissue was defined as everything caudal to, but not including, the primary bronchi. Trachea tissue was defined as that caudal to the prospective larynx and cranial to the bifurcation; this strategy defined two spatially distinct tissue populations. Tissues were lysed in 4 M guanidinium isothiocyanate and centrifuged over a cushion of 5.7 M CsCl; the purified RNA was then transferred to the Affymetrix GeneChip Core Facility at Cincinnati Children’s Hospital Medical Center for oligonucleotide microarray hybridization. Affymetrix Murine Genome U74v2A chips were used according to the manufacturer’s protocol to compare lung and trachea gene expression. Affymetrix Microarray Suite version 5.0 was used to scan and quantify the gene chips. Three separate hybridizations of paired tissue samples (lung and trachea) were used to generate the results. Data from all chips were applied to two-step normalization to remove or minimize systemic sources of variation at both the chip and gene level. Specifically, we chose the 50th percentile (median of all genes distribution) as the baseline for per chip normalization, followed by normalizing each gene to the median of this gene across pair-wise designed experiments. Genes that were differentially expressed between lung and trachea were identified by the combination of distribution of distribution analysis (JMP4, SAS Institute, Cary, NC) and/or Welch ANOVA with the cutoff p value < 0.05 (Gene Spring 4.2.1, Silicon Genetics, Redwood City, CA). Candidates were further filtered by their reproducibility and their absolute intensity. The coefficient variation (CV) was calculated for replicates of each gene and replicates with a CV > 50% were removed. Genes displaying extremely low signals in both control and sample groups were eliminated as experimental noise.
SP-C/MIA vs Control
Lung cRNA from SP-C/MIA (described below) and control mice was generated and hybridized to Affymetrix murine genome MOE430 chips according to the manufacturer’s protocol. We analyzed cRNA from three independent SP-C/MIA and three control lungs. Affymetrix Microarray Suite 5.0 was used to scan and quantify the gene chips under default scan settings. Normalization was performed using the Robust Multichip Average model (Irizarry et al., 2003a; Irizarry et al., 2003b). Data were further analyzed using AfflmGUI (Linear Models for Microarray Data) and Genespring GX 7.3 (Silicon genetics)) (Smyth, 2004). Differentially expressed genes were selected with the threshold of T-Test P ≤ 0.05, False Discovery Rate (FDR) ≤ 10% and fold change ≥ 1.5. Differentially expressed genes were subjected to functional classification according to the Biological Process terms in Gene Ontology database. Gene Ontology Analysis was preformed using public available web-based tool David (database for annotation, visualization, and integrated discovery) (Dennis et al., 2003). Overrepresented biological processes were selected at the threshold of Fisher Exact Test P-Value ≤ 0.05 and minimum gene counts belonging to an annotation term ≥ 5.
Explant cultures
In experiments testing the effects of retinoic acid on Mia1 expression, lungs from E11.5 FVB/N embryos were cultured on 8-μm Whatman nucleopore filters (Whatman International, Clifton, NJ) placed in dissecting dishes. Lungs were cultured for up to 4 days in serum-free BGJb medium (GIBCO/Invitrogen, Carlsbad, CA) in the presence or absence of all-trans retinoic acid (Sigma Chemical Co., St. Louis, MO) at concentrations of 10−6 and 10−5 M. In experiments evaluating FGF signaling, E12.5 lungs were cultured for 1 day in medium containing either 10 μM SU5402, or 20 μM LY294002, or 20 μM UO126 (all from Calbiochem, La Jolla, CA), or DMSO as a vehicle control. In experiments testing the effects of exogenous MIA on epithelial differentiation, lungs from E18.5 embryos were isolated, cut into 1 mm3 explants and cultured for 48 hours in serum-free Waymouth’s medium (GIBCO/Invitrogen) in the presence or absence of 500 ng/ml recombinant human MIA. Recombinant human MIA (Gly25-Gln131) was generated in E. coli, renatured, and purified by chromatography and gel filtration as previously described (Bauer et al., 2006). Protein purity was checked by SDS-PAGE and HPLC and was shown to be 95% pure.
Tissue recombinations
Tissue recombinants composed of lung mesenchyme plus either lung epithelium or tracheal epithelium were prepared as previously described (Shannon et al., 1998). Briefly, day E11.5 distal lung tips were removed and incubated with dispase (BD Biomedical) for 30 minutes at 37°C, then separated into purified epithelial and mesenchymal components using tungsten needles. Day E11.5 tracheae were bisected, incubated for 45 minutes at 37°C in DMEM/F12 containing 0.05% collagenase IV (Worthington Biochemicals, Freehold, NJ) and 1% FBS (Sigma), then separated into epithelial and mesenchymal components using tungsten needles. Both lung and tracheal epithelial rudiments were treated a second time with dispase for 10 minutes to remove any residual adherent mesenchymal cells. Tissues were recombined on the surface of a semisolid medium consisting of DMEM/F12 containing 0.5% agarose and 5% FBS. Dishes containing tissue recombinants were placed in humidified 150 mm diameter dishes and cultured for up to 5 days.
Mesenchyme-free culture of embryonic lung epithelium
Distal lung epithelium was isolated from the tips of E12.5 lungs by digestion with dispase followed by dissection with tungsten needles as previously described (Deterding and Shannon, 1995). The epithelial rudiments were enrobed in Matrigel (Invitrogen) and cultured for 7 days in DMEM/F12 medium containing 5% charcoal-stripped FBS, insulin 10 μg/ml, cholera toxin 1 μg/ml, human recombinant epidermal growth factor (EGF) 25 ng/ml, human recombinant hepatocyte growth factor (HGF) 10 ng/ml, FGF7 (Peprotech) 25 ng/ml and FGF1 100 ng/ml. This culture system stimulates embryonic lung epithelial proliferation and differentiation (Shannon et al., 1999). Experimental cultures were treated with SU5402 for the last 24 hours.
Transgenic mice
All mice used in these studies were generated on an FVB/N background. Mice bearing transgenes that allowed the inducible expression of FGF7 (Tichelaar et al., 2000) and FGF10 (Clark et al., 2001) were kindly provided by Dr. Jeff Whitsett (Cincinnati Children’s Hospital). Briefly, permanent lines of activator mice have been established in which the 3.7 kb human SFTPC promoter or 2.3 kb rat CCSP promoter drive expression of a reverse tetracycline responsive transactivator (hereafter SP-C/rtTA or CCSP/rtTA, respectively). Two separate responder lines were also established, bearing either a (tetO)7/cmv/FGF7 or a (tetO)7/cmv/FGF10 transgene. These transgenes consist of seven copies of the tet operator, a minimal cmv promoter, either the human FGF7 or the mouse FGF10 coding sequence, and a SV40 polyadenylation signal. Activator and responder mice were bred to generate bitransgenic fetuses (hereafter SP-C/FGF7 or CCSP/FGF10 mice) and doxycycline (0.5 mg/ml) was administered to pregnant dams via drinking water beginning on day E10.5. Fetuses were harvested on day E18.5. Doxycycline crossed the placenta and allowed the rtTA fusion protein to bind the tet operator, activating the cmv promoter, which in turn drove expression of FGF7 or FGF10 in bitransgenic fetal lungs.
In order to study the effect of continued MIA expression during late lung development, we generated mice bearing an inducible Mia1 transgene. The transgene comprised 430 bp of murine Mia1 cDNA followed by a Flag tag (DYKDDDDK), which was cloned into the tet-responsive vector pTRE-HA vector (Clontech). Prior to injection, a 2.2 kb fragment containing the (tetO)7/cmv promoter, Mia1 cDNA, Flag tag and the β-globin poly A tail was released from the vector by AatII/SatI digestion and gel-purified. Purified DNA was microinjected into fertilized FVB/N mouse eggs by the Cincinnati Children’s Hospital Transgenic Core Facility. Founder mice were identified by two sets of PCR amplification of mouse tail DNA using 5′-primers specific to either the cmv promoter (5′-TTTTGACCTCCATAGAAGACACCG-3′) or the Mia1 coding sequence (5′-TTTCCCCAGTAGCATTGTCCG-3′) and a 3′-primer specific to the β-globin poly A tail (5′-CTACATCCTGGTAATCATCCTGCC-3′) to generate 1050 bp or 600 bp products, respectively. Five founder lines of tetO/MIA responder mice were generated; we selected the line that displayed the most robust expression of MIA for further study. The tetO/MIA responder mice were mated with SP-C/rtTA activator mice to generate bitransgenic progeny (hereafter SP-C/MIA mice) expressing MIA in a doxycycline-inducible manner. Dox was added on day E15.5 to ensure MIA expression in distal epithelia on day E16.5 and later. Single transgenic littermates (those expressing only the SP-C/rtTA or tetO/MIA transgene) served as controls.
Generation of lung tumors
Lung tumors were produced by treating 8 week old strain A/J mice with a single intraperitoneal injection of urethane (Sigma) dissolved in 0.9% NaCl at a dose of 1 mg/g body weight (Malkinson and Beer, 1983). Mice were sacrificed 44 weeks following urethane injection and tumors harvested. Uninvolved tissue adjacent to the tumors and lung tissue from age-matched vehicle treated animals served as controls.
In situ hybridization
The murine cDNAs for Mia1 (580 bp) and Sftpc (758 bp) were used as templates for generating riboprobes. Digoxigenin-labeled probes were used for whole mount in situ hybridization (ISH), which was performed as described by Wilkinson (Wilkinson, 1992). Tissue section ISH was performed as previously described (Deterding and Shannon, 1995), with the exception that 33P-UTP was used for labeling the probe.
Quantitative RT-PCR
Mia1 expression was assessed by quantitative real-time PCR (qRT-PCR). Template cDNAs were obtained by reverse transcriptase reaction (cDNA Cycle kit, Invitrogen) of total RNAs from E11.5 lung and trachea. Amplification was carried out using the DNA master SYBR green I mix (Roche Molecular Biochemicals). The template was mixed with 1x SYBR mix, 2.5 mM MgCl2 and 0.25 μM each probe. All reactions were performed in a Smart Cycler (Cepheid, Sunnyvale, CA). Reaction conditions were 95°C for 150 sec, followed by 40 cycles of amplification (95°C for 10 sec, 58–60°C (depending on primer set) for 15 sec and 72°C for 20 sec). Data were normalized to β-actin or the amount of input cDNA.
The sequences of the primers used were as follows:
Mia1, sense: 5′-TCCTTGGCATCGTCGTCTTGTC-3′,
antisense: 5′-CCATAGTAACCTCCCTGAACACTGC-3′;
Sftpa, sense: 5′-AGTCCTCAGCTTGCAAGGATC-3′,
antisense: 5′-CGTTCTCCTCAGGATTCCTCG-3′;
Sftpc, sense: 5′-CATCGTTGTGTATGACTACCAGCG-3′,
antisense: 5′-GAATCGGACTCGGAACCAGTATC;
Sftpb, sense: 5′-CTGTGCCAAGAGTGTGAGGA-3′,
antisense: 5′-TTGGGGTTAATCTGGCTCTG-3′;
Abca3, sense: 5′-TTCAACAACCAGGCATACCA-3′,
antisense: 5′-AGCAAATTGAGGGCAATGTC-3′;
Dusp9, sense: 5′-TTCCTCCTCCCCCAGATGTC-3′,
antisense: 5′-CCCCAAACCAACAGTGGCTG-3′
β-actin, sense: 5′-TGGAATCCTGTGGCATCCATGAAAC-3′,
antisense: 5′-TAAAACGCAGCTCAGTAACAGTCCG-3′
Quantification of Mia1 expression in urethane-induced lung tumors and in the SU5402-treated lung epithelium experiments was done using an Applied Biosystems 7300 instrument and TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA), as was quantification of Ptch1 in the E11.5 lung and trachea. Reaction conditions were 50°C for 120 sec, 95°C for 10 min, then 40 cycles of amplification (95°C for 15 sec and 60°C for 60 sec). Mia1 expression was normalized to β-actin.
Western blotting
Lungs were snap frozen in liquid nitrogen, ground with mortar and pestle in ice-cold 1x PBS containing 1x protease inhibitor (Roche) and 1% NP-40 (Sigma), followed by sonication. Samples were centrifuged and protein concentration determined by BCA analysis. Immunoblots of total lung homogenate from SP-C/MIA and control littermates were prepared by electrophoresing 20 μg of total protein from lung homogenates by SDS-PAGE and transferring it to nitrocellulose. Western blots were performed using antiserum 28031, which is directed against mature SP-B (Lin et al., 1996), at a dilution of 1:15,000.
Immunohistochemistry
Paraffin sections were immunostained with the following antibodies: anti-FLAG (1:2000; Sigma), anti-T1α (1:500; Developmental Studies Hybridoma Bank, Iowa City, IA), and anti-SP-B (antibody 28031, 1:1000). Biotinylated anti-rabbit secondary antibody was used along with Vectastain ABC kit (Vector Laboratories) to visualize staining.
Morphometry
Morphometric measurements were performed on E18.5 lungs from four SP-C/MIA and four single transgenic fetuses. The overall proportion (% fractional area) of respiratory parenchyma and airspace was determined by using point counting method (Bolender et al., 1993; Wert et al., 2000) on two sections taken at intervals throughout the left or right lobes. Images (fields) were transferred by video camera to a computer screen using Metamorph imaging software (Universal Imaging, West Chester, PA). A computer-generated, 121-point lattice grid was superimposed on each field, and the number of intersections (points) falling over respiratory parenchyma (alveoli and alveolar ducts) or airspace was counted. Points falling over bronchioles, large vessels and smaller arterioles and venules were excluded from the study. Fractional areas (% fractional area) were calculated by dividing the number of points for each compartment (n) by the total number of points contained within the field (N), and then multiplying by 100: % fractional area = n/N X 100. Epithelial and mesenchymal cells were counted manually. Ten to twenty fields per section were analyzed to gather the data; more than 500 cells were analyzed for each tissue compartment.
Phospholipid analysis
Lungs were homogenized in 0.9% NaCl and then the protease inhibitor P8340 (1:100, Sigma) was added to the homogenate. Lipids were extracted from the homogenate (Bligh and Dyer, 1959), and phospholipid composition was determined by 2-D thin-layer chromatography (Ikegami et al., 2003). Saturated phosphatidylcholine was isolated from lipid extracts using the osmium tetroxide method (Mason et al., 1976) followed by a phosphorus content assay (Bartlett, 1959).
Statistics
All data were statistically analyzed using GraphPad Prizm version 4.0 (GraphPad Software, San Diego, CA). Data from SU5402-treated epithelium cultures, saturated phosphatidylcholine content, and morphometry were analyzed using unpaired t-tests. Data from lung explant cultures treated with RA, or with DMSO, SU5402, LY294002 and UO126, or with recombinant MIA were analyzed by one-way ANOVA, as were the lung tumor Mia1 content experiments.
Results
Differential gene expression in the E11.5 lung
Statistical comparison of the genes expressed in the E11.5 mouse lung vs trachea resulted in the identification of 51 genes that were upregulated in the lung. These comprised 48 known genes and 3 ESTs. As shown in Table 1, the known genes included transcription factors (7), signal transduction proteins (5), proteins associated with development (8), cell adhesion proteins (5), proteins involved in processing of nucleic acids and proteins (11), and other proteins (12) with various functions. One of the proteins identified as lung-specific was surfactant protein C (Sftpc), which has been shown by a variety of techniques to be expressed only in the lung epithelium (Kalina et al., 1992; Wert et al., 1993). qRT-PCR confirmed the differential expression of SP-C, along with two other genes identified in the microarray, Dusp9 and Ptch1 (data not shown). Taken together, these results indicated that the microarray hybridization was performing as intended.
Table 1.
Genes Upregulated in Embryonic Mouse Lung vs Trachea
| GenBank Accession #
|
Fold Change
|
Symbol
|
Gene
|
|---|---|---|---|
| Transcription Factors | |||
| NM_008269 | 6.0 | Hoxb6 | Homeobox B6 |
| NM_010426 | 2.5 | FoxF1a | Forkhead box F1a |
| NM_009538 like 1 | 2.1 | Plagl1 | Pleiomorphic adenoma gene |
| NM_010703 factor 1 | 2.1 | Lef1 | Lymphoid enhancer binding |
| NM_011415 | 2.1 | Snai2 | Snail homolog 2 |
| NM_009331 | 1.8 | Tcf7 | Transcription factor 7 |
| NM_008584 | 1.5 | Meox2 | Mesenchyme homeobox 2 |
| Development | |||
| NM_009152 | 4.2 | Sema3a | Semaphorin 3a |
| NM_010025 | 3.2 | Dcx | Doublecortin |
| NM_008957 | 2.2 | Ptch1 | Patched homolog 1 |
| NM_053089 gene 1 | 2.1 | Narg1 | NMDA receptor regulated |
| NM_009510 | 2.1 | Vil2 | Villin 2 |
| NM_010060 Chain 11 | 1.7 | Dnahc11 | Dynein, axonemal, heavy |
| NM_011199 1 | 1.6 | Pthr1 | Parathyroid hormone receptor |
| NM_007555 5 | 1.6 | Bmp5 | Bone morphogenetic protein |
| Cell Adhesion | |||
| NM_019934 1 | 12.9 | Mia1 | Melanoma inhibitory activity |
| NM_008485 | 3.0 | Lamc2 | Laminin, gamma 2 |
| NM_009902 | 2.2 | Cldn3 | Claudin 3 |
| AF041409 | 2.0 | Itga8 | Integrin alpha 8 |
| NM_018777 | 1.9 | Cldn6 | Claudin 6 |
| Signal Transduction | |||
| NM_010305 protein | 2.0 | Gnai1 | Guanine nucleotide binding |
| NM_053272 reductase | 2.0 | Dhcr24 | 24-dehydrocholesterol |
| NM_011945 kinase | 1.7 | Map3k1 | Mitogen activated protein |
| NM_133235 | 1.7 | Khdrbs2 | kinase kinase 1 KH domain containing, RNA binding, signal transduction Associated 2 |
| NM_010579 protein | 1.5 | Itgb4bp | Integrin beta 4 binding |
| Protein Metabolism | |||
| NM_009183 neuraminide | 2.4 | St8sia4 | ST8 alpha-N-acetyl-alpha-2,8-sialyltransferase 4 |
| NM_025553 protein L11 | 2.4 | Mrpl11 | Mitochondrial ribosomal |
| NM_029352 9 | 2.2 | Dusp9 | Dual specificity phosphatase |
| NM_152804 (Drosophila) | 1.9 | Plk2 | Polo-like kinase 2 |
| NM_026130 receptor | 1.9 | Srpr | Signal recognition particle |
| NM_025338 protein 1 | 1.7 | Aurkaip1 | Aurora kinase A interacting |
| NM_026332 | 1.6 | Dnajc19 | DnaJ homolog, subfamily C, Member 19 |
| DNA/RNA Metabolism | |||
| NM_009104 | 2.0 | Rrm2 | Ribonucleotide reductase M2 |
| NM_010931 PHD and | 1.9 | Uhrf1 | Ubiquitin-like, containing |
| NM_019870 homolog | 1.5 | Ard1 | RING finger domains, 1 N-acetyltransferase ARD1 |
| NM_138586 | 1.5 | Exosc5 | Exosome component 5 |
| Other Proteins | |||
| NM_013492 | 8.7 | Clu | Clusterin |
| NM_011359 C | 5.9 | Sftpc | Surfactant associated protein |
| NM_021301 member 2 | 2.9 | Slc15a2 | Solute carrier family 15, |
| NM_008471 | 2.4 | Krt19 | Keratin 19 |
| NM_013820 | 2.2 | Hk2 | Hexokinase 2 |
| NM_010284 | 2.0 | Ghr | Growth hormone receptor |
| NM_026323 domain 2 | 1.9 | Wfdc2 | WAP four-disulfide core |
| NM_133706 | 1.9 | Temem97 | Transmembrane protein 97 |
| NM_010390 region 1 | 1.6 | H2-Q1 | Histocompatibility 2, Q |
| NM_009171 hydroxymethyltransferase 1 | 1.6 | Shmt1 | Serine |
| NM_016737 phosphoprotein 1 | 1.6 | Stip1 | Stress-induced |
| NM_177420 aminotransferase 1 | 1.5 | Psat1 | Phosphoserine |
| ESTs | |||
| BB645708 | 2.3 | B430320C24 | |
| AK008780 | 1.9 | 2210039B01Rik | |
| NM_026063 | 1.5 | 2900010M23Rik | |
One of the largest differences in gene expression we observed in the E11.5 lung vs trachea was melanoma inhibitory activity (Mia1) (Blesch et al., 1994), also known as cartilage-derived retinoic acid sensitive protein (CD-RAP) (Dietz and Sandell, 1996). MIA is a secreted protein, which suggested that it could play a role in mediating cell-cell interactions. Furthermore, Mia1 expression is regulated by RA, which is known to play a major role in lung morphogenesis (Mendelsohn et al., 1994; Mollard et al., 2000). We therefore examined Mia1 expression during lung development.
Ontogeny of Mia1 in the lung
Previous studies had noted Mia1 expression in the embryonic lung (Bosserhoff et al., 1997b; Xie et al., 2000), and characterized the site of expression as being in cartilaginous lung bronchi (Bosserhoff et al., 1997b). As shown by whole mount ISH in Figure 1, we found that Mia1 is expressed in the endodermal epithelium of embryonic lung buds on day E10.5 (Fig. 1A). By day E11.5 (Fig. 1B) expression was restricted to the lung parenchyma, and was particularly intense in the most distal lung tips. Expression in the bronchi and trachea was not observed, a result that was confirmed by qRT-PCR (data not shown). This pattern of expression continued through day E12.5 (Fig. 1C). On day E13.5 (Fig. 1D) Mia1 expression persisted in the lung epithelium, but was also noted in the incipient cartilaginous rings that were now developing in the trachea and bronchi (Fig. 1D, arrowheads). Tissue section ISH confirmed epithelial expression of Mia1 on days E14.5 and E15.5 (Fig. 1E–H), and further revealed that distribution was not uniform. The distal epithelium expressed Mia1, while the proximal epithelium did not (arrows, Fig. 1H). An abrupt change in the pattern of Mia1 expression was seen on day E16.5, when it was seen only in the most distal epithelial tubules lying immediately under the pleural surface (Fig. 1I,J). This pattern of expression continued until day E18.5, when another change was seen: distal epithelial expression of Mia1 was extinguished, and expression in the large proximal airways was detected, albeit at low levels (Fig. 1K,L). This pattern persisted through birth (Fig. 1M,N). In agreement with previous observations (Dietz and Sandell, 1996) we found that Mia1 was essentially undetectable in the adult lung by ISH (Fig. 1O,P).
Figure 1.
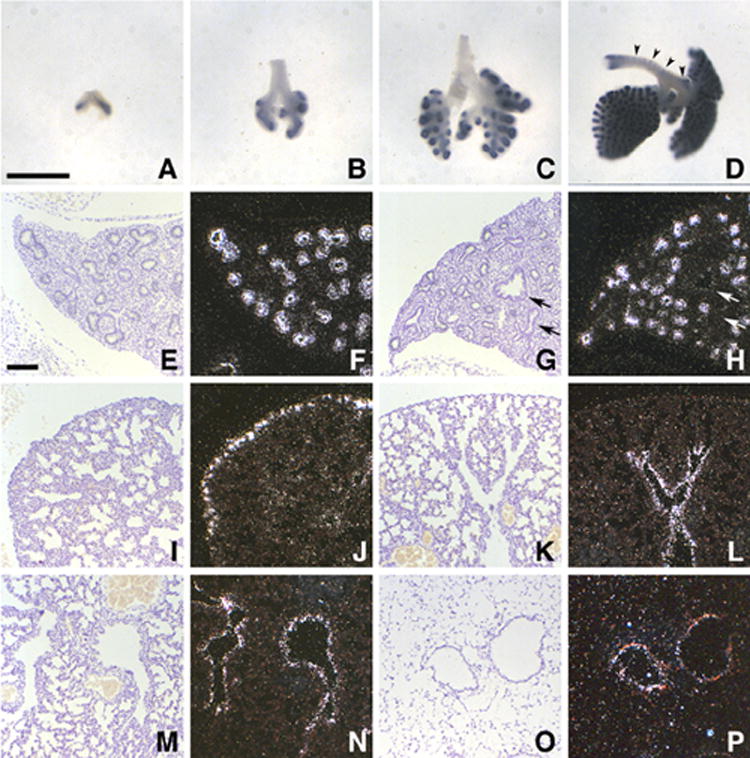
Ontogeny of Mia1 expression in the developing lung. Whole mount in situ hybridization (ISH) shows that Mia1 is expressed in lung distal epithelium on days E10.5 (A), E11.5 (B), and E12.5 (C), but is not detected in the tracheal epithelium. Epithelial expression continues on day E13.5 (D), but signal is also present in incipient tracheal cartilage (arrowheads). Panels A–D are at the same magnification; bar = 590 μm. Tissue section ISH shows that Mia1 is expressed in the distal but not proximal (arrows) epithelium on days E14.5 (E, F) and E15.5 (G, H). On day E16.5 the expression pattern changes, and only the most distal epithelium is positive for Mia1 (I,J). On day E18.5 signal for Mia1 is seen in the proximal epithelium (K,L), a pattern that continues on postnatal day 1 (M,N). Mia1 is essentially undetectable in the adult lung (O,P). Panels E–P are at the same magnification; bar = 100 μm.
RA suppresses Mia1 in developing lung explants in vitro
We next determined the effect of RA on Mia1 expression in cultured E11.5 lung explants. We treated explants with RA at final concentrations of 10−6 M and 10−5 M, which have been previously shown to disrupt branching morphogenesis and arrest epithelial maturation in cultured embryonic lungs (Cardoso et al., 1995; Malpel et al., 2000). E11.5 lungs showed continued dichotomous and lateral branching when cultured in serum-free medium for 4 days (Fig. 2B). These explants showed maintenance of Mia1 expression, which was most intense at the distal tips (Fig. 2C). Lung explants treated with 10−6 M RA showed reduced branching (Fig. 2E), but the overall pattern was still lung-like. Mia1 expression was still evident in the distal epithelium of explants treated with 10−6 M RA (Fig. 2F), although the intensity of signal appeared reduced in comparison to controls. Lungs treated with 10−5 M RA were smaller than controls and lung branching was clearly reduced (Fig. 2H). The decreases in branching seen in 10−5 M RA-treated explants were accompanied by a clear diminution in Mia1 expression (Fig. 2I). These graded effects of RA on lung morphogenesis were consistent with what has been previously reported (Cardoso et al., 1995). The decrease in Mia1 expression we observed in response to RA was detectable by day 2 of culture when measured by qRT-PCR (Fig. 2J).
Figure 2.

Retinoic acid (RA) suppresses Mia1 expression. Day E11.5 lungs (A,D,G) were grown for 4 days in BGJb medium containing no additions (B,C), 1 μM all-trans RA (E,F), or 10 μM RA (H,I). RA attenuates normal branching morphogenesis at 1 μM (E), and significantly disrupts branching at 10 μM (H). Whole mount ISH shows that the distal epithelium of lungs cultured with no additions express high levels of Mia1 (C). RA diminishes Mia1 expression at 1 μM (F), and nearly abolishes expression at 10 μM (I). All panels are at the same magnification; bar = 300 μm. qRT-PCR shows that Mia1 expression is significantly (*, p < 0.05 vs control and 1 μM RA) reduced in lungs cultured in medium containing 10 μM RA (J). Data are plotted as means ± SE (n = 3).
Mia1 expression is induced by lung mesenchyme
Our ISH and qRT-PCR data both demonstrated that Mia1, like Sftpc, is not expressed in the developing tracheal epithelium. We have previously shown that embryonic tracheal epithelium recombined with distal lung mesenchyme was reprogrammed to express both morphological and molecular markers of differentiated distal lung epithelium, including Sftpc (Shannon et al., 1998). We therefore examined tissue recombinations to determine whether Mia1 expression was also induced when tracheal epithelium had been reprogrammed to a distal lung epithelial cell phenotype. The epithelium of intact E11.5 distal lung tips cultured for 3 days branched within the mesenchyme and expressed Mia1 (Fig. 3A,B). Recombinants of distal tip lung epithelium and mesenchyme, which served as surgery controls, showed normal epithelial branching and Mia1 expression in the distal tips (Fig. 3C,D). Tracheal epithelium also branched in a lung-like pattern when recombined with lung mesenchyme. The reprogrammed tracheal epithelium was induced to express Mia1, and the level of expression appeared identical to that seen in recombinants of distal tip lung epithelium and mesenchyme (Fig. 3E,F). Like the early lung in vivo, Mia1 expression was localized to the distal epithelial tips in these recombinants. Identical to our previous experiments (Shannon et al., 1998), recombination of tracheal epithelium with lung mesenchyme also induced Sftpc expression in the distal tips at a level equivalent to that seen in recombinants of lung epithelium and mesenchyme (data not shown). These data demonstrate that Mia1 expression is dependent on factors produced by the mesenchyme.
Figure 3.
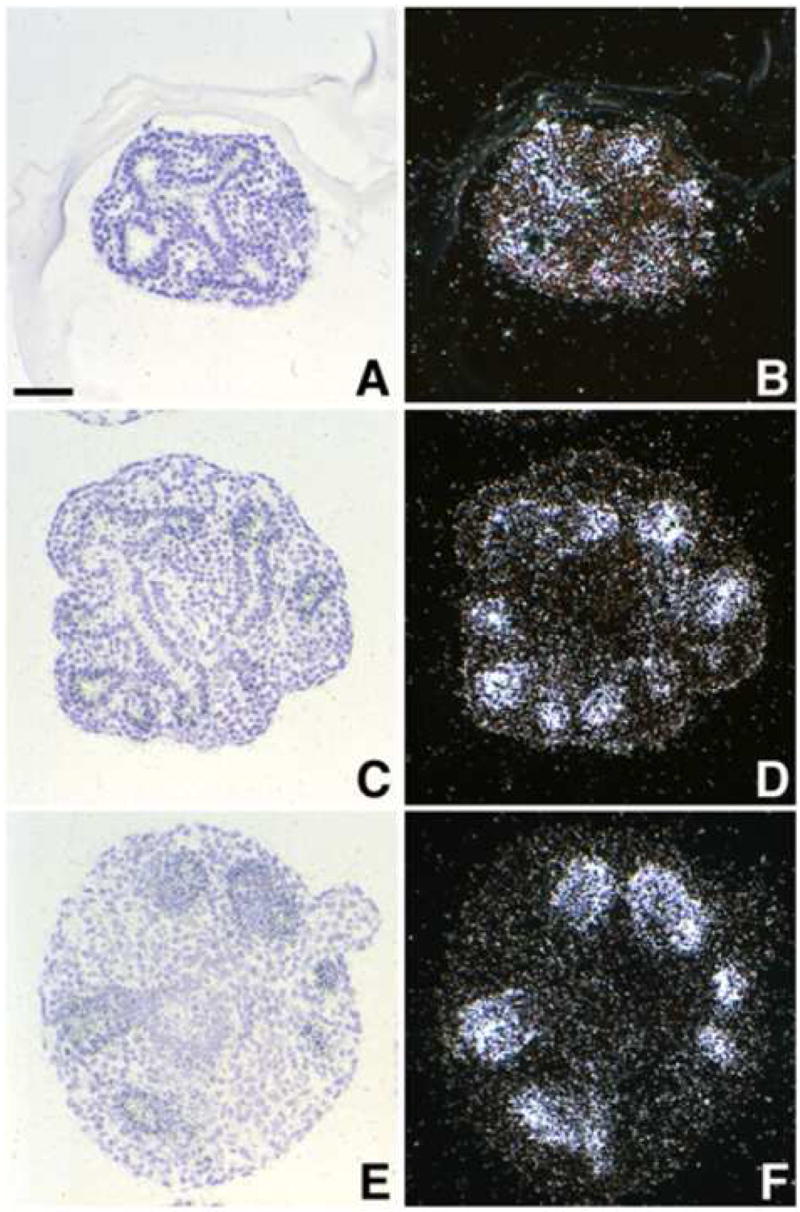
Induction of Mia1 in embryonic tracheal epithelium. Intact distal lung tips, recombinations of E11.5 lung mesenchyme and lung epithelium, or recombinations of E11.5 lung mesenchyme and tracheal epithelium were prepared and cultured for 3 days. ISH of intact E11.5 distal lung tips (A,B) and lung mesenchyme + lung epithelium recombinants (C,D) demonstrates that Mia1 expression was sustained by lung epithelium in vitro. Recombination of lung mesenchyme with E11.5 tracheal epithelium, which was negative for Mia1 at the time of isolation (see Figure 1), results in the induction of Mia1 in the reprogrammed tracheal epithelium (E,F). All panels are at the same magnification; bar = 45 μm.
Mia1 expression is FGF dependent
To determine if Mia1 is regulated by FGFs, we treated E12.5 lung explants for 24 hours with the FGF receptor antagonist SU5402. Using qRT-PCR, we found that blockade of FGF signaling caused a significant decrease in Mia1 expression (Fig. 4A). The two downstream signaling pathways of FGF signaling that have been most thoroughly studied are the PI3-Kinase/Akt and MAP Kinase/Erk pathways. To gain some insight into the downstream effectors involved in regulating Mia1 expression, we cultured E12.5 lungs for 1 day in the presence of either SU5402, or LY294002 (a PI3K/AKT signaling inhibitor), or UO126 (a MAPK/ERK signaling inhibitor). We found that all three inhibitors significantly reduced the level of Mia1 expression (Fig. 4A). We also cultured purified E12.5 distal lung epithelial rudiments for 7 days in a complex medium containing FGF7 and FGF1, which we have previously shown (Shannon et al., 1999) to support the differentiation of embryonic lung endoderm. Treating these rudiments with SU5402 for the last 24 hours of culture reduced Mia1 expression by 50% (Fig. 4B), indicating that FGFs directly regulate Mia1 expression.
Figure 4.
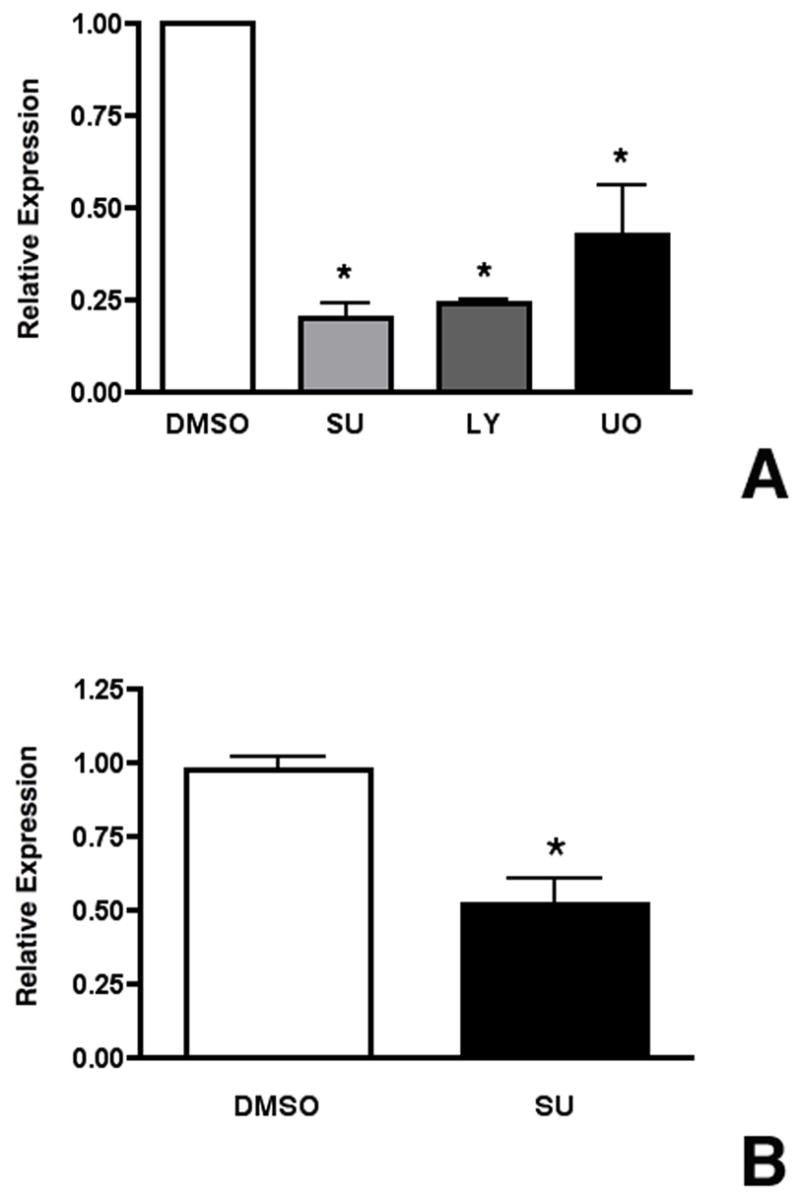
Mia1 expression is directly regulated by FGFs. (A) Day E12.5 lungs were grown for 1 day in BGJb medium containing DMSO, SU5402, LY294002, or UO126. qRT-PCR shows that Mia1 expression is significantly (*, p < 0.001 vs DMSO) reduced by SU5402, LY294002 and UO126. (B) Purified day E12.5 lung epithelial rudiments were grown for 7 days then treated with SU5402 for 1 day. qRT-PCR demonstrates that SU5402 significantly (*, p = 0.004) reduced Mia1 expression. Data are plotted as means ± SE (n = 4 for both A and B).
Mia1 is expressed in the epithelium of embryonic lungs with abnormal morphogenesis
Our ontogeny data showed that Mia1 expression was highest during the embryonic and pseudoglandular stages of lung development, when the epithelium is most actively invading and ramifying within the mesenchyme. Mia1 expression was extinguished in the lung parenchyma by the saccular stage on day E18.5, when lung patterning has been largely completed. These observations suggested that Mia1 expression might be associated with epithelial invasion of surrounding mesenchyme. Because our in vitro data showed that Mia1 expression was FGF dependent, we examined this possibility in lungs from bitransgenic fetuses in which normal epithelial patterning had been compromised by misexpression of FGF7 or FGF10. Lungs from E18.5 SP-C/FGF7 fetuses showed an increase in overall size and generalized epithelial hyperplasia (Fig. 5C) that resembled cystadenomatoid malformation (Tichelaar et al., 2000). In contrast to control littermates, in which Mia1 expression was limited to the proximal epithelium at this stage (Fig. 5A,B), virtually all of the epithelial cells in the lungs of SP-C/FGF7 mice were positive for Mia1 (Fig. 5D). Conditional expression of high levels of secreted FGF10 by the lung epithelium of CCSP/FGF10 mice gave a different morphology. Although there was some widespread epithelial hyperplasia, there were also focal adenoma-like lesions (Fig. 5E) interspersed with normal lung parenchyma (Clark et al., 2001). The adenomatous structures in these lungs were strongly positive for Mia1 (Fig. 5F), whereas surrounding normal tissue remained negative (Fig. 5F, asterisk). These observations confirmed that Mia1 was FGF dependent in vivo, and that its expression correlated with areas of active morphological changes.
Figure 5.
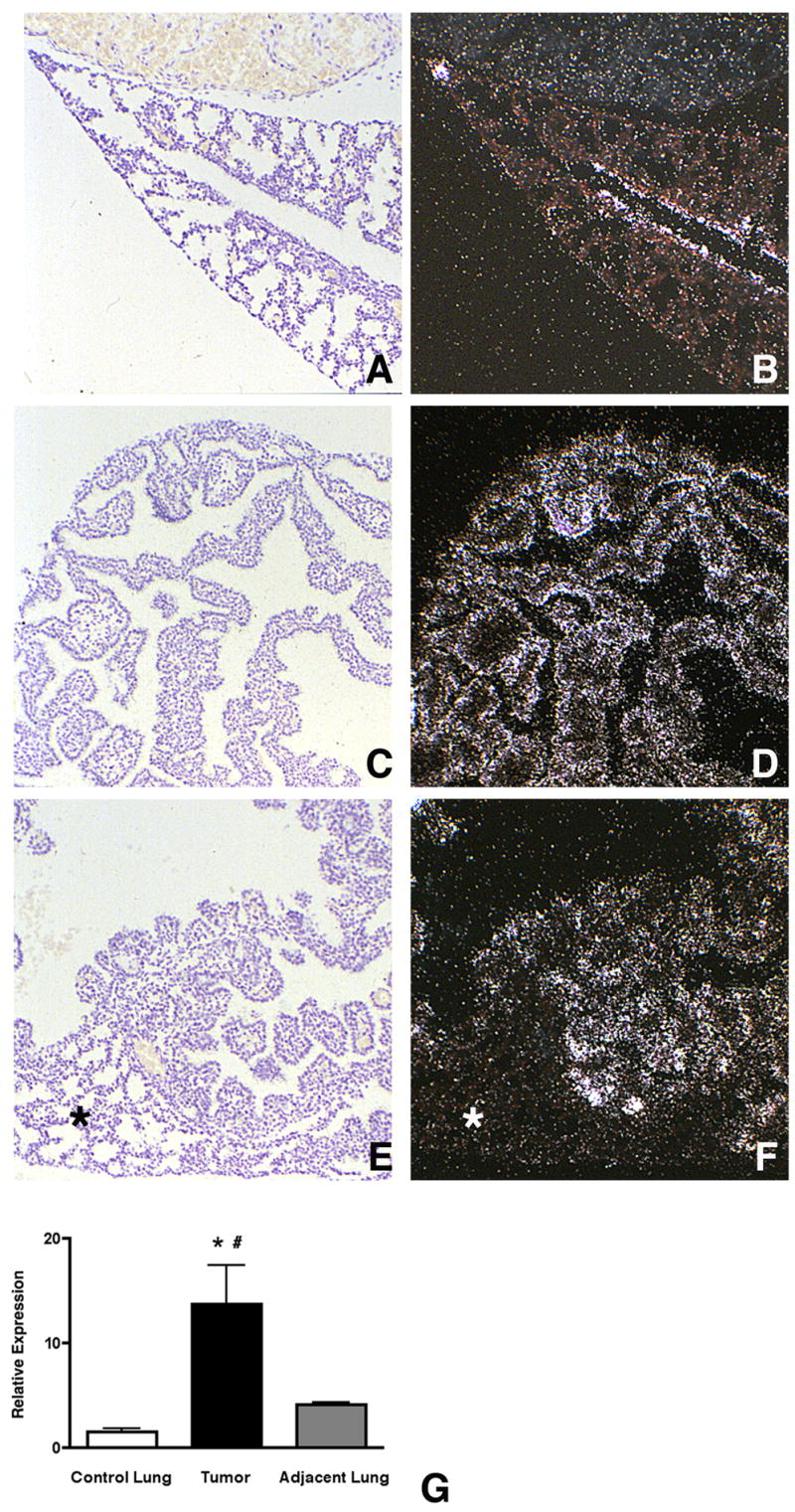
Mia1 is expressed in regions of aberrant morphogenesis. Mia1 expression is restricted to the proximal epithelium in wild type E18.5 mice (A,B). Inducible misexpression of FGF7 in the lung epithelium causes widespread epithelial cell hyperplasia in E18.5 lungs (C), which is accompanied by sustained expression of Mia1 in virtually all epithelial cells (D). Inducible misexpression of FGF10 also causes epithelial cell hyperplasia (E), but in some areas focal adenomatous lesions are surrounded by normal tissue (asterisk). Mia1 is present in the areas of abnormal growth (F), but is absent in normal tissue. All panels are at the same magnification. qRT-PCR shows that Mia1 expression is significantly (*, p < 0.01 vs control lung, #, p < 0.01 vs adjacent lung) higher in independent lung tumors induced by urethane injection compared to lung tissue from control animals injected with saline, and to uninvolved lung tissue adjacent to the tumors (G). Data are plotted as means ± SE (n = 5).
In order to determine if Mia1 expression was affected in independent lung tumors, we measured Mia1 content in urethane-induced lung adenomas (Fig. 5G). We found that Mia1 expression was increased over 10-fold compared to lung tissue from vehicle treated adults. We also measured Mia1 content in tissue adjacent to the adenomas where no obvious tumors were apparent. Although Mia1 expression appeared to be increased over controls in this tissue, these differences did not reach statistical significance (n = 5).
MIA inhibits type II cell differentiation and lung maturation in vitro
The fact that Mia1 is absent in the distal epithelium in late gestation suggested the possibility that silencing of Mia1 may be required for lung maturation. Previous studies have shown that MIA inhibits the interaction of integrins with fibronectin and laminin (Bauer et al., 2006; Bosserhoff et al., 2003b). Furthermore, a number of studies have shown that type II cell differentiation requires interaction with ECM components, particularly laminin (Nguyen et al., 2005; Olsen et al., 2005; Shannon et al., 1990). We speculated that excessive production of MIA in late gestation could prevent type II cells from interacting with laminin and therefore inhibit type II cell differentiation. To test this possibility, we cultured E18.5 lung explants in the presence or absence of recombinant human MIA to determine if the expression of several distal epithelial differentiation markers was affected. Explants treated with 500 ng/ml MIA appeared identical to controls, and we observed no morphological changes indicating toxicity. We found (Fig. 6) that MIA significantly reduced expression of Sftpa (p = 0.02, n = 4), Sftpb (p = 0.02, n = 3), Sftpc (p = 0.04, n = 4), and Abca3 (p = 0.05, n = 4), suggesting that MIA inhibited type II cell differentiation.
Figure 6.
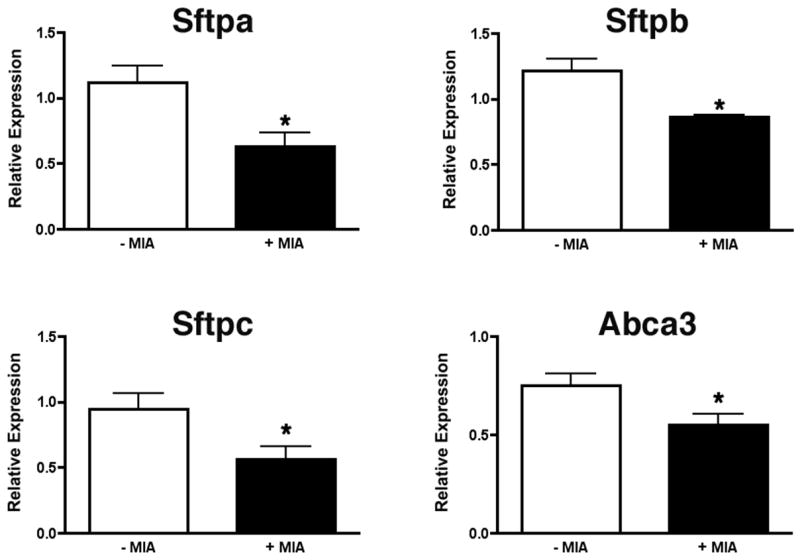
MIA downregulates surfactant gene expression. Explants of E18.5 lungs were cultured for 2 days in the presence or absence of 500 ng/ml recombinant MIA. qRT-PCR demonstrates that expression of Sftpa, Sftpb, Sftpc and Abca3 is significantly (*, p < 0.05) reduced in lungs cultured in medium containing MIA. Data are plotted as means ± SE (n = 4).
Mice misexpressing MIA die from respiratory failure
To test whether excess MIA could similarly affect type II cell differentiation in vivo, we generated bitransgenic mice that inducibly expressed MIA under the control of the Sftpc promoter. Misexpressing MIA in the lung epithelium of SP-C/MIA mice from day E16.5 onwards, when Mia1 expression is normally extinguished, resulted in death from respiratory failure in 100% of the neonates. Animals were cyanotic at birth and died within minutes. No deaths were observed in neonates bearing single SP-C/rtTA or Otet/MIA transgenes, or in SP-C/MIA neonates whose mothers were not given Dox. Histology of the lungs from E18.5 fetuses (Fig. 7A,B) revealed pronounced mesenchymal thickening and decreased alveolar space, observations that were confirmed morphometrically (Fig. 7C,D). We observed no morphological abnormalities in SP-C/rtTA or Otet/MIA single transgenic mice. Histochemical staining of lung sections from SP-C/MIA and control mice showed that the epithelial cells of the double transgenic mice contained copious amounts of glycogen (data not shown). Since the content of glycogen, which serves as a substrate for surfactant phospholipid biosynthesis, decreases in late gestation (Bourbon et al., 1982), these data were consistent with impaired maturation of the surfactant system.
Figure 7.
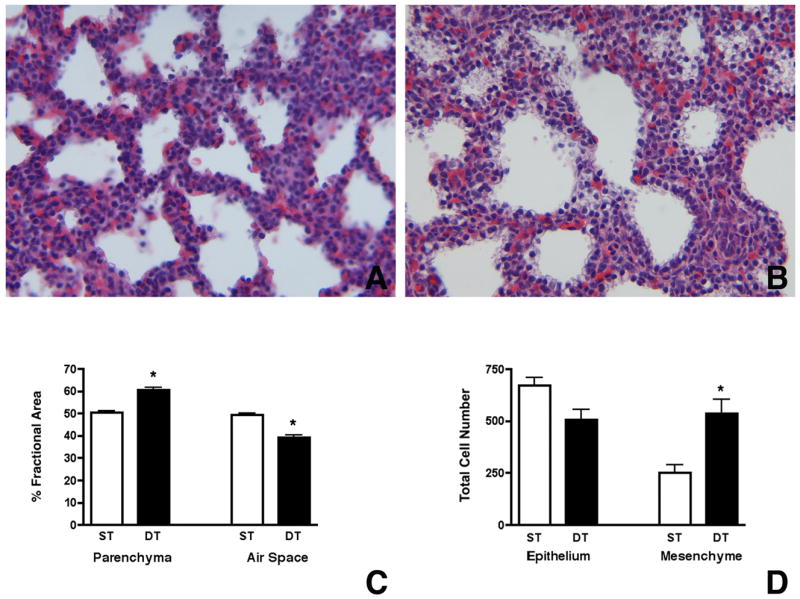
Misexpression of Mia1 results in mesenchymal thickening and reduced alveolar space. Mia1 expression was induced in the epithelium of SP-C/MIA mice beginning on E16.5 and continued until sacrifice at E18.5. Histology shows that the lung mesenchyme in SP-C/MIA double transgenic fetuses (B) appears thickened compared to single transgenic (expressing only the tetO/MIA transgene) littermates (A). Comparison of changes in fractional areas of airspace and respiratory parenchyma in the E18.5 lungs show that SP-C/MIA double transgenic (DT) mice have significantly (*, p < 0.001) more parenchyma and less airspace compared to single transgenic (ST) mice (C). Comparison of total cell number show that total mesenchymal cells are significantly (*, p < 0.01) higher in SP-C/MIA mice compared to controls while no significant differences are observed in total epithelial cells between the two groups (D). Data are plotted as means ± SE (n = 4).
Immunostaining demonstrated that mature SP-B protein was significantly reduced (Fig. 8C,D) in the distal epithelium of bitransgenic mice in areas where transgene expression was detected by FLAG immunostaining (Fig. 8A,B). Western blot analysis confirmed that mature SP-B was decreased in bitransgenic mice compared to controls (wild type mice and mice singly expressing either a SP-C/rtTA or tetO/MIA transgene) (Fig. 8G). Immunostaining for T1α, a type I cell marker, also appeared unaffected in the lung epithelium (Fig. 8E,F). We assessed the overall phospholipid composition in the lungs of SP-C/MIA mice and control littermates and found that there were no significant differences between the two groups (data not shown). However, comparison of the content of saturated phosphatidylycholine (SatPC), the predominant phospholipid of pulmonary surfactant, in the lungs of SP-C/MIA mice and control littermates showed (Fig. 8H) that the lungs of bitransgenic mice contained only 40% of the SatPC found in controls. These data indicated that misexpression of MIA in the distal epithelium after day E16.5 inhibited several aspects of maturation of the surfactant system.
Figure 8.

Gene expression in SP-C/MIA mice. Immunostaining of E18.5 lung sections for FLAG identifies epithelial cells misexpressing Mia1 (B), whereas single transgenic controls show no signal (A). Immunostaining for mature SP-B shows widespread expression in single transgenic (expressing only the tetO/MIA transgene) controls (C). SP-C/MIA mice, however, show greatly reduced SP-B staining (D), a finding that is confirmed by Western blot (G). Immunostaining for the type I cell marker T1α appears similar in both single transgenic and SP-C/MIA mice (E,F). All panels are at the same magnification. Comparison of the content of saturated phosphatidylcholine (SatPC) in the lungs of SP-C/MIA mice and controls demonstrates that the lungs of SP-C/MIA mice contain only 40% (*, p < 0.0001) of the SatPC found in single transgenic controls. Data are plotted as means ± SE (n = 6 for SP-C/MIA mice and 7 for controls).
To assess the effect of misexpressing MIA on overall lung gene expression, we analyzed RNA from day E18.5 lungs of bitransgenic and control littermates by microarray (Affymetrix MOE430 chip). We found 102 genes that were decreased at least 1.5-fold and 87 genes that were increased for more than 1.5-fold in bitransgenic lungs (Table 2). Downregulated genes potentially relevant to the phenotype of respiratory failure included surfactant proteins (Sftpa, Sftpb), proteins involved in phospholipid production (ABCA3, Aytl2, and Scd2), proteins related to other aspects of lipid metabolism (Acoxl, Pon1, Gdpd2, Dlk1), and Cebpa. Upregulated genes potentially relevant to the phenotype included Mia1 (30-fold), along with genes involved in cell adhesion (Dsg2, Dcbld2, Cdh1, Cdh8, Cdh16). The overall gene expression profile was consistent with retarded lung maturation.
Table 2.
Genes Changed by Misexpression of MIA
| Genbank Accession #
|
Fold Change
|
Symbol
|
Gene
|
|---|---|---|---|
| Transcription Factors | |||
| NM_007678 Protein | −1.7 | Cebpa | CCAAT/enhancer Binding (C/EBP), alpha |
| NM_010234 Oncogene | −1.6 | Fos | FBJ osteosarcoma related |
| NM_009331 Specific | −1.6 | Tcf7 | Transcription factor 7, T-cell |
| NM_010703 factor 1 | −1.6 | Lef1 | Lymphoid enhancer binding |
| NM_008259 | 1.6 | Foxa1 | Forkhead box A1 |
| NM_016768 transcription | 1.7 | Pbx3 | Pre B-cell leukemia factor 3 |
| NM_207215 | 1.7 | Phr1 | Pam, highwire, rpm 1 |
| NM_010574 | 1.7 | Irx2 | Iroquois related homeobox 2 |
| NM_177338 | 2.8 | Hmbox1 | Homeobox containing 1 |
| Development | |||
| NM_177905 (Drosophila) | −3.5 | Piwil4 | Piwi-like homolog 4 |
| XM_897426 | −2.9 | Plekhk1 | Pleckstrin homology domain containing, family K member |
| 1 | |||
| NM_133245 | −1.9 | Eraf | Erythroid associated factor |
| NM_011313 | −1.8 | S100a6 | S100 calcium binding protein |
| A6 | |||
| NM_008970 peptide | −1.7 | Pthlh | Parathyroid hormone-like |
| NM_010103 discoidin I-like | −1.7 | Edil3 | EGF-like repeats and domains 3 |
| NM_007447 | −1.7 | Ang1 | Angiogenin 1 |
| NM_054077 leucine- | −1.6 | Prelp | Proline arginine-rich end rich repeat |
| NM_009114 | −1.6 | S100a9 | S100 calcium binding protein |
| A9 NM_009627 | −1.5 | Adm | Adrenomedullin |
| XM_134149 homolog | 1.5 | Fath | Fat tumor suppressor |
| NM_172537 | 1.5 | Sema6d | Semaphorin 6D |
| NM_011350 | 1.6 | Sema4f | Semaphorin 4f |
| NM_010164 | 1.7 | Eya1 | Eyes absent 1 homolog |
| NM_031166 | 1.9 | Idb4 | Inhibitor of DNA binding 4 |
| NM_008520 factor | 1.9 | Ltbp3 | Latent transforming growth beta binding protein 3 |
| NM_007963 | 1.9 | Evi1 | Ecotropic viral integration |
| site 1 | |||
| NM_010181 | 2.4 | Fbn2 | Fibrillin 2 |
| NM_011674 | 2.5 | Ugt8 | UDP-glucuronosyltransferase |
| 8 | |||
| Signal Transduction | |||
| NM_008731 | −4.20 | Npy2r | Neuropeptide Y receptor Y2 |
| NM_007413 | −2.9 | Adora2b | Adenosine A2b receptor |
| NM_023635 | −2.6 | Rab27a | RAB27A |
| NM_013875 | −2.2 | Pde7b | Phosphodiesterase 7B |
| NM_144547 | −2.0 | Amhr2 | Anti-Mullerian hormone type receptor |
| 2 | |||
| NM_007592 | −2.0 | Car8 | Carbonic anhydrase 8 |
| NM_010407 | −1.8 | Hck | Hemopoietic cell kinase |
| NM_009217 | −1.7 | Sstr2 | Somatostatin receptor 2 |
| NM_021409 homolog | −1.6 | Pard6b | Partitioning defective 6 beta |
| NM_009062 signaling 4 | −1.6 | Rgs4 | Regulator of G-protein |
| NM_021099 | −1.6 | Kit | Kit oncogene |
| NM_025730 | −1.5 | Lrrk2 | Leucine-rich repeat kinase 2 |
| NM_138650 gamma | −1.5 | Dgkg | Diacylglycerol kinase, |
| NM_019725 split 2 | −1.5 | Tle2 | Transducin-like enhancer of homolog |
| NM_020505 | −1.5 | Vav3 | Vav 3 oncogene |
| NM_145566 like | 1.5 | Hrbl | HIV-1 Rev binding protein- |
| NM_013846 orphan | 1.6 | Ror2 | Receptor tyrosine kinase-like receptor 2 |
| XM_130346 155 | 1.7 | Gpr155 | G protein-coupled receptor |
| NM_019688 exchange | 1.7 | Rapgef4 | Rap guanine nucleotide factor 4 |
| NM_022721 | 1.7 | Fzd5 | Frizzled homolog 5 |
| NM_008965 | 1.7 | Ptger4 | Prostaglandin E receptor 4 |
| NM_009827 | 1.9 | Cckar | Cholecystokinin A receptor |
| NM_172815 | 2.9 | Rspo2 | R-spondin 2 homolog |
| Cell Adhesion | |||
| NM_001010937 channel | −4.4 | Gjb6 | Gap junction membrane protein beta 6 |
| NM_018857 | −2.3 | Msln | Mesothelin |
| XM_356226 homolog 3 | −1.9 | Fat3 | Fat tumor suppressor |
| NM_133721 | −1.7 | Itga9 | Integrin alpha 9 |
| NM_007734 | −1.6 | Col4a3 | Procollagen, type IV, alpha 3 |
| NM_009868 | −1.5 | Cdh5 | Cadherin 5 |
| NM_009903 | 1.7 | Cldn4 | Claudin 4 |
| NM_028078 | 1.7 | Jam4 | Junction adhesion molecule 4 |
| NM_007667 | 1.7 | Cdh8 | Cadherin 8 |
| NM_178382 | 1.8 | Flrt3 | Fibronectin leucine rich transmembrane protein 3 |
| NM_172862 matrix | 1.8 | Frem2 | Fras1 related extracellular protein 2 |
| NM_009864 | 1.9 | Cdh1 | Cadherin 1 |
| NM_007883 | 2.3 | Dsg2 | Desmoglein 2 |
| NM_028523 domain | 2.3 | Dcbld2 | Discoidin, CUB and LCCL containing 2 |
| NM_007663 | 2.9 | Cdh16 | Cadherin 16 |
| NM_019394 | 30.3 | Mia1 | Melanoma inhibitory activity |
| 1 | |||
| Transport | |||
| NM_009701 | −10.8 | Aqp5 | Aquaporin 5 |
| XM_130308 receptor- | −2.9 | Lrp2 | Low density lipoprotein related protein 2 |
| NM_133969 | −2.7 | Cyp4v3 | Cytochrome P450, family 4, subfamily v, polypeptide 3 |
| NM_013855 family A | −2.5 | Abca3 | ATP-binding cassette, sub-member 3 |
| NM_172621 | −2.0 | Clic5 | Chloride intracellular channel |
| 5 | |||
| NM_027491 | −1.9 | Rragd | Ras-related GTP binding D |
| NM_029310 gene | −1.8 | RIKEN cDNA 1700008G05 | |
| NM_011403 member 1 | −1.8 | Slc4a1 | Solute carrier family 4 |
| XM_130038 | −1.6 | Cubn | Cubilin |
| NM_153522 gated, type | −1.6 | Scn3b | Sodium channel, voltage-III, beta |
| NM_009197 member 2 | 1.5 | Slc16a2 | Solute carrier family 16 |
| NM_027052 member 4 | 1.5 | Slc38a4 | Solute carrier family 38, |
| NM_025383 associated 2 | 1.6 | Necap2 | NECAP endocytosis |
| NM_013569 channel, | 1.6 | Kcnh2 | Potassium voltage-gated subfamily H |
| NM_011985 | 1.7 | Mmp23 | Matrix metalloproteinase 23 |
| NM_172778 | 1.9 | Maob | Monoamine oxidase B |
| NM_053079 member 1 | 1.9 | Slc15a1 | Solute carrier family 15, |
| NM_018732 gated, type | 2.1 | Scn3a | Sodium channel, voltage-III, alpha polypeptide |
| NM_172613 | 2.4 | Atp13a4 | ATPase type 13A4 |
| NM_008557 ion | 2.4 | Fxyd3 | FXYD domain-containing transport regulator 3 |
| Lipid Metabolism | |||
| NM_009127 desaturase 1 | −30.3 | Scd1 | Stearoyl-Coenzyme A |
| NM_028765 like | −10.8 | Acoxl | Acyl-Coenzyme A oxidase- |
| NM_011134 | −8.8 | Pon1 | Paraoxonase 1 |
| NM_010052 | −4.1 | Dlk1 | Delta-like 1 homolog |
| NM_145376 | −4.1 | Aytl2 | Acyltransferase like 2 |
| NM_147779 | −2.7 | Sftpb | Surfactant associated protein |
| B | |||
| NM_023608 | −2.7 | Gdpd2 | Glycerophosphodiester phosphodiesterase domain containing 2 |
| NM_009128 desaturase 2 | −1.9 | Scd2 | Stearoyl-Coenzyme A |
| NM_008149 mitochondrial | −1.5 | Gpam | Glycerol-3-phosphate acyltransferase, |
| NM_175556 | 1.5 | Plch2 | Phospholipase C, eta 2 |
| NM_011921 family 1, | 1.6 | Aldh1a7 | Aldehyde dehydrogenase subfamily A7 |
| NM_146106 | 1.6 | Lyplal1 | Lysophospholipase-like 1 |
| NM_001004762 | 2.1 | Pla2g4c | Phospholipase A2, group (cytosolic, calcium-independent) |
| IVC | |||
DISCUSSION
In this study we compared transcriptomes in the E11.5 lung vs trachea in order to identify differentially expressed genes. This choice of these two tissues was based on our previous studies (Shannon et al., 1998), which showed that the entire respiratory endoderm at this stage will adopt the fate specified by the mesenchyme with which it is associated. We reasoned that number of genes differentially expressed between these tissues would be limited, and that those genes found to be upregulated in the E11.5 lung might be important in defining the lung phenotype at this stage in development. Among the diverse panel of genes listed in Table 1, one notable gene we found that was expressed in high abundance in the embryonic lung was Mia1.
MIA was originally isolated from a human melanoma cell line as a protein that was able to both suppress proliferation and change the morphology of malignant melanoma cells in vitro (Blesch et al., 1994). MIA is a small protein that is translated as a 131 amino acid precursor, which is subsequently processed to 107 amino acids by N-terminal cleavage of a hydrophobic signal peptide (Blesch et al., 1994). MIA is the prototypical member of a family of four proteins that share sequence and structural homology, including conserved cysteine residues and Src homology 3 (SH3)-like domains (Lougheed et al., 2001; Stoll et al., 2001). Whereas MIA was initially identified as an inhibitor of melanoma cell proliferation (Blesch et al., 1994; Bogdahn et al., 1989), further investigation revealed that MIA was elevated in malignant melanoma cells in vivo (Bosserhoff et al., 1999) and in vitro (van Groningen et al., 1995). Furthermore, high serum MIA levels correlated with advanced stages of melanoma (Bosserhoff et al., 1997a; Stahlecker et al., 2000) and with a poor prognosis following surgery (Guba et al., 2002). Mia1 was also identified as the gene CD-RAP in a differential display screen comparing differentiating bovine chondrocytes with those in which differentiation was suppressed by RA (Dietz and Sandell, 1996). Expression of CD-RAP coincides with the initiation of chondrogenesis and remains high throughout development of the cartilaginous skeleton (Bosserhoff et al., 1997b).
We observed that Mia1 was abundantly expressed in the epithelium during early lung patterning. One potential explanation for this pattern of expression can be drawn from observations made on metastatic melanoma cells. It has been shown that MIA binds to type III fibronectin repeats, thereby making these sites unavailable for binding to cell surface integrins (Bosserhoff et al., 2001; Stoll et al., 2001). MIA interacts with integrins α4β1 and α5β1, downregulates their activity, and reduces MAP Kinase signaling (Bauer et al., 2006). The predicted result of such competition was that the melanoma cells could detach from the extracellular matrix and metastasize (Bosserhoff et al., 2003b). Indeed, overexpression of MIA enhanced the metastatic and invasive properties of hamster melanoma cells, and antisense inhibition of Mia1 reduced their metastatic potential (Guba et al., 2000). MIA also increased the invasiveness of pancreatic cancer cells without affecting their proliferation (El Fitori et al., 2005). Considered with these results, our observations are consistent with a model in which MIA facilitates epithelial invasion of the mesenchyme by preventing cells from attaching to ECM molecules. Mia1 expression was highest during the embryonic and pseudoglandular stages of lung development, when the epithelium is actively invading the surrounding mesenchyme to pattern the pulmonary tree. This phase of lung development in the mouse ends with the initiation of the canalicular phase, which occurs on day E16.5 (Ten Have-Opbroek, 1991). Distal epithelial morphogenesis continues during the canalicular phase, with terminal bronchioles lengthening and subdividing into short acinar tubules consisting of cuboidal epithelial cells that are beginning to differentiate into type I and type II alveolar cells. Continued expansion of the most distal epithelium into the mesenchyme would explain the persistence of Mia1 expression seen in these acini on days E16.5-17.5 (Fig. 1I,J).
The E11.5 tracheal epithelium does not express Mia1, but did so when it was induced to branch by recombination with distal lung mesenchyme. Thus Mia1, like Sftpc, was expressed in epithelial cells as a specific response to inductive cues produced by lung mesenchyme. These include members of the FGF family, since treatment of intact lung explants with the FGF receptor antagonist SU5402 drastically reduced Mia1 expression. Our observation that SU5402 also inhibited Mia1 expression in mesenchyme-free lung epithelial rudiments cultured with FGFs demonstrated that FGFs directly affect Mia1 expression. The MAP Kinase/Erk and PI3-Kinase/Akt pathways are both used to propagate FGF signaling. We found that either U0126 (a MEK inhibitor) or LY294002 (a PI3-Kinase inhibitor) significantly inhibited Mia1 expression in whole lung explants. This could mean that either FGF stimulation of Mia1 is mediated by both pathways, or that Mia1 is also regulated by another factor(s) that signals through either of these pathways.
The fact that Mia1 expression was highest in the morphogenetically active signaling centers of the most distal epithelium further supports a role for MIA in mediating epithelial-mesenchymal interactions and facilitating epithelial expansion. A role for MIA in epithelial expansion may not be restricted to the lung, since we also observed that the branching epithelia of the kidney, submandibular salivary gland, and pancreas all expressed abundant Mia1 on day E15.5 (data not shown). Mia1 is also expressed in the branching epithelium of the developing mammary gland (Xie et al., 2000). Interestingly, the pattern of Mia1 expression in the developing mammary gland is dynamically similar to what we observed in the lung—expression begins on day E11.5 and ceases on E16.5. The observation that Mia1 is also expressed in carcinogen-induced mammary tumors (Lu et al., 1997) has led to speculation that Mia1 may act as a fetal oncogene (Xie et al., 2000).
Our results showing that the hyperplastic epithelial growths induced by FGF7 or FGF10 express Mia1 support the hypothesis that MIA plays a role in epithelial expansion. The lungs of E18.5 SP-C/FGF7 mice showed diffuse epithelial cell hyperplasia in the parenchyma, which causes an increase in overall lung size and volume (Tichelaar et al., 2000). Virtually every epithelial cell in this actively proliferating population was positive for Mia1. The results with CCSP/FGF10 fetuses were even more informative. In these lungs we observed focal adenoma-like regions where Mia1 was intensely expressed, whereas surrounding areas of normal parenchyma showed no signal. Therefore only those cells that were engaged in morphogenetic activity, albeit abnormal, expressed Mia1. Recent studies using a soluble dominant-negative form of the FGFR2b (Hokuto et al., 2003), or misexpression of the FGF signaling antagonist Sprouty-4 (Perl et al., 2003), have shown that FGF signaling is most critical during the embryonic and pseudoglandular stages of lung development, when Mia1 expression is highest. Misexpression of FGF7 and FGF10 may therefore cause epithelial cells to remain in a state of embryonic/pseudoglandular patterning.
The possibility that Mia1 may act as a fetal oncogene in the mammary gland epithelium should also be considered for the lung. Although we observed that Mia1 is expressed in the adenoma-like growths in the lungs of CCSP/FGF10 mice, it is important to note that these hyperplastic regions are not independent tumors, because they regress when doxycycline is withdrawn and FGF10 stimulation subsequently ends (Clark et al., 2001). We therefore examined urethane-induced lung tumors and found that Mia1 expression was increased more than 10-fold in these progressive neoplasms. These observations support the possibility that Mia1 acts as a fetal oncogene. We do not currently know if this increase in Mia1 is restricted to lung tumors induced by chemical carcinogens, or if these observations extend to human lung tumors.
Treating cultured lung explants with RA disrupted normal branching and concomitantly suppressed Mia1 expression, which also correlates with a role for Mia1 in lung branching morphogenesis. Previous studies on its effects on cultured lung explants showed that RA suppresses expression of epithelial differentiation markers Sftpa, Sftpb, Sftpc, and “proximalizes” the lung (Cardoso et al., 1996). Since our ontogeny study showed that Mia1 expression was highest in the distal lung epithelium, the RA-induced decrease in Mia1 may therefore reflect a suppression of distal epithelial differentiation in general.
While MIA appears to be involved in epithelial expansion during patterning of the pulmonary epithelium, its silencing may be required for normal lung development to proceed, particularly the differentiation of alveolar type II cells. Our observation that exogenous MIA caused significant decreases in expression of the type II cell differentiation markers Sftpa, Sftpb, Sftpc and Abca3 in cultured lung explants led us to generate transgenic mice in which we induced continued MIA expression after E16.5. We did not observe expanded morphogenesis in these fetal lungs, but instead saw decreased formation of acini and a lack of the mesenchymal thinning that normally occurs towards the end of gestation. This may indicate that MIA by itself is not able to facilitate morphogenesis and must work in concert with other factors. It is also possible that the levels of MIA expressed in SP-C/MIA mice were so high that normal cell-ECM interactions were disrupted.
Importantly, we found that continued expression of MIA resulted in neonatal death from respiratory failure, which was accompanied by changes in the expression of many genes. The observed decreases in Sftpb, Abca3, and Cebpa are notable, because deletion of any of these genes results in respiratory failure at birth (Ban et al., 2007; Clark et al., 1995; Fitzgerald et al., 2007; Martis et al., 2006). Furthermore, we found that the amount of SatPC, the most important phospholipid for lowering surface tension at the air-liquid interface in the alveolus, was only 40% of normal levels, which is consistent with a phenotype of respiratory failure (Ikegami et al., 1980). The reduced amount of SatPC correlated with decreased expression of the Aytl2 gene, which encodes a lysophosphatidylcholine acyltransferase (Chen et al., 2006) involved in the critical deacylation/reacylation pathway of SatPC synthesis (Mason and Nellenbogen, 1984).
One possible reason for the inhibition of type II cell differentiation seen when MIA was misexpressed is that MIA prevented the cells from interacting with critical ECM molecules. MIA has been shown to directly interact with both fibronectin and laminin (Bosserhoff et al., 2003b; Stoll et al., 2001), and prevents melanoma cells from adhering to these matrix proteins (Bosserhoff et al., 2003b). MIA has also been shown to interact with integrins α4β1 and α5β1, which bind fibronectin, to reduce their activity and signaling through the MAP Kinase/Erk pathway (Bauer et al., 2006). The ability of MIA to prevent cells from interacting with laminin and other ECM molecules presents one possible explanation for the observed phenotype of respiratory failure in SP-C/MIA mice. Several studies have shown that type II cells isolated from late-gestation fetuses (Chelly et al., 1999) or adult animals (Rice et al., 2002; Shannon et al., 1990; Shannon et al., 1987) require a laminin-rich substratum in order to functionally differentiate in vitro. Furthermore, targeted deletion of laminin α5 in lung epithelial cells caused a marked reduction in the number of differentiated type II cells (Nguyen et al., 2005). Mice with a specific ablation of the nidogen binding site in the laminin γ1 chain exhibited immature lungs that contained reduced numbers of air saccules and thickened mesenchyme (Willem et al., 2002). Neonatal mice with a compound genetic ablation of nidogen 1 and 2 also showed immature lung morphology, and likewise died at birth from respiratory failure (Bader et al., 2005). We observed the same immature lung morphology in SP-C/MIA mice that died at birth, which was correlated with reductions in several key molecular components of the pulmonary surfactant system. Although this possibility will require more experimentation for its validation, we speculate that the 30-fold increase in Mia1 expression in the lungs of SP-C/MIA mice may act in a dominant-negative fashion to block a critical type II cell-laminin interaction. Alternatively, high levels of MIA may disrupt the interaction of the nidogens with laminin, which is required to produce a basement membrane capable of supporting type II cell differentiation.
Whatever its role in branching morphogenesis of the lung and other epithelia may be, Mia1 by itself is not essential for normal development. Mice with a targeted deletion of Mia1 were viable and showed no major organ abnormalities, although the lung was not specifically examined. These mice did, however, exhibit subtle defects in cartilage formation at the ultrastructural level (Moser et al., 2002). The lack of phenotype in Mia1 null mice might be attributed to the fact that MIA is a member of a family of four homologous proteins that includes Otor (Mial, Fdp) (Cohen-Salmon et al., 2000; Robertson et al., 2000), Mia2 (Bosserhoff et al., 2003a), and Tango (Bosserhoff and Buettner, 2002). Since these proteins are secreted, it is possible that one or more of them serves a compensatory role in Mia1 null mice. However, we did not detect Otor and Mia2 in the E12.5 lung by RT-PCR (data not shown). Our data suggest that silencing of Mia1 may precede type II cell differentiation in late gestation. Deletion of Mia1 might therefore lead to premature type II cell differentiation, a phenotype that might not be deleterious to the neonates.
In summary, we have used DNA microarray technology to identify a panel of genes that are differentially expressed in the embryonic lung vs the trachea. We have shown that one of these genes, Mia1 exhibits a dynamic pattern of expression over the course of lung development. Mia1 encodes an ECM protein that is regulated by FGFs and is highly expressed in lung epithelial cells that are undergoing FGF-induced dysmorphogenesis. Mia1 is also increased in urethane-induced independent lung tumors. Misexpression of Mia1 results in neonatal death from respiratory failure secondary to immaturity of the surfactant system, suggesting the possibility that the decrease in Mia1 observed during late gestation may be required to facilitate type II cell differentiation.
Supplementary Material
Acknowledgments
This work was supported by Franklin Delano Roosevelt fellowship 19-FY02-234 from the March of Dimes (JMS), NHLBI R01 HL084376 (JMS), CA33497 (AMM), and the German Research Foundation (AKB). The authors gratefully acknowledge the excellent technical assistance of Kathleen Shannon, Kalpana Srivastava and Xiaofei Shangguan, and Jason Fritz for preparing the lung tumor samples.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bader BL, et al. Compound genetic ablation of nidogen 1 and 2 causes basement membrane defects and perinatal lethality in mice. Mol Cell Biol. 2005;25:6846–56. doi: 10.1128/MCB.25.15.6846-6856.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban N, et al. ABCA3 as a Lipid Transporter in Pulmonary Surfactant Biogenesis. J Biol Chem. 2007;282:9628–34. doi: 10.1074/jbc.M611767200. [DOI] [PubMed] [Google Scholar]
- Bartlett GR. Phosphorus assay in column chromatography. J Biol Chem. 1959;234:466–8. [PubMed] [Google Scholar]
- Bauer R, et al. Regulation of integrin activity by MIA. J Biol Chem. 2006;281:11669–77. doi: 10.1074/jbc.M511367200. [DOI] [PubMed] [Google Scholar]
- Blesch A, et al. Cloning of a novel malignant melanoma-derived growth-regulatory protein, MIA. Cancer Res. 1994;54:5695–701. [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Bogdahn U, et al. Autocrine tumor cell growth-inhibiting activities from human malignant melanoma. Cancer Res. 1989;49:5358–63. [PubMed] [Google Scholar]
- Bolender RP, et al. Lung morphometry: a new generation of tools and experiments for organ, tissue, cell and molecular biology. Am J Physiol. 1993;265:L521–L548. doi: 10.1152/ajplung.1993.265.6.L521. [DOI] [PubMed] [Google Scholar]
- Bosserhoff AK, Buettner R. Expression, function and clinical relevance of MIA (melanoma inhibitory activity) Histol Histopathol. 2002;17:289–300. doi: 10.14670/HH-17.289. [DOI] [PubMed] [Google Scholar]
- Bosserhoff AK, et al. Functional role of melanoma inhibitory activity in regulating invasion and metastasis of malignant melanoma cells in vivo. Melanoma Res. 2001;11:417–21. doi: 10.1097/00008390-200108000-00013. [DOI] [PubMed] [Google Scholar]
- Bosserhoff AK, et al. Melanoma-inhibiting activity, a novel serum marker for progression of malignant melanoma. Cancer Res. 1997a;57:3149–53. [PubMed] [Google Scholar]
- Bosserhoff AK, et al. Mouse CD-RAP/MIA gene: structure, chromosomal localization, and expression in cartilage and chondrosarcoma. Dev Dyn. 1997b;208:516–25. doi: 10.1002/(SICI)1097-0177(199704)208:4<516::AID-AJA7>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Bosserhoff AK, et al. In situ expression patterns of melanoma-inhibiting activity (MIA) in melanomas and breast cancers. J Pathol. 1999;187:446–54. doi: 10.1002/(SICI)1096-9896(199903)187:4<446::AID-PATH267>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Bosserhoff AK, et al. Specific expression and regulation of the new melanoma inhibitory activity-related gene MIA2 in hepatocytes. J Biol Chem. 2003a;278:15225–31. doi: 10.1074/jbc.M212639200. [DOI] [PubMed] [Google Scholar]
- Bosserhoff AK, et al. Active detachment involves inhibition of cell-matrix contacts of malignant melanoma cells by secretion of melanoma inhibitory activity. Lab Invest. 2003b;83:1583–94. doi: 10.1097/01.lab.0000097191.12477.5d. [DOI] [PubMed] [Google Scholar]
- Bourbon JR, et al. Utilization of glycogen for phospholipid synthesis in fetal rat lung. Biochim Biophys Acta. 1982;712:382–389. doi: 10.1016/0005-2760(82)90356-3. [DOI] [PubMed] [Google Scholar]
- Cardoso WV, Lu J. Regulation of early lung morphogenesis: questions, facts and controversies. Development. 2006;133:1611–24. doi: 10.1242/dev.02310. [DOI] [PubMed] [Google Scholar]
- Cardoso WV, et al. Retinoic acid alters the expression of pattern-related genes in the developing rat lung. Dev Dyn. 1996;207:47–59. doi: 10.1002/(SICI)1097-0177(199609)207:1<47::AID-AJA6>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Cardoso WV, et al. Retinoic acid induces changes in the pattern of airway branching and alters epithelial cell differentiation in the developing lung in vitro. Am J Respir Cell Mol Biol. 1995;12:464–476. doi: 10.1165/ajrcmb.12.5.7742011. [DOI] [PubMed] [Google Scholar]
- Chelly N, et al. Keratinocyte growth factor enhances maturation of fetal rat lung type II cells. Am J Respir Cell Mol Biol. 1999;20:423–32. doi: 10.1165/ajrcmb.20.3.3201. [DOI] [PubMed] [Google Scholar]
- Chen X, et al. Identification and characterization of a lysophosphatidylcholine acyltransferase in alveolar type II cells. Proc Natl Acad Sci U S A. 2006;103:11724–9. doi: 10.1073/pnas.0604946103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark JC, et al. FGF-10 disrupts lung morphogenesis and causes pulmonary adenomas in vivo. Am J Physiol Lung Cell Mol Physiol. 2001;280:L705–15. doi: 10.1152/ajplung.2001.280.4.L705. [DOI] [PubMed] [Google Scholar]
- Clark JC, et al. Targeted disruption of the surfactant protein B gene disrupts surfactant homeostasis, causing respiratory failure in newborn mice. Proc Natl Acad Sci U S A. 1995;92:7794–8. doi: 10.1073/pnas.92.17.7794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Salmon M, et al. Fdp, a new fibrocyte-derived protein related to MIA/CD-RAP, has an in vitro effect on the early differentiation of the inner ear mesenchyme. J Biol Chem. 2000;275:40036–41. doi: 10.1074/jbc.M002876200. [DOI] [PubMed] [Google Scholar]
- Dennis G, Jr, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- Deterding RR, Shannon JM. Proliferation and differentiation of fetal rat pulmonary epithelium in the absence of mesenchyme. J Clin Invest. 1995;95:2963–2972. doi: 10.1172/JCI118004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz UH, Sandell LJ. Cloning of a retinoic acid-sensitive mRNA expressed in cartilage and during chondrogenesis. J Biol Chem. 1996;271:3311–6. doi: 10.1074/jbc.271.6.3311. [DOI] [PubMed] [Google Scholar]
- El Fitori J, et al. Melanoma Inhibitory Activity (MIA) increases the invasiveness of pancreatic cancer cells. Cancer Cell Int. 2005;5:3. doi: 10.1186/1475-2867-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald ML, et al. ABCA3 inactivation in mice causes respiratory failure, loss of pulmonary surfactant, and depletion of lung phosphatidylglycerol. J Lipid Res. 2007;48:621–32. doi: 10.1194/jlr.M600449-JLR200. [DOI] [PubMed] [Google Scholar]
- Guba M, et al. Overexpression of melanoma inhibitory activity (MIA) enhances extravasation and metastasis of A-mel 3 melanoma cells in vivo. Br J Cancer. 2000;83:1216–22. doi: 10.1054/bjoc.2000.1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guba M, et al. Elevated MIA serum levels are predictors of poor prognosis after surgical resection of metastatic malignant melanoma. Oncol Rep. 2002;9:981–4. [PubMed] [Google Scholar]
- Hokuto I, et al. Prenatal, but not postnatal, inhibition of fibroblast growth factor receptor signaling causes emphysema. J Biol Chem. 2003;278:415–21. doi: 10.1074/jbc.M208328200. [DOI] [PubMed] [Google Scholar]
- Hyatt BA, et al. FGF-10 induces SP-C and Bmp4 and regulates proximal-distal patterning in embryonic tracheal epithelium. Am J Physiol Lung Cell Mol Physiol. 2004;287:L1116–26. doi: 10.1152/ajplung.00033.2004. [DOI] [PubMed] [Google Scholar]
- Ikegami M, et al. The quantity of natural surfactant necessary to prevent the respiratory distress syndrome in premature lambs. Pediatr Res. 1980;14:1082–5. doi: 10.1203/00006450-198009000-00013. [DOI] [PubMed] [Google Scholar]
- Ikegami M, et al. Alveolar lipoproteinosis in an acid sphingomyelinase-deficient mouse model of Niemann-Pick disease. Am J Physiol Lung Cell Mol Physiol. 2003;284:L518–25. doi: 10.1152/ajplung.00258.2002. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, et al. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003a;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003b;4:249–64. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Kalina M, et al. Surfactant protein C is expressed in alveolar type II cells but not in Clara cells of rat lung. Am J Respir Cell Mol Biol. 1992;6:595–600. doi: 10.1165/ajrcmb/6.6.594. [DOI] [PubMed] [Google Scholar]
- Lin S, et al. Structural requirements for intracellular transport of pulmonary surfactant protein B (SP-B) Biochim Biophys Acta. 1996;1312:177–85. doi: 10.1016/0167-4889(95)00201-4. [DOI] [PubMed] [Google Scholar]
- Lougheed JC, et al. Structure of melanoma inhibitory activity protein, a member of a recently identified family of secreted proteins. Proc Natl Acad Sci U S A. 2001;98:5515–20. doi: 10.1073/pnas.091601698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, et al. Gene expression changes associated with chemically induced rat mammary carcinogenesis. Mol Carcinog. 1997;20:204–15. doi: 10.1002/(sici)1098-2744(199710)20:2<204::aid-mc7>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Malkinson AM, Beer DS. Major effect on susceptibility to urethan-induced pulmonary adenoma by a single gene in BALB/cBy mice. J Natl Cancer Inst. 1983;70:931–6. [PubMed] [Google Scholar]
- Malpel S, et al. Regulation of retinoic acid signaling during lung morphogenesis. Development. 2000;127:3057–67. doi: 10.1242/dev.127.14.3057. [DOI] [PubMed] [Google Scholar]
- Martis PC, et al. C/EBPalpha is required for lung maturation at birth. Development. 2006;133:1155–64. doi: 10.1242/dev.02273. [DOI] [PubMed] [Google Scholar]
- Mason RJ, Nellenbogen J. Synthesis of saturated phosphatidylcholine and phosphatidylglycerol by freshly isolated rat alveolar type II cells. Biochim Biophys Acta. 1984;794:392–402. doi: 10.1016/0005-2760(84)90005-5. [DOI] [PubMed] [Google Scholar]
- Mason RJ, et al. Isolation of disaturated phosphatidylcholine with osmium tetroxide. J Lipid Res. 1976;17:281–284. [PubMed] [Google Scholar]
- Mendelsohn C, et al. Function of the retinoic acid receptors (RARs) during development (II). Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development. 1994;120:2749–71. doi: 10.1242/dev.120.10.2749. [DOI] [PubMed] [Google Scholar]
- Mollard R, et al. Stage-dependent responses of the developing lung to retinoic acid signaling. Int J Dev Biol. 2000;44:457–62. [PubMed] [Google Scholar]
- Moser M, et al. Ultrastructural cartilage abnormalities in MIA/CD-RAP-deficient mice. Mol Cell Biol. 2002;22:1438–45. doi: 10.1128/mcb.22.5.1438-1445.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen NM, et al. Epithelial laminin alpha5 is necessary for distal epithelial cell maturation, VEGF production, and alveolization in the developing murine lung. Dev Biol. 2005;282:111–25. doi: 10.1016/j.ydbio.2005.02.031. [DOI] [PubMed] [Google Scholar]
- Olsen CO, et al. Extracellular matrix-driven alveolar epithelial cell differentiation in vitro. Exp Lung Res. 2005;31:461–82. doi: 10.1080/01902140590918830. [DOI] [PubMed] [Google Scholar]
- Perl AK, et al. Temporal effects of Sprouty on lung morphogenesis. Dev Biol. 2003;258:154–68. doi: 10.1016/s0012-1606(03)00106-4. [DOI] [PubMed] [Google Scholar]
- Rice WR, et al. Maintenance of the mouse type II cell phenotype in vitro. Am J Physiol Lung Cell Mol Physiol. 2002;283:L256–64. doi: 10.1152/ajplung.00302.2001. [DOI] [PubMed] [Google Scholar]
- Robertson NG, et al. A novel conserved cochlear gene, OTOR: identification, expression analysis, and chromosomal mapping. Genomics. 2000;66:242–8. doi: 10.1006/geno.2000.6224. [DOI] [PubMed] [Google Scholar]
- Rudnick D. Developmental capacities of the chick lung in chorioallantoic grafts. J Exp Zool. 1933;66:125–154. [Google Scholar]
- Shannon JM. Induction of alveolar type II cell differentiation in fetal tracheal epithelium by grafted distal lung mesenchyme. Develop Biol. 1994;166:600–614. doi: 10.1006/dbio.1994.1340. [DOI] [PubMed] [Google Scholar]
- Shannon JM, et al. Effect of a reconstituted basement membrane on expression of surfactant apoproteins in cultured adult rat alveolar type II cells. Am J Respir Cell Mol Biol. 1990;2:183–192. doi: 10.1165/ajrcmb/2.2.183. [DOI] [PubMed] [Google Scholar]
- Shannon JM, et al. Induction of alveolar type II cell differentiation in embryonic tracheal epithelium in mesenchyme-free culture. Development. 1999;126:1675–88. doi: 10.1242/dev.126.8.1675. [DOI] [PubMed] [Google Scholar]
- Shannon JM, Hyatt BA. Epithelial-mesenchymal interactions in the developing lung. Annu Rev Physiol. 2004;66:625–45. doi: 10.1146/annurev.physiol.66.032102.135749. [DOI] [PubMed] [Google Scholar]
- Shannon JM, et al. Functional differentiation of alveolar type II epithelial cells in vitro: effects of cell shape, cell-matrix interactions, and cell-cell interactions. Biochim Biophys Acta. 1987;931:143–156. doi: 10.1016/0167-4889(87)90200-x. [DOI] [PubMed] [Google Scholar]
- Shannon JM, et al. Mesenchyme specifies epithelial differentiation in reciprocal recombinants of embryonic lung and trachea. Dev Dyn. 1998;212:482–94. doi: 10.1002/(SICI)1097-0177(199808)212:4<482::AID-AJA2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- Spooner BS, Wessells NK. Mammalian lung development: Interactions in primordium formation and bronchial morphogenesis. J Exp Zool. 1970;175:445–454. doi: 10.1002/jez.1401750404. [DOI] [PubMed] [Google Scholar]
- Stahlecker J, et al. MIA as a reliable tumor marker in the serum of patients with malignant melanoma. Anticancer Res. 2000;20:5041–4. [PubMed] [Google Scholar]
- Stoll R, et al. The extracellular human melanoma inhibitory activity (MIA) protein adopts an SH3 domain-like fold. Embo J. 2001;20:340–9. doi: 10.1093/emboj/20.3.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ten Have-Opbroek A. Lung development in the mouse embryo. Exp Lung Res. 1991;17:111–130. doi: 10.3109/01902149109064406. [DOI] [PubMed] [Google Scholar]
- Tichelaar JW, et al. Conditional expression of fibroblast growth factor-7 in the developing and mature lung. J Biol Chem. 2000;275:11858–64. doi: 10.1074/jbc.275.16.11858. [DOI] [PubMed] [Google Scholar]
- van Groningen JJ, et al. Identification of melanoma inhibitory activity and other differentially expressed messenger RNAs in human melanoma cell lines with different metastatic capacity by messenger RNA differential display. Cancer Res. 1995;55:6237–43. [PubMed] [Google Scholar]
- Warburton D, et al. Growth factor signaling in lung morphogenetic centers: automaticity, stereotypy and symmetry. Respir Res. 2003:4–5. doi: 10.1186/1465-9921-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wert SE, et al. Transcriptional elements from the human SP-C gene direct expression in the primordial respiratory epithelium of transgenic mice. Dev Biol. 1993;156:426–443. doi: 10.1006/dbio.1993.1090. [DOI] [PubMed] [Google Scholar]
- Wert SE, et al. Increased metalloproteinase activity, oxidant production, and emphysema in surfactant protein D gene-inactivated mice. Proc Natl Acad Sci U S A. 2000;97:5972–7. doi: 10.1073/pnas.100448997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson DG. Whole mount in situ hybridization of vertebrate embryos. In: Wilkinson DG, editor. In Situ Hybridization, A Practical Approach. IRL Press; New York: 1992. pp. 75–83. [Google Scholar]
- Willem M, et al. Specific ablation of the nidogen-binding site in the laminin gamma1 chain interferes with kidney and lung development. Development. 2002;129:2711–22. doi: 10.1242/dev.129.11.2711. [DOI] [PubMed] [Google Scholar]
- Xie WF, et al. The 2.2-kb promoter of cartilage-derived retinoic acid-sensitive protein controls gene expression in cartilage and embryonic mammary buds of transgenic mice. Matrix Biol. 2000;19:501–9. doi: 10.1016/s0945-053x(00)00087-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.