

SUMMARY
Challenges in the production of integral membrane proteins for structural studies include low expression levels, incorrect membrane insertion, aggregation and instability. In this report, we describe a “funnel approach” to overcome these difficulties, and demonstrate its efficacy in a case study of 36 prokaryotic P-type transporters. A diverse ensemble of modified constructs are generated and tested for expression in E. coli, membrane localization, detergent extraction, and homogeneity. High throughput methodologies are implemented throughout the process to facilitate identification of promising targets. We find that the choice of promoter, the choice of source organism providing the cloned gene, and most importantly the position of the affinity tag have a great effect on successful production. The latter had pronounced effects at all tested levels, from expression levels observed in whole cells, to extent of membrane insertion, and even on protein function. Following the initial stream lined screening, we were able to fine tune and produce 9 of the 36 targets as material suitable for crystallization or other structural studies.
Keywords: membrane protein, crystallization, P, type ATPase
INTRODUCTION
The passage of most biologically relevant molecules across the permeability barrier created by the cell membrane is mediated by specialized membrane proteins known as transporters. The significance of transport processes to cellular metabolism is emphasized by the observation that over 550 families of transporters have been identified through biochemical and genomic analyses1 (http://www.tcdb.org) Despite recent successes and advances2, structural studies of transporters remain a formidable challenge. The relative paucity of structural information stands in clear contrast to the interest and importance of these proteins as pivotal participants in all physiological processes. Moreover, their membrane localization makes them attractive pharmaceutical targets, and an increasing number of transporters have been directly implicated in human diseases3–8. An example of the physiological significance of membrane proteins may be provided by the P-type ATPases, a family of transporters characterized by unique signature motifs, functional, and structural features9. Members of this family include the Na+/K+-ATPase, the gastric H+-ATPase, and the sarcoplasmic reticulum (SR) Ca+2-ATPase. The hallmark of this family of pumps is the formation of a phospho-enzyme intermediate (hence the name P-type ATPase), by the transfer of the γ-phosphate from ATP to a conserved aspartic residue. To date, only the structure of the SR Ca+2-ATPase has been solved due to pioneering work by Toyoshima and colleagues10. Structures of this pump in multiple conformations, representing various intermediates in the catalytic cycle of the enzyme, have provided an explicit transport mechanism for the SR Ca+2-ATPase and other P-type ATPases11–13.
Transition metal transporters that catalyze the extrusion of metals such as copper, zinc, lead, mercury, cadmium, manganese and magnesium, constitute a sub-class of P-type ATPases loosely referred to as “heavy-metal” or type P1B 14. While transition metals are crucial participants in many enzymatic reactions, intra-cellular concentrations of these metals are carefully controlled since elevated levels are toxic. Transition metal P-type ATPases participate in this process by pumping the cognate metals across the membrane; malfunctions of these proteins can lead to pathologies as manifested in the Wilson and Menkes diseases15. In view of their physiological functions, we have focused on this sub-group of transporters for structural analysis.
A variety of obstacles litter the path towards successful crystallization of a transporter or any other membrane protein. Not only do these proteins need to be over-expressed, correctly folded and inserted into the plasma membrane in vivo, they need to be subsequently extracted from it in vitro. Following extraction, the detergent solubilized protein must be purified while retaining stability. To overcome these obstacles, we have developed a “funnel approach” based on the screening of multiple constructs to identify those suitable for structural studies16. This is the same strategy utilized by Kendrew in the original structure determination of myoglobin17, except that rather than obtaining homologous proteins from different natural sources, one can now amplify the desired targets from genomic DNA. The basic idea is that if there is a 90% chance that a given protein will fail to crystallize, the probability that two different proteins will both fail is reduced to 0.92 = 81%; i.e., success is more likely the more proteins that are tried. As practically implemented for prokaryotic transporters18, ~30 homologous genes of interest are chosen from a variety of host genomes. A diverse pool of constructs is generated from these genes, where each gene is represented by several slightly modified constructs. This pool of constructs is then subjected to a series of sequential tests aimed at judging their suitability for structural studies. Promising candidates proceed to the next stage, while the rest of the ensemble is discarded. The number of potential targets dwindle as more tests are applied, therefore the funnel effect. The goal is to start with enough proteins so that one or more will survive to the end.
In the present work, we have amplified genes of 36 P-type ATPases from 11 different genomes and sub-cloned each gene into four different expression vectors (Table 1). The expression vectors harbor a poly-histidine affinity tag at either the N-terminus or C-terminus position, and several constructs were also fused to an additional affinity tag. Further diversity is achieved by utilizing either the T7 or arabinose promoters. An initial collection of 144 constructs was introduced into five different E. coli strains, and expression in whole cells lysates was evaluated using high throughput techniques. Highly expressed proteins were then tested for membrane localization followed by detergent extraction screens. We find that the identity and location of the fused affinity tag has a significant effect at all stages, from expression levels observed in whole cells, through membrane localization of the expressed proteins, detergent extraction, protein aggregation and stability, and even on protein function in-vivo. N-terminal tagging is generally preferable to C-terminal tagging, and transcriptional control by the T7 promoter often results in higher expression than by the arabinose promoter. By using a combinatorial approach of multiple genes, constructs, and host strains, we were able to over-produce 24 out of 36 chosen targets. 20 of these proteins were amenable to detergent extraction, and were purified using single step metal-affinity chromatography. Following purifications, candidate proteins were assayed for mono-dispersity and stability. By combining high throughput methodology with standard biochemical methods, 25% (9/36) of the target proteins could be prepared in purified, mono-disperse and stable form suitable for crystallographic trials.
Table 1.
List of genes described in this work. Proteins that expressed well in the membrane fraction are denoted with an asterix preceding the Accession number. Proteins that expressed well and survived to the end of the funnel are preceded with an additional asterix. Within the Phylum Proteobacteria, E. coli, C. jejuni, H. pylori, R. radiobacter, P. aeruginosa, R. metallidurans and P. mirabilis are in the γ–, ε–, ε–, α–, γ–, β–, α–, proteobacteria classes, respectively. S. pneumoniae and L. plantarum belong to the Phylum Firmicutes, while P. furiosus, T. volcanium and A. fulgidus belong to the Domain Archaea, Phylum Euryarchaeota (reference 44).
| Organism | Accession number | Organism | Accession number |
|---|---|---|---|
| Escherichia coli | *P37617 | Streptococcus pneumoniae | P35597 |
| Escherichia coli | *NA1 | Thermoplasma volcanium | Q978Z8 |
| Escherichia coli | *P0ABB8 | Pseudomonas aeruginosa | **Q9HXV0 |
| Escherichia coli | *P03960 | Pseudomonas aeruginosa | Q9HX93 |
| Campylobacter jejuni | **Q9PNE0 | Pseudomonas aeruginosa | *Q9I147 |
| Campylobacter jejuni | Q9PND4 | Pseudomonas aeruginosa | *Q9I3G8 |
| Campylobacter jejuni | Q7AR91 | Pseudomonas aeruginosa | **Q9HUY5 |
| Pyrococcus furiosus | Q8TH11 | Archaeoglobus fulgidis | **O29777 |
| Helicobacter pylori | P55989 | Archaeoglobus fulgidis | **O30085 |
| Helicobacter pylori | O26033 | Ralstonia metallidurans | **Q1LEH0 |
| Helicobacter pylori | Q59465 | Ralstonia metallidurans | *Q1LKZ44 |
| Rhizobium radiobacter | *Q8UH42 | Ralstonia metallidurans | **Q1LKZ45 |
| Rhizobium radiobacter | **Q8UG47 | Ralstonia metallidurans | *Q1LKZ46 |
| Rhizobium radiobacter | **Q8UGU8 | Ralstonia metallidurans | *Q1LAJ7 |
| Rhizobium radiobacter | *Q8UF71 | Proteus mirabilis | *O33448 |
| Streptococcus pneumoniae | Q97RR4 | Human-E. coli chimera | *NA2 |
| Streptococcus pneumoniae | Q97NE2 | Human-E. coli chimera | *NA3 |
| Streptococcus pneumoniae | *Q97PQ2 | Lactobacillus plantarum | Q88VW3 |
NA: Not applicable.
Δ-ZntA, Cu(6)-ZntA, and Cu(1–6)-ZntA, respectively, from 35.
Alternative ORF’s starting at the 1st, 51st, or 103rd amino acid respectively.
RESULTS
Amplification and molecular cloning of target genes
It has been our experience and those of others19–21 that the expression level and properties of a given gene product are often affected by the type and position of the fused affinity tag. Accordingly, as detailed in the Materials and Methods section, we created a collection of constructs where each target gene is represented by several slightly modified clones. A custom multiple cloning site was engineered and inserted in place of the original multiple cloning sites of pET19b, pET21d+ (Novagen, CA), pBAD/HisA and pBAD Myc-his-C (Invitrogen, CA). The modified vectors contained only the His-tag at either the N-terminal or C–terminal positions. All other fusions/additions originally present in these vectors were removed. Due to the identity of the multiple cloning sites in all vectors, each target gene was amplified using only one set of oligonucleotides. Our final collection of clones consisted of 144 constructs representing different versions of 36 genes. In general (with a few exceptions), the following variants were constructed for each of the target genes: N-terminal His tag, C-terminal His tag, transcriptional control via the T7 promoter, and transcriptional control via the arabinose promoter. In several cases, a second affinity tag was also introduced.
Testing expression in whole cells
To simplify expression testing, plasmid DNA for the 144 clones was stored in 96 well plates. Similarly, chemically competent cells of the host strains were prepared in 96 well plate format. Standard transformation protocols were used, and transformed cells were plated onto an LB agar tray divided to a grid of 48 squares (Q-trays, Genetix USA). The optical densities of 150μl cultures were continuously monitored in an automated plate reader (Saffire II, Tecan Austria), which facilitated mid log-phase induction (figure 1A). The growth curves of the cultures harboring the different pBAD (arabinose promoter) constructs were more uniform relative to those harboring the pET (T7 promoter) constructs (compare figure 1A left and right panels, respectively). Post induction, the pBAD constructs generally continued to grow normally, while the growth of many of the pET constructs stopped, with some cultures lysing. The initial expression studies for all clones utilized E. coli strain BL21 (DE3) GOLD™ (Stratagene). Expression was tested at three different temperatures (37°, 28°, 23°), along with variations in duration of induction times and inducer concentrations (see Materials and Methods for details). To semi-quantitatively assess the results, expression levels were categorized relative to the expression level of the vitamin B12 transporter BtuCD that was previously found to express and purify well18. The His-tagged protein content of each culture well was analyzed using dot-blot techniques (Figure 1B). 62 of the 144 clones showed expression levels equal to or higher than BtuCD and were defined as High level expressors; 29 clones showed expression levels 2–3 fold lower than BtuCD (i.e. Medium level), and 52 clones had very low expression levels (i.e. Low level).
Figure 1.
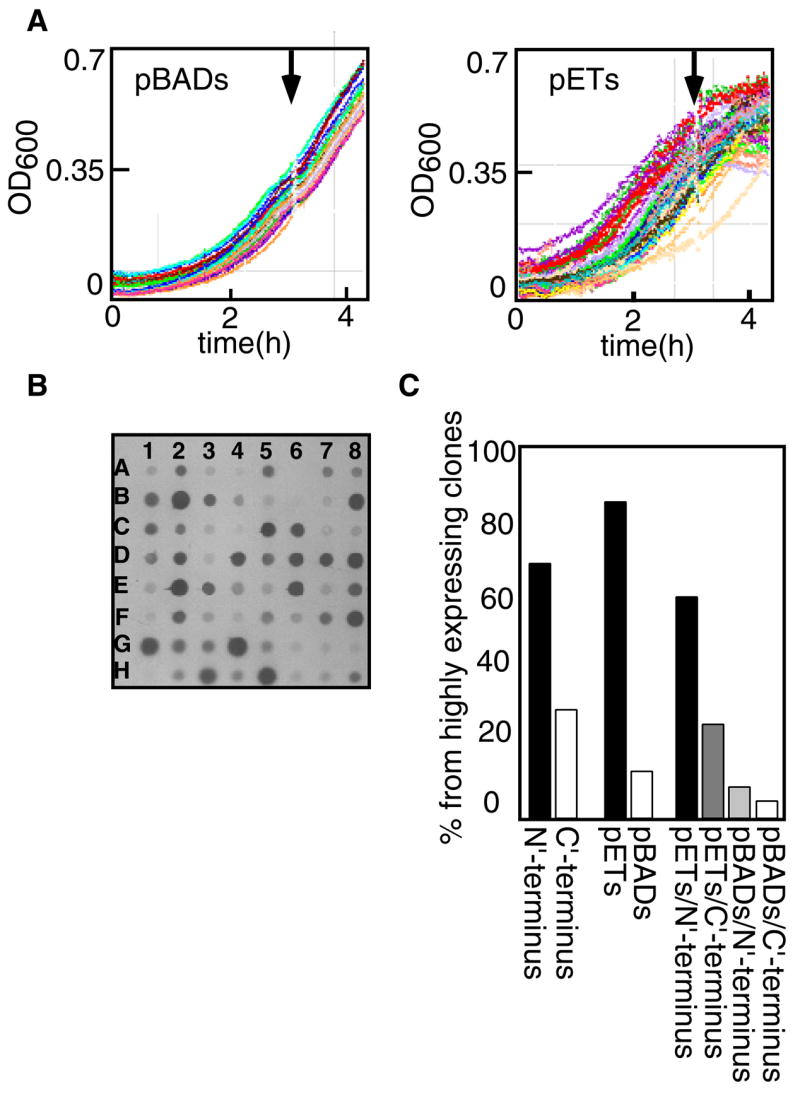
Expression in whole cell lysates: (A) Growth in an automated plate reader of E. coli BL21 (DE3) Gold expressing 48 different pBAD constructs (left panel) and 48 different pET constructs (Right panel). Arrows indicate time of induction. (B) Dot blot analysis of whole cell lysates of 64 pBAD constructs. Wells C6 and C7: ORF Q9HXV0 His-tagged at the N or C-terminus, respectively. Wells F6 and F7: Cu(1–6)-ZntA His-tagged or Myc-His-tagged, respectively. (C) Distribution of highly expressing clones according to tag position and vector type.
The most evident observation that emerges from this set of experiments is the significant influence of the affinity tag location. In many instances, an N-terminal His-tagged protein expresses well, while the same protein tagged at its C-terminal did not (figure 1B, compare well C6 to well C7). The opposite situation was also observed, although at lower frequency (figure 1C). More striking are incidents where the presence of a second tag, adjacent to the His-tag, changes the expression level of a given protein (figure 1B, compare well F6 to well F7). The choice of promoter is also important, as the T7 promoter typically resulted in higher expression than the arabinose promoter, although again there are exceptions (figure 1C). Another factor that influences expression levels is the temperature of induction. In 138 clones out of the tested 144 (~96%), the best results were obtained when inducing for two to three hours at 28°.
We next explored whether we could improve expression by using different E. coli strains. All constructs that expressed poorly in the BL21 (DE3) GOLD strain were introduced into four additional strains: BL21(DE3)RIPL™, BL21(DE3)Star™, BL21(DE3)C41/pLysS and BL21(DE3) C43/pLysS. The BL21(DE3)RIPL™ strain carries a plasmid encoding t-RNAs for codons rarely used in E. coli; however, none of the constructs tested in this strain showed elevated expression. 16% of tested clones showed somewhat elevated expression with the BL21(DE3)C41 pLysS and BL21(DE3)C43 pLysS strains that were selected for enhanced membrane protein expression22. The biggest improvement was observed with the BL21(DE3)Star™ strain containing a mutation in the rne131 gene. This strain is characterized by greater mRNA stability and indeed, 60% of the tested clones showed increased levels of expression. Overall, by using this combination of constructs and host strains, we were able to overexpress 82 constructs, representing 35 of the 36 target genes.
Membrane localization of the expressed proteins
While overexpressed membrane proteins can successfully integrate into the plasma membrane23–25, in other cases, they may not correctly incorporate into the plasma membrane and instead form insoluble aggregates or inclusion bodies26; 27. Despite the time consuming nature of membrane fraction preparations, we wished to examine the correlation between expression levels observed in the whole cells lysates and expression levels in the membrane. We thus prepared membrane vesicles from 63 of our highest expressing clones, representing 35 different genes. Surprisingly, only 29 of the 63 (46%) constructs found to express at high levels in whole cell lysates also highly expressed in the membrane fraction (again, relative to the corresponding levels of BtuCD). 8 of these 63 (13%) were present at low levels, despite their high presence in the whole cell lysates. 26 of the 63 (41%) were completely absent from the membrane fraction and were only found in the pellet of the low-speed centrifugation following cell disruption, suggesting that they were present as inclusion bodies or aggregates. 7 of the 34 clones (20%) that localized poorly to the membrane of the BL21(DE3) GOLD strain could be expressed at higher levels in the BL21(DE3)Star™ strain. Overall, 36 constructs representing 24 different genes were found to express in the membrane at levels higher than or equal to the expression level of BtuCD (Table 1).
As observed in lysates of whole cells, the type and position of the affinity tag can also influence the extent of membrane incorporation. When examining pairs of constructs of the same gene that showed similar expression levels in the whole cell lysates, several cases were observed with pronounced differences in membrane fraction content (figure 2A). Differences were not only manifested in expression levels, but also by the presence of additional bands in SDS-PAGE analysis. These bands, often of smaller size, presumably represent degradation products or products of incomplete translation. We also noted that in several of the examined pairs, a second affinity-tag (adjacent to the His-tag) resulted in alteration of the extent of membrane incorporation and in the appearance of additional bands (figure 2A). As with whole cell expression, N-terminal tagging also proved advantageous with respect to levels of membrane expression, since 64% of the clones that were highly expressed in the membrane were tagged at their N-terminus (figure 2B).
Figure 2.

Membrane-associated expression: (A) Western blot analysis of his-tagged protein expression in the membrane fraction. 1: C- and N- terminal his tagged versions of ORF O29777. 2: C- and N- terminal his tagged versions of ORF Q1LKZ4. 3: C-V5-His and C-His tagged versions of ORF Q9PNE0. 4: C-terminal his and C-terminal Myc-his tagged versions of Cu(6)-ZntA (B) Distribution of highly expressed membrane-fraction constructs according to tag position and vector type. (C) Correlation between expression levels observed in whole cells and those observed in the membrane fraction. Left panel; pBAD constructs, Right panel pET constructs. In both panels, each point is an individual protein, and the linear fit is depicted as a solid line. In the right panel only, the dashed line represents the linear fit when only data up to 8μg protein per ml of culture is included.
At the whole cell level, transcription regulation by the T7 promoter resulted in higher expression levels than those achieved by the arabinose promoter (figure 1C). This difference was less pronounced in membranes, however, as 40% of the highly expressing proteins in this case were expressed as pBAD constructs, relative to only 13% at the whole cell level (compare figure 1C to figure 2B). In general, the correlation between expression levels in whole cells and in the membrane was better for the pBAD constructs than that of the pET constructs. Figure 2C shows a semi-quantitative analysis (see Material and Methods for details) of this effect for a subset of 24 pBAD constructs and 24 pET constructs representing a wide range of expression levels (0.75 to 6.8 μg/ml for the pBADs, 2.6 to 17.2 μg/ml for the pETs). The correlation between whole cell and membrane expression for the pBAD constructs is essentially linear over the whole expression range. In contrast, the correlation between whole cell and membrane expression is much less evident for the pET constructs (figure 2C right panel, solid line). Interestingly, for the subset of pET constructs expressing at lower levels (in the range of 2.6 to 8 μg/ml), a linear fit is clearly evident (figure 2C right panel, dashed line) and similar to the fit of the complete set of pBAD constructs.
Detergent extraction of membrane proteins
An essential step in crystallographic studies of membrane proteins is their detergent-mediated extraction from the membrane. The choice of detergent is crucial, since it has significant implications for protein oligomeric state, integrity and function. Changes in head group size, chemical nature, and chain length can result in inactivation of the target protein, a highly undesirable consequence28–30. Moreover, detergent molecules may also effect crystal formation, and often a membrane protein that will crystallize in one detergent will not crystallize in another. Of the plethora of commercially available detergents, we opted to test only the following four: N,N-dimethyldodecylamine-N-oxide (LDAO), octyl-β-D-glucopyranoside (OG), dodecyl-β-D-maltoside (DDM), and dodecyloctaoxyethylene (C12E8). These detergents have been successfully used in the past in functional and crystallographic studies of numerous membrane proteins (for a comprehensive list see http://blanco.biomol.uci.edu/Membrane_Proteins_xtal.html, http://www.mpdb.ul.ie, and http://www.mpibp-frankfurt.mpg.de/michel/public/memprotstruct.html); furthermore C12E8 was used in the crystallization of rabbit SR Ca+2-ATPase10. Membrane preparations of each protein were placed in a 96-well plate and the test detergent was added directly to the wells. Following agitation, soluble and insoluble fractions were separated by brief ultracentrifugation. Figure 3A shows the results of such an extraction assay. 29 of the 36 constructs (representing 20 genes) could be extracted in soluble form with at least one of the test detergents, and often with more than one. The most efficient detergent was DDM, closely followed by LDAO. Overall, OG resulted in lower extraction levels than with either DDM or LDAO, while the least efficient detergent for extraction was C12E8. When the membranes were treated with the more aggressive detergent Fos-choline 14 (FC-14), much higher extraction levels were observed. In some instances, proteins that could not be extracted at all with the four “mild” detergents were efficiently extracted by FC-14 (Figure 3B).
Figure 3.

Western blot analysis of detergent extraction assays. (A) Membrane vesicles of 11 different proteins (accession numbers given on top of each panel) were incubated with 1% dodecyl-β-D-maltoside (DDM). T: total membrane fraction content. S: soluble fraction following detergent extraction. (B) Soluble fractions following detergent extraction of ORF’s Q9HXV0, Q1LKZ4, and Q9PNE0 as indicated. 1% of either Fos-choline 14 (Fch), octyl-β-D-glucopyranoside (OG), dodecyl-β-D-maltoside (DDM), N,N-dimethyldodecylamine-N-oxide (LDAO) were used.
Purification and stability of the chosen targets
We next proceeded to purify twenty proteins that showed adequate levels of membrane-associated expression and could be extracted with at least one of the mild detergents. Most of these proteins (17/20) could be purified to 80–90% homogeneity using single step metal affinity chromatography. Figure 4 shows an ensemble of such purifications, representing different levels of purity and yields. In general, yields varied between 0.5 and 2 mg of pure protein per liter of culture. Since our goal at this stage of the process is the initial identification of promising targets, we did not attempt to further refine these crude purifications. Rather, we proceeded to examine the mono-dispersity and stability of the selected targets. For this purpose, each of the purified proteins was concentrated to 10–20 mg/ml and injected onto a gel-filtration column (Superdex 200, GE Healthcare). These concentrated protein preparations were stored either at 4°C or room temperature, or snap-frozen in liquid nitrogen and stored at −80 °C. Following 7–10 days of storage at the different temperatures, the protein samples were re-analyzed by gel filtration chromatography. Figure 5 shows the elution profiles of several proteins, both at the time of preparation and following storage. While certain proteins displayed an elution profile that was independent of storage length or temperatures, others that initially exhibited a high level of mono-dispersity developed additional peaks corresponding to higher molecular weight species upon storage. In some of the latter cases, a high molecular-weight peak eluting in the void volume of the column (representing protein aggregation) could be removed by ultracentrifugation (Figure 5B). Mono-dispersity is often affected by the type of detergent used for extraction, and this choice may need to be re-evaluated at this stage (Figure 5C). Of the 17 proteins subjected to this analysis, 9 were found to retain mono-dispersity under all tested conditions and were consequently categorized as suitable targets for crystallization trials.
Figure 4.
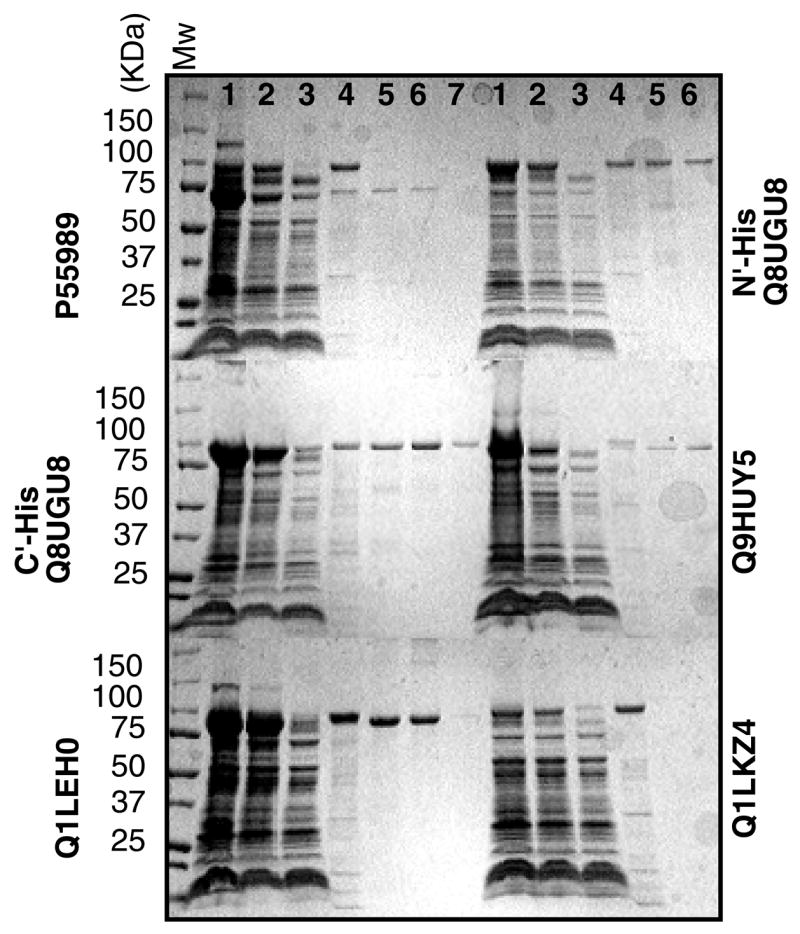
Coomassie-Blue stained SDS gel electrophoresis analysis of protein purification through Ni-NTA affinity chromatography. Six purifications are shown, with accession numbers indicated adjacent to each panel. Lanes 1: Total membrane protein, Lanes 2: Soluble fraction following detergent extraction, Lanes 3: Unbound material. Lanes 4: Wash with 60mM imidazole, Lanes 5: Wash with 100mM imidazole, Lanes 6: Wash with 200mM imidazole, Lanes 7 (were applicable): Wash with 350mM imidazole.
Figure 5.

Elution profiles following size exclusion chromatography of different protein preparations, with accession numbers indicated adjacent to each panel. Profiles shown are at time of preparation (fresh protein), or following 10-day storage at either 4°, room temperature (RT), or liquid nitrogen (N2). (B) Elution profile of ORF Q9HUY5 before or after ultracentrifugation. (C) Elution profiles of ORF Q9HUY5 when either dodecyl-β-D-maltoside (DDM) or octyl-β-D-glucopyranoside (OG) were used for extraction.
DISCUSSION
In recent years, two general approaches for the structural analysis of membrane proteins have emerged of. The first approach capitalizes on intimate knowledge of a specific protein of interest that often derives from years of comprehensive research. Functional, structural and genetic data are combined to find conditions that will allow crystallization of a single, specific membrane protein. Notable examples of membrane embedded transporters whose structures have been so determined are the lactose permease31 and the sodium/proton transporter NhaA32 from E. coli. In contrast, the second approach targets a related family of proteins, rather than a specific protein, taking advantage of the large number of homologues that have been identified through genomic sequencing studies to identify particular proteins amenable to structural study. This funnel approach to membrane protein structure was employed, for example, in our structural analysis of the mechanosensitive channel of large conductance16.
The fundamental basis to the funnel approach is diversity and selection. For prokaryotic targets, 30 to 40 homologues are typically cloned and inserted into multiple expression vectors. This pool of constructs is then tested for expression in several E. coli strains. As expression levels observed in whole cells do not necessarily correspond to expression in the membrane fraction, the second stage of our screen is to test the highly expressing clones for membrane localization of the protein targets. Once membrane-fraction localization is confirmed, several mild detergents are assayed for their extraction ability. Metal-affinity chromatography is then used for initial purification, and the mono-dispersity of the purified proteins is evaluated using gel-filtration chromatography. Proteins exhibiting a high level of mono-dispersity are then re-examined following storage at different temperatures. Only those proteins that express at adequate levels in the membrane fraction, can be extracted using a mild detergent, purified to near homogeneity, and display mono-dispersity over time are considered promising targets for crystallization trials.
In the present report, we describe the feasibility and analyze the various stages of the funnel approach as exemplified in a study of 36 metal P-type transporters.
Target selection
Our initial selection of targets was based primarily on the availability of genetic material. Nevertheless, a closer look at the results suggests that a more rational choice at the initial stage of such a project may lead to higher success rates. In the study described here, proteins found to express well in the membrane fraction were clustered according to their taxonomy. Membrane-associated expression was highest (see Table 1) for proteins from organisms belonging to the same phylum as E. coli, the proteobacteria. Even within this phylum, notable differences between classes were apparent. In addition to the three E. coli proteins, almost all of α, β, or γ subdivisions proteins expressed at high levels (13 out of 14 tested). In contrast, only 1 of the 6 tested ε-subdivision proteins expressed well. Success rates were also low (1 out of 4) for the Streptococcus pneumoniae proteins, a bacterium that belongs to a different phylum (Firmicutes) and thus further removed from E. coli. Due to the relative low number of subjects, the statistical significance of these observations is questionable. (This is also true for the archaeal genes where only 1–2 genes from a given organism were tested.) However, it seems that successful expression may be influenced by the taxonomic proximity between the host organism and the source organism. This proposal may be supported in part by the observation that a substantial majority of polytopic α-helical membrane proteins whose structures of have been solved to date have been produced from their native tissues. No structure of an ε-subdivision membrane protein has been solved, despite the considerable attention that Helicobacter pylori has received in recent years. In comparison, several structures of membrane proteins derived from γ-proteobacteria have been reported, including those of transporters. In all these cases, the proteins have been produced in E. coli33. In this regard, it is interesting to note that the membrane composition of E. coli and other γ-proteobacteria is quite similar, yet very different from that of ε-subdivision bacteria (Campylobacter jejuni, Helicobacter pylori)34–37. Clearly, more detailed studies are necessary in order to establish such a connection, but the hypothesis that genetic proximity and/or compatible membrane compositions may effect successful heterologous expression is appealing.
Choice of promoter
Each of the tested proteins was expressed under control of the T7 or arabinose promoters. When expression level was estimated by analyzing protein content of whole cell lysates, the T7 promoter appeared to be greatly advantageous (Figure 1C). However, when expression level was estimated by analyzing protein content in the membrane fraction, this apparent advantage was greatly diminished (Figure 2B). The correlation between expression level observed in whole cell lysates and that observed in the membrane fraction was better for proteins expressed under the control of the arabinose promoter (Figure 2C). The observation that proteins expressed under control of the T7 promoter more often resulted in insoluble aggregates may be due in part to very high levels of expression that exceed the capacity of the membrane. It may also be that the rate of translation exceeds the maximal rate of insertion. To partially solve the problem of membrane incorporation, one may try to slow down the rate of translation. This can be achieved by either lowering the temperature (post induction) and by varying the concentration of the inducer. In our hands, almost without exception, improved membrane integration was achieved this way without compromising total yields.
Effects of the affinity tag position
The location and identity of the affinity tag had a dramatic effect on expression levels (Figure 1) and on the extent of membrane insertion (Figure 2). Generally, N-terminal tagging resulted in higher levels of membrane-inserted protein. Nevertheless, for about 1/3 of the proteins C-terminal tagging was preferable. The magnitude of the “tag effect” was quite surprising, sometimes altering expression by 100 fold (Figures 1&2). Due to the extent of these effects, and since the goal of X-ray crystallography is to provide information on correctly folded and functionally relevant proteins, we extended our investigation of the “tag effect” to functional studies. Activity as a function of tag position was determined for 4 proteins using in-vivo metal resistance assays38. Essentially, the proteins were tested for their ability to rescue the growth of E. coli GG48, a Zn+2/Cd+2 sensitive strain39. In 3 out of the 4 test cases, significant differences between activities of the N-terminus and C-terminus versions were observed (Figure 6A–C). For example, at 60μM ZnCl2 the C-terminus version of Ralstonia metallidurans ZntA did not support growth any better than the empty vector, while the growth of cells expressing the N-terminus version was inhibited only by 40% (Figure 6A). The opposite was true for Ralstonia metallidurans CadA (Figure 6B), where the C-terminus version performed better. The most obvious differences were observed in the case of Cu(6)–ZntA, the human-E. coli chimera (Table 1). Regardless of metal concentration, the C-terminus version conferred no growth advantage relative to the empty vector, whereas cells expressing the N-terminus version grew substantially better (Figure 6C). Only in the case of Eschericia coli ZntA, the growth phenotypes seemed independent of the position of the tag (Figure 6D). The observed differences could not be attributed to differences in expression levels, since with both Ralstonia metallidurans CadA and Cu(6)–ZntA, the more active form was the one that expressed to a lesser extent. For many transport proteins, simple growth rescue experiments can be performed at the early stages of the screening process. For importers, that assay may be based on growth rescue under conditions of a limiting concentration of a relevant metabolite. For exporters, increasing concentrations of a toxic compound can be used, as was shown here and extensively performed with multidrug transporters. The simplicity of these assays, combined with the abundance of E. coli deletion strains that can be used as hosts, make these approaches readily accessible. Early identification of the active/inactive form of the target protein may save much labor and time. In retrospect, our efforts aimed at the large-scale production, purification and stabilization of the C-terminus version of Ralstonia metallidurans ZntA (Figure 4) probably would have been better directed towards the N-terminus tagged version of the same protein.
Figure 6.

Metal-sensitivity growth experiments. E. coli W3110 (Wt strain, insensitive to metals) or E. coli GG48 (mutant strain, sensitive to Cd+2 and Zn+2) were cultured in the presence of the indicated metal concentrations. In all panels: Squares: W3110/pBAD N (empty vector), circles GG48/pBAD N (empty vector), triangles: GG48 cells expressing the N-terminal tagged version of the indicated protein, crosses: GG48 cells expressing the C-terminal tagged version of the indicated protein. (A) Ralstonia metallidurans ZntA; (B) Ralstonia metallidurans CadA; (C) Human-Eschericia coli chimera Cu(6)-ZntA; (D) Eschericia coli ZntA
A recent study by von Heijne and colleagues 40 demonstrated the effect of point mutations of charged residues on membrane protein topology and membrane insertion. In light of these findings, it is not surprising that addition of multiple positively charged imidazoles at either the N- or C-termini could have profound effects on overall expression, membrane incorporation, and even function.
Choice of detergent
Once membrane integration of the target proteins is established, a suitable detergent must be identified for membrane extraction. In the absence of prior functional or structural knowledge of the protein, a good starting point is to employ detergents used previously in structural studies. Three detergents stand out with respect to successful crystallization of polytopic inner membrane proteins: N,N-dimethyldodecylamine-N-oxide (LDAO), octyl-β-D-glucopyranoside (OG) and dodecyl-β-D-maltoside (DDM); dodecyloctaoxyethylene (C12E8) was also tested since it was used in the crystallization of the Ca+-ATPase. With these four detergents, 20 of the 24 membrane proteins (29 of 36 constructs) could be extracted from the membrane, with DDM displaying the highest efficiency (Table 1). When mild detergents fail, more aggressive ones may be more effective. Treatment with Fos-14 resulted in much higher levels of extracted protein, and almost all of the tested proteins could be extracted using this detergent (figure 3). For reasons mentioned above, however, caution should be exercised when using harsh detergents. Fos-14 for example, has been reported to be used in the structural analysis of only one protein, MscS41, and it is likely that the structure may not correspond to the closed, resting state present in the membrane42.
A summary of the various stages of the funnel approach presented in this work is depicted in Figure 7. Starting with 144 constructs (representing 36 genes), expression is first tested in whole cells. Each of the constructs was introduced into five different E. coli strains and tested for expression at different temperatures. Despite the large number of combinations (~2000) this step is quite brief, as high throughput methodology allows for rapid identification of promising candidates43. This relatively rapid stage allowed us to identify favorable conditions for expression of 82 constructs, and to eliminate poorly expressing constructs from further consideration. As a next step, membrane fractions were prepared, and membrane-associated expression was evaluated. 36 constructs (representing 24 genes) proved to be adequately incorporated in the plasma membrane, and were tested in detergent-extraction assays. 29 of these constructs (representing 20 genes) were sufficiently extracted using mild detergents, while the complete set could be extracted using harsher ones. A subset of 20 constructs, representing 20 genes, was subsequently purified using single step metal chromatography. 17 of these were purified to 80–90% with yields of 0.5–2 mg of pure protein per liter of culture. Of these 17 proteins, 9 displayed the desirable single-peak appearance when analyzed by gel filtration chromatography, and maintained the same appearance over time (Table 1). The overall yield reported here (9/36; 25%), combined with the increasing number of potential targets identified through genomic sequencing, provides a basis for some optimism that the structural characterization of prokaryotic membrane proteins is a tractable objective.
Figure 7.
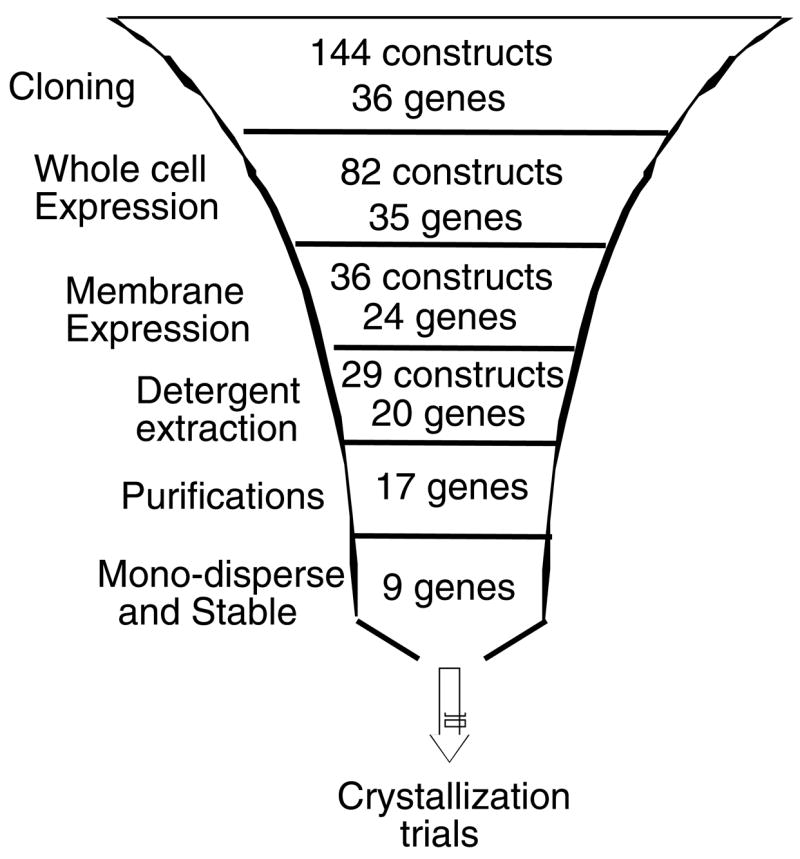
Summary of the funnel approach presented in this work, with the number of gene products remaining at each stage indicated.
MATERIALS AND METHODS
Unless otherwise stated, all chemicals were purchased from Sigma, and detergents from Anatrace.
Bacterial strains and molecular cloning techniques
E. coli strains BL21 (DE3) GOLD and BL21(DE3)RIPL were from Stratagene. Strain BL21(DE3)Star was from Invitrogen and strains BL21(DE3)C41/pLysS and BL21(DE3) C43/pLysS were purchased from Lucigen Corporation. Restriction enzymes were from Roche diagnostics, and DNA polymerase was purchased from Finnzymes. Sequences of oligonucleotides used for amplification of target genes are available upon request. The original multiple cloning sites (MCS) of pET19b, pET21d+ (Novagen, CA), pBAD/HisA, and pBAD Myc-his-C (Invitrogen, CA) were replaced with the following MCS: GAATTCGGTACCGTACGCTCGAGCTCTTCGAA.
Plasmid DNA was stored in 96-well format as were chemically competent cells of the host strains. Standard protocols were used to perform transformations in 96-well format.
Expression testing in whole cells
Single colonies were used to inoculate 150 μl of Luria Bertani medium in 96-well plates. To avoid precipitation, overnight cultures were grown at 30° under vigorous agitation (300 rpm). Cultures were then diluted 50 fold into 150 μl of Terrific Broth media and grown in an automated plate reader (Saffire II, Tecan Austria). Cell growth was conducted at temperatures of 23°, 28°, or 37°. Depending on the growth temperature, expression was induced as follows: 23°: 0.1 mM IPTG or 0.02% L-arabinose for 16 hours. 28°: 0.1 mM IPTG or 0.02% L-arabinose for 2–3 hours. 37°: 1 mM IPTG or 0.2% L-arabinose for 1 hour. To evaluate expression, 10 μl from each culture-well were added to 200 μl of 20mM Tris-HCl, 150 mM NaCl. 1%SDS, pH 7.5 in a deep-well block. This block was then sealed with adhesive aluminum tape and heated to 95° for 10 minutes. Samples were cooled to room temperature before application onto nitrocellulose membrane using a 96-well vacuum manifold (Biorad). For visualization of His-tagged protein content, a single step nickel-HRP conjugate was used (His-probe, Pierce).
Membrane associated expression
20ml cultures were grown according to previously identified conditions. Cells were disrupted by sonication and debris was removed by centrifugation at 10,000xg for 5 minutes. Membranes were collected by ultra-centrifugation at 150,000xg for 20 minutes. His-tagged protein content of the membrane fraction was analyzed either by dot-blot or by SDS-PAGE.
For semi-quantitative analysis of His-tagged protein content, micrographs were scanned using an Alpha Innotech imager. Protein bands (or dots) were quantified using the NIH Image 1.62 software.
Detergent extraction
Detergents were added to a final concentration of 1% to a 1mg/ml membrane protein suspension. Samples were agitated for 30 minutes at 4°, and insoluble material was removed by ultra-centrifugation at 100,000xg for 20 minutes. His-tagged protein content of the soluble fraction was analyzed either by dot-blot or by SDS-PAGE.
Purifications
1 liter cultures were grown according to previously identified conditions. Cells (typically 3–5 grams per liter of culture) were collected by centrifugation and disrupted using a microfluidizer processor (Microfluidics, MA, USA) at ~18,000 psi. Prior to disruption, DNAse I (Sigma) was added at 30 μg/ml to reduce the viscosity of the suspension. Unbroken cells and debris were removed by centrifugation at 8,000xg for 10 minutes, and membranes were collected by ultra-centrifugation. The soluble fraction following detergent extraction was directly added to 1.5 ml of Ni-NTA super-flow resin (Qiagen) or 1.5 ml of Talon resin (ClonTech). Following 16 hours of gentle agitation at 4°, the resin was collected by brief centrifugation (2 minutes at 700xg) and moved to disposable 10 ml columns. Contaminants were removed by washing with 20 column volumes of 20mM Tris-HCl pH 8, 500mM NaCl, 5mM β-ME, 20mM imidazole and detergent at a final concentration of 1–3 X the CMC. Additional washes with 40–100mM imidazole were used for some proteins, and elution of the His-tagged targets was observed at 100–350mM imidazole (protein specific). Purity of eluted fractions was visualized by coomassie-blue staining of SDS-PAGE gels.
Gel filtration analysis and stability assays
Purified proteins were concentrated to using a 100KDa concentrator (Amicon-Ultra, Millipore) and washed once by a ten-fold addition of buffer without imidazole. Protein concentrations were measured by absorbance at 280 nm or with Bio-rad’s RC DC Protein Assay kit. Typically, 100μl of concentrated protein (10–20 mg/ml) were injected onto a Superdex 200 gel filtration column (GE Healthcare). Samples were injected either at time of preparation or following 7–10 days of storage in different conditions, as indicated.
Metal sensitivity assays
Cultures of E. coli W3110 or E. coli GG4839. expressing the indicated proteins were diluted to optical density (600nm) of 0.05 in 150 μl of Luria Bertani medium supplemented with 0.01% L-arabinose. Cells were grown in 96-well plates in an automated plate reader (Saffire II, Tecan Austria) with increasing amounts of ZnCl2 or CdCl2.
Acknowledgments
The authors would like to thank members of the Rees group for their help in preparation of this manuscript. We thank Dietrich H. Nies for the generous gift of plasmid DNA of R. metallidurans genes; Chris Rensing for strains W3110 and GG48, David B. Wilson for plasmid DNA of L. plantarum mntA; B. Sarkar for plasmid DNA of Cu(6)-zntA and Cu(1–6)-zntA; Bharati Mitra for E. coli zntA; and Gianluca Quintini for H. pylori CadA. This work was supported in part by grants to O.L. from the Fulbright foundation and the Jane Coffin Childs Memorial Fund for Medical Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Busch W, Saier MHJ. The transporter classification (TC) system. Crit Rev Biochem Mol Biol. 2002;27:287–337. doi: 10.1080/10409230290771528. [DOI] [PubMed] [Google Scholar]
- 2.Locher KP, Bass RB, Rees DC. Breaching the barrier. Science. 2003;301:603–604. doi: 10.1126/science.1088621. [DOI] [PubMed] [Google Scholar]
- 3.Everett LA, Green ED. A family of mammalian anion transporters and their involvement in human genetic diseases. Hum Mol Genet. 1999;8:1883–91. doi: 10.1093/hmg/8.10.1883. [DOI] [PubMed] [Google Scholar]
- 4.Borst P, Elferink RO. Mammalian ABC transporters in health and disease. Annu Rev Biochem. 2002;71:537–92. doi: 10.1146/annurev.biochem.71.102301.093055. [DOI] [PubMed] [Google Scholar]
- 5.Hilgemann DW, Yaradanakul A, Wang Y, Fuster D. Molecular control of cardiac sodium homeostasis in health and disease. J Cardiovasc Electrophysiol. 2006;17(Suppl 1):S47–S56. doi: 10.1111/j.1540-8167.2006.00383.x. [DOI] [PubMed] [Google Scholar]
- 6.Camacho A, Massieu L. Role of glutamate transporters in the clearance and release of glutamate during ischemia and its relation to neuronal death. Arch Med Res. 2006;37:11–8. doi: 10.1016/j.arcmed.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 7.MacDonald KD, McKenzie KR, Zeitlin PL. Cystic fibrosis transmembrane regulator protein mutations: ‘class’ opportunity for novel drug innovation. Paediatr Drugs. 2007;9:1–10. doi: 10.2165/00148581-200709010-00001. [DOI] [PubMed] [Google Scholar]
- 8.Bressler JP, Olivi L, Cheong JH, Kim Y, Maerten A, Bannon D. Metal transporters in intestine and brain: their involvement in metal-associated neurotoxicities. Hum Exp Toxicol. 2007;26:221–9. doi: 10.1177/0960327107070573. [DOI] [PubMed] [Google Scholar]
- 9.Axelsen KB, Palmgren MG. Evolution of substrate specificities in the P-type ATPase superfamily. J Mol Evol. 1998;46:84–101. doi: 10.1007/pl00006286. [DOI] [PubMed] [Google Scholar]
- 10.Toyoshima C, Nakasako M, Nomura H, Ogawa H. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 A resolution. Nature. 2000;405:647–55. doi: 10.1038/35015017. [DOI] [PubMed] [Google Scholar]
- 11.Sorensen TL, Olesen C, Jensen AM, Moller JV, Nissen P. Crystals of sarcoplasmic reticulum Ca(2+)-ATPase. J Biotechnol. 2006;124:704–16. doi: 10.1016/j.jbiotec.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 12.Toyoshima C. Ion pumping by calcium ATPase of sarcoplasmic reticulum. Adv Exp Med Biol. 2007;592:295–303. doi: 10.1007/978-4-431-38453-3_25. [DOI] [PubMed] [Google Scholar]
- 13.Jorgensen PL, Hakansson KO, Karlish SJ. Structure and mechanism of Na, K-ATPase: functional sites and their interactions. Annu Rev Physiol. 2003;65:817–49. doi: 10.1146/annurev.physiol.65.092101.142558. [DOI] [PubMed] [Google Scholar]
- 14.Arguello JM. Identification of ion-selectivity determinants in heavy-metal transport P1B-type ATPases. J Membr Biol. 2003;195:93–108. doi: 10.1007/s00232-003-2048-2. [DOI] [PubMed] [Google Scholar]
- 15.Cox DW, Moore SD. Copper transporting P-type ATPases and human disease. J Bioenerg Biomembr. 2002;34:333–8. doi: 10.1023/a:1021293818125. [DOI] [PubMed] [Google Scholar]
- 16.Chang G, Spencer RH, Lee AT, Barclay MT, Rees DC. Structure of the MscL homolog from Mycobacterium tuberculosis: a gated mechanosensitive ion channel. Science. 1998;282:2220–6. doi: 10.1126/science.282.5397.2220. [DOI] [PubMed] [Google Scholar]
- 17.Kendrew JC, Parrish RG. The crystal structure of myoglobin. III. Sperm-whale myoglobin. Proc Roy Soc. 1957;A238:305–324. [Google Scholar]
- 18.Locher KP, Lee AT, Rees DC. The E. coli BtuCD structure: a framework for ABC transporter architecture and mechanism. Science. 2002;296:1091–8. doi: 10.1126/science.1071142. [DOI] [PubMed] [Google Scholar]
- 19.Kashino Y. Separation methods in the analysis of protein membrane complexes. J Chromatogr B. 2003;797:191–216. doi: 10.1016/s1570-0232(03)00428-8. [DOI] [PubMed] [Google Scholar]
- 20.Mohanty AK, Wiener MC. Membrane protein expression and production: effects of polyhistidine tag length and position. Prot Expr Purif. 2004;33:311–325. doi: 10.1016/j.pep.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 21.Esposito D, Chatterjee DK. Enhancement of soluble protein expression through the use of fusion tags. Curr Opin Biotech. 2006;17:353–358. doi: 10.1016/j.copbio.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 22.Miroux B, Walker JE. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol. 1996;260:289–98. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- 23.Drew DE, von Heijne G, Nordlund P, de Gier JW. Green fluorescent protein as an indicator to monitor membrane protein overexpression in Escherichia coli. FEBS Lett. 2001;507:220–4. doi: 10.1016/s0014-5793(01)02980-5. [DOI] [PubMed] [Google Scholar]
- 24.Korepanova A, Gao FP, Hua Y, Qin H, Nakamoto RK, Cross TA. Cloning and expression of multiple integral membrane proteins from Mycobacterium tuberculosis in Escherichia coli. Protein Sci. 2005;14:148–58. doi: 10.1110/ps.041022305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Surade S, Klein M, Stolt-Bergner PC, Muenke C, Roy A, Michel H. Comparative analysis and “expression space” coverage of the production of prokaryotic membrane proteins for structural genomics. Protein Sci. 2006;15:2178–89. doi: 10.1110/ps.062312706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fiermonte G, Walker JE, Palmieri F. Abundant bacterial expression and reconstitution of an intrinsic membrane-transport protein from bovine mitochondria. Biochem J. 1993;294 (Pt 1):293–9. doi: 10.1042/bj2940293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kiefer H, Krieger J, Olszewski JD, Von Heijne G, Prestwich GD, Breer H. Expression of an olfactory receptor in Escherichia coli: purification, reconstitution, and ligand binding. Biochemistry. 1996;35:16077–84. doi: 10.1021/bi9612069. [DOI] [PubMed] [Google Scholar]
- 28.Borths EL, Poolman B, Hvorup RN, Locher KP, Rees DC. In vitro functional characterization of BtuCD-F, the Escherichia coli ABC transporter for vitamin B12 uptake. Biochemistry. 2005;44:16301–9. doi: 10.1021/bi0513103. [DOI] [PubMed] [Google Scholar]
- 29.Lacapere JJ, Robert JC, Thomas-Soumarmon A. Efficient solubilization and purification of the gastric H+, K+-ATPase for functional and structural studies. Biochem J. 2000;345(Pt 2):239–45. [PMC free article] [PubMed] [Google Scholar]
- 30.Powell LD, Cantley LC. Structural changes in (Na+ + K+)-ATPase accompanying detergent inactivation. Biochim Biophys Acta. 1980;599:436–47. doi: 10.1016/0005-2736(80)90189-3. [DOI] [PubMed] [Google Scholar]
- 31.Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S. Structure and mechanism of the lactose permease of Escherichia coli. Science. 2003;301:610–615. doi: 10.1126/science.1088196. [DOI] [PubMed] [Google Scholar]
- 32.Hunte C, Screpanti E, Venturi M, Rimon A, Padan E, Michel H. Structure of a Na+/H+ antiporter and insights into mechanism of action and regulation by pH. Nature. 2005;435:1197–202. doi: 10.1038/nature03692. [DOI] [PubMed] [Google Scholar]
- 33.Raman P, Cherezov V, Caffrey M. The Membrane Protein Data Bank. Cell Mol Life Sci. 2006;63:36–51. doi: 10.1007/s00018-005-5350-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clarke K, Gray GW, Reaveley DA. The cell walls of Pseudomonas aeruginosa. General composition. Biochem J. 1967;105:749–54. doi: 10.1042/bj1050749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ames GF. Lipids of Salmonella typhimurium and Escherichia coli: structure and metabolism. J Bacteriol. 1968;95:833–43. doi: 10.1128/jb.95.3.833-843.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Inamoto Y, Ariyama S, Hamanaka Y, Okita K, Kaneta Y, Nagate T, Kondou I, Takemoto T. Lipid analysis of Helicobacter pylori. J Clin Gastroenterol. 1993;17(Suppl 1):S136–9. doi: 10.1097/00004836-199312001-00024. [DOI] [PubMed] [Google Scholar]
- 37.DiRusso CC, Nystrom T. The fats of Escherichia coli during infancy and old age: regulation by global regulators, alarmones and lipid intermediates. Mol Microbiol. 1998;27:1–8. doi: 10.1046/j.1365-2958.1998.00645.x. [DOI] [PubMed] [Google Scholar]
- 38.Hou ZJ, Narindrasorasak S, Bhushan B, Sarkar B, Mitra B. Functional analysis of chimeric proteins of the Wilson Cu(I)-ATPase (ATP7B) and ZntA, a Pb(II)/Zn(II)/Cd(II)-ATPase from Escherichia coli. J Biol Chem. 2001;276:40858–63. doi: 10.1074/jbc.M107455200. [DOI] [PubMed] [Google Scholar]
- 39.Grass G, Fan B, Rosen BP, Franke S, Nies DH, Rensing C. ZitB (YbgR), a member of the cation diffusion facilitator family, is an additional zinc transporter in Escherichia coli. J Bacteriol. 2001;183:4664–7. doi: 10.1128/JB.183.15.4664-4667.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rapp M, Seppala S, Granseth E, von Heijne G. Emulating membrane protein evolution by rational design. Science. 2007;315:1282–4. doi: 10.1126/science.1135406. [DOI] [PubMed] [Google Scholar]
- 41.Bass RB, Strop P, Barclay M, Rees DC. Crystal structure of Escherichia coli MscS, a voltage-modulated and mechanosensitive channel. Science. 2002;298:1582–7. doi: 10.1126/science.1077945. [DOI] [PubMed] [Google Scholar]
- 42.Sukharev S, Akitake B, Anishkin A. The bacterial mechanosensitive channel MscS: Emerging principles of gating and modulation. Curr Topics Memb. 2007;58:235–267. [Google Scholar]
- 43.Eshaghi S, Hedren M, Nasser MI, Hammarberg T, Thornell A, Nordlund P. An efficient strategy for high-throughput expression screening of recombinant integral membrane proteins. Protein Sci. 2005;14:676–83. doi: 10.1110/ps.041127005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garrity GM, Bell JA, Lilburn TG. Bergey’s Manual of Systematic Bacteriology release 5.0. Vol. 2. Springer-Verlag; New York: 2004. Taxonomic Outline of the Prokaryotes. [DOI] [Google Scholar]