

Abstract
The need for biochemical quantities of nonmuscle actin has been increased by observations that actin isoform composition of a cell influences the cell’s motile and structural properties. In addition, the number of actin binding proteins that exhibit different binding interactions with β- and γ-actin compared to α-actin from skeletal muscle is growing. We report a procedure designed to purify actin from nonmuscle tissues employing extraction of monomeric actin from tissues with high concentrations of Tris, chromatography on DE-53 cellulose, and affinity chromatography of DNase I-agarose. The preparation is easy to perform and yields quantities of nonmuscle actin sufficient for biochemical and cell biological assays. Actin from bovine erythrocytes and from brains of adult and embryonic chickens was obtained using this method, which can be readily used with other sources of tissue. Coomassie-Blue-stained SDS gels of the purified actin show no contaminants; capping protein, a common contaminant of actin preparations, is absent by immunoblotting. This method for purifying nonmuscle actin will be useful to investigate functional differences in the biology of actin isoforms or their regulating proteins.
Keywords: actin isoforms, nonmuscle sources, purification
INTRODUCTION
An understanding of the biochemical properties of the different actin isoforms in actin assembly and in interactions with actin binding proteins is essential for understanding how actin provides for specific cellular functions. Functional differences among the actin isoforms is suggested by the highly specific temporal and spatial patterns of expression of actin isoforms and their mRNAs in different cells and tissues [reviewed in Gunning et al., 1997; Herman, 1993; Janmey and Chaponnier, 1995]. The fact that many actin binding proteins also are expressed as tissue-specific isoforms or are localized to distinct structures in a single cell supports the idea that diversity in the binding properties of actin binding protein isoforms to different isoforms of actin provides for selective functions. In some cases, changes in actin isoform expression in cells in culture leads to differences in the structure and motility of the cells [Ronnov-Jessen and Petersen, 1996]. Furthermore, alterations in the actin isoform content in mouse heart results in severe structural and functional perturbations in cardiac tissue [Kumar et al., 1997].
Actin isoforms exhibit different biophysical and biochemical properties under some experimental conditions in vitro [Allen et al., 1996; Gordon et al., 1977; Just et al., 1994]. These studies include the findings that some actin binding proteins (i.e., profilin [Ohshima et al., 1989], thymosin β4 [Weber et al., 1992], ezrin [Shuster and Herman, 1995] and plastin [Prassler et al., 1997]) exhibit preferences for binding muscle and nonmuscle actins. Thus, studies of regulators of actin assembly should include the appropriate actin isoform. Nonetheless, most studies of actin assembly or actin binding use skeletal muscle α-actin because of the ease of obtaining the actin in high yield [Spudich and Watt, 1971]. Actin from nonmuscle sources is rarely used for biochemical analyses, even though several protocols for purifying nonmuscle actin have been described [Gordon et al., 1977; Puszkin et al., 1978; Schaier, 1992; Segura and Lindberg, 1984; Weir and Frederiksen, 1982]. One reason for this may be that many of the published methods, which are based on the method for purifying actin from skeletal muscle, are not suited for use with tissues where actin is less abundant.
We have developed a new procedure for isolating biochemical quantities of actin in high purity from nonmuscle vertebrate cells and tissues. This procedure combines several methods described in the literature, including extraction and dissociation of actin complexes in cells with high concentrations of Tris [Pinder et al., 1995], anion-exchange chromatography [Gordon et al., 1976] and affinity chromatography on DNase I-agarose [Kron et al., 1992]. Actin from adult chicken brain, bovine erythrocytes and chick embryo brain was successfully purified by this method. The method can be easily adapted for use with a variety of tissues or cultured cells.
METHODS AND MATERIALS
Reagents
Reagents were purchased from Sigma Chemical Company (St. Louis, MO) or Fisher Chemical Company (Pittsburgh, PA), except as noted.
Extraction of Actin from Bovine Erythrocytes
Approximately 6 L of bovine blood was collected using 20 ml/dl whole blood acid citrate/dextrose (0.085 M sodium citrate, 0.065 M citric acid 2% dextrose) as anticoagulate. Erythrocytes were obtained by centrifugation for 10 min at 4,300g and washed three times in Tris-buffered saline (10 mM Tris-HCl, pH 7.4, 146 mM NaCl and 0.01 mM PMSF); the white blood cell (WBC)-rich buffy coat was removed after the first wash. 500 ml of packed red cells was lysed by addition of an equal volume of 2X extraction buffer (2 M Tris-HCl, pH 7.0, 1.2 M KCl, 1 mM ATP, 2 mM DTT, 1 mM MgCl2, 2 mg/ml Tween-20, 7.5% (w/v) Triton X-100, and 0.2 mM PMSF) with gentle stirring; the mixture was incubated at 30°C for 30 min with occasional stirring. The extract was dialyzed against Buffer D (10 mM Tris-HCl, pH 8.0, 0.2 mM CaCl2, 0.5 mM ATP, 0.1 mM [DTT], 100 mM KCl, 0.1 mM [PMSF], and applied batchwise to approximately 1.5 L of DE-53 cellulose (Whatman, Maidstone, England) as described below.
Extraction of Actin from Chicken Brain
Approximately 200 g of frozen adult chicken brain tissue (Pel-Freez, Rogers, AR) was thawed in 2 L of detergent-free extraction buffer (1 M Tris-HCl, pH 7.0, 0.6 M KCl, 0.5 mM ATP, 1 mM DTT, 0.5 mM MgCl2 and 0.1 mM PMSF) by swirling the frozen tissue in buffer in a 37°C water bath. As soon as the tissue was thawed, it was homogenized using a Polytron. Triton X-100 and Tween-20 were added to the homogenate at final concentrations of 4% (v/v) and 1 mg/ml, respectively, and the mixture was homogenized briefly to mix. The homogenate was stirred on ice for 30 min and centrifuged at 25,400g for 30 min and then centrifuged at 100,000g for 1 hr. The supernatant fraction was filtered through cheesecloth to remove lipid and dialyzed against 10 L of cold water for 4 hr, followed by dialysis overnight with two changes of Buffer D. Precipitate that formed during dialysis was removed by centrifugation at 25,400g for 30 min.
Chromatography on DE-53 Cellulose
DE-53 cellulose (1.5 L) was equilibrated in Buffer D and saturated with ATP as described [Gordon et al., 1976] before use. After batchwise absorption of either erythrocyte or brain extract, the resin was stirred for 30 min, transferred to a column 10 cm in diameter, and washed with 3 L of Buffer D. The column was eluted with a 4 L gradient from 100 mM KCl to 500 mM KCl in Buffer D. Actin-containing fractions were identified by dot-blot using mAb C4 (see below), pooled, and applied to a 2.5 × 7-cm column of DNase I-agarose.
Affinity Chromatography on DNase I-agarose
Affinity chromatography on DNase I-agarose was based on the method for preparation of yeast actin described by Kron et al. [1992], using a DNase I-agarose column connected in tandem to a Sephadex G-25-150 column equilibrated in Buffer G (1 mM Tris-HCl, pH 8.0, 0.2 mM ATP, 0.2 mM CaCl2, 0.1 mM DTT, and 0.005% NaN3). The DNase I-agarose column (2.5 cm × 7 cm) was prepared as described [Kron et al., 1992] and equilibrated in Buffer G. The DE-53 cellulose pool was applied to the column, the flowthrough collected and saved and the column washed with Buffer G until the O.D. at 280 nm was < 0.01. The column was washed sequentially with 60 ml each of Buffer G containing 10% formamide, Buffer G containing 10% formamide and 0.2 M NH4Cl, and finally Buffer G. The outlet of the DNase I-agarose column was connected to the inlet of a 2.5 × 44-cm column of Sephadex G-25 equilibrated in Buffer G and the actin was eluted from the DNase I-agarose with ice-cold Buffer G containing 50% formamide. After 60 ml of the elution buffer had entered the DNase I-agarose column, the DNase I-agarose column was disconnected from the gel-filtration column. Elution of the actin from the Sephadex G-25 column was continued using Buffer G. Fractions (4 ml) were collected and monitored at 290 nm to identify the protein.
If desired, the actin obtained from the DNase I-agarose column can be concentrated by absorption to a 1-ml aliquot of DE-53 cellulose equilibrated in Buffer D and saturated with ATP as described above. After absorption, the resin is placed in a column and the actin is eluted with Buffer D containing 0.4 M NaCl. Alternatively, chromatography on a MonoQ column with elution in a steep gradient from 0.1 M NaCl to 0.4 M NaCl in Buffer D can be used in place of the batchwise DE-53 cellulose step. The actin also can be concentrated by polymerization induced by addition of 1/9 volume of 10 × MKEI buffer (1 M KCl, 20 mM MgCl2, 10 mM EGTA, and 0.2 M imidazole-HCl, pH 7.0) and incubated overnight at room temperature. Actin was pelleted by centrifugation at 132,00g in a Ti75 rotor (Beckman, Palo Alto, CA). The pellet was resuspended using a small Dounce homogenizer in a minimal volume of G-buffer (2 mM Tris-HCl, pH 8.0, 0.2 mM ATP, 0.1 mM DTT, 0.2 mM CaCl2, 0.005% NaN3) and dialyzed against several changes of G-buffer. The sample was clarified by centrifugation at 70,000 rpm in a TLA-100.3 rotor (Beckman), and the upper three-fourths of the supernatant was collected. The actin was stored on ice or flash frozen in aliquots using liquid N2.
Electrophoresis, Western Blotting, and Dot-Blots
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed using 10% acrylamide gels as described [Laemmli, 1970]. Western blots were performed as described [Schafer et al., 1994, Towbin et al., 1979]. Dot-blots to identify actin in the DE-53 column fractions were performed by spotting 2 μl of each fraction tested to a sheet of nitrocellulose. After the samples were air dried, the nitrocellulose was soaked for 15 min in 0.039 M glycine, 0.048 M Tris base, 0.0375% SDS, and 20% methanol. After rinsing the blot in TTBS (0.3 M NaCl, 20 mM Tris-HCl, pH 7.8, 0.1% (v/v) Tween-20 and 0.01% NaN3), the nitrocellulose was reacted with monoclonal antibody (mAb) C4 [Lessard, 1988], followed by reaction with alkaline phosphatase-conjugated goat anti-mouse secondary antibody. MAb 5B12, and mAb 3F2 [Schafer et al., 1996] were used to assess the amount of capping protein in samples throughout the purification. Two-dimensional gels were run as described [Rubenstein and Spudich, 1977].
RESULTS AND DISCUSSION
Actin from chicken brain and from bovine erythrocytes was prepared using a method based on a combination of extraction of monomeric actin from the tissue with high concentrations of Tris [Pinder and Gratzer, 1982], chromatography on DE-53 cellulose [Gordon et al., 1976] and affinity chromatography on DNase I-agarose [Kron et al., 1992]. The final affinity chromatography on DNase I-agarose yields a single protein with an Mr identical to that of actin on SDS gels. The purity of the actin prepared from chicken brain and from bovine erythrocytes are documented in Figure 1. Both actin preparations are free of protein contaminants as detected by Coomassie Blue staining of the SDS gels; no minor contaminants are observed in overloaded samples (lane 5, Fig. 1). A two-dimensional gel of brain actin (Fig. 2) further documents the purity of the actin. Capping protein, a common contaminant in actin prepared from skeletal muscle [Casella et al., 1995], is absent, as detected by quantitative immunoblots using mAb 3F2, the most sensitive reagent for detecting nonmuscle capping protein. The molar ratio of actin to capping protein in the sample from the DNase I-agarose column was greater than ~ 11,000:1. Absence of capping protein is important because trace amounts of capping protein will affect the kinetics of actin polymerization, the length of filaments and the binding of ligands to actin filament barbed ends. The molar ratio of capping protein and actin is similar in all tissues [Hart et al., 1997], thus, evaluation of the level of capping protein contamination in actin preparations clearly is warranted.
Fig. 1.
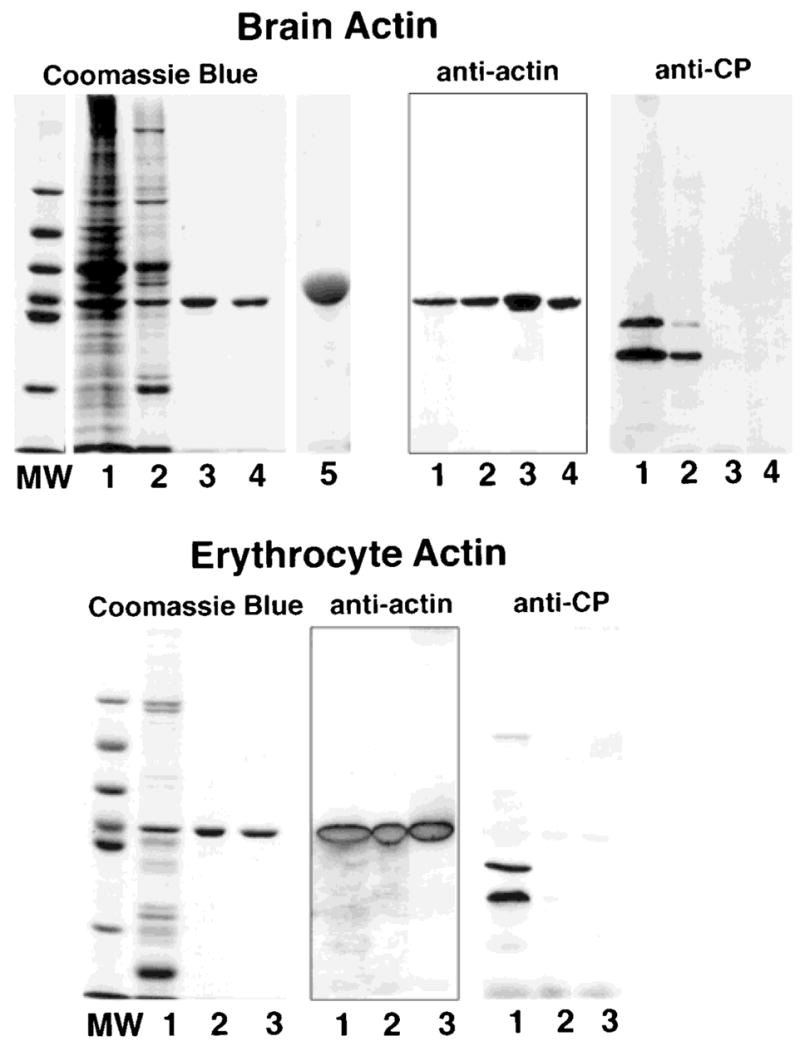
SDS-PAGE and Western blots of actin purified from chicken brain (top) and from bovine erythrocytes (bottom). Samples from each preparation were subjected to SDS-PAGE in 10% gels and stained with Coomassie Blue or transferred to nitrocellulose and probed with mAb C4 to identify actin and with mAbs 5B12 and 3F2 to identify capping protein. Samples from the brain actin preparation: lane 1, brain homogenate after dialysis; lane 2, pool from the DE-53 cellulose column; lane 3, pool from DNase I column; lane 4, pool from a MonoQ column used to concentrate the actin; lane 5, overloaded lane containing 30 μg of brain actin to document the absence of minor contaminants. Samples from the erythrocyte actin preparation: lane 1, pool from the DE-53 cellulose column; lane 2, pool from the DNase I column; lane 3, pool from the MonoQ column. A single band that binds mAb C4 is observed on the purified actin preparation obtained from the DNase I column; the samples are free of capping protein. Lanes labeled MW contain the molecular weight standards with the following molecular weights, from top to bottom: 97 kDa, 66 kDa, 55 kDa, 43 kDa, 40 kDa and, 31 kDa.
Fig. 2.

Two-dimensional gel of brain actin stained with Coomassie Blue. A major spot corresponding in pI to γ-actin (pI 5.3) and two minor spots corresponding in pI to β-actin (pI 5.28) and smooth muscle α-actin (pI 5.2) are resolved.
The yield of nonmuscle actin is sufficient for several types of biochemical assays, including kinetic analyses of binding of actin binding proteins [Schafer et al., 1996] and comparable to yields obtained from other methods for purifying nonmuscle actin (Gordon et al., 1977; Weir and Frederiksen, 1982]. Approximately 3 mg of actin was obtained from 200 g of chicken brain, ~ 2 mg of erythrocyte actin was obtained from 6 L of bovine blood, and ~ 1 mg of actin was obtained from 8 g of chick embryo brain; the increased yield of actin/g tissue from chick embryo brain may result because the Tris-Triton extract was run directly on the DNase I affinity column, bypassing the DE-53 column. Yields can be increased by running the flowthrough material from the DNase I affinity column through the DNase I column. Overall, the procedure is efficient and yields actin of high purity. The advantages of this new protocol are the increased efficiency of extracting actin from tissues and the ability to use the method on a variety of cells and tissues, including small samples.
This protocol for preparing actin exploits the most useful steps from several different described protocols for isolating cytoskeletal components. The combination of methods used here results in a protocol that is ideally suited for use with a variety of tissues and is especially useful for isolation of actin from small tissue samples or from cells grown in culture. Extraction of monomeric actin from nonmuscle tissues using high concentrations of Tris [Pinder et al., 1995] was more efficient at disrupting cytoskeletal elements than the extraction used in most of the published protocols that employ a low-ionic-strength extraction as described for isolation of actin from skeletal muscle [Spudich and Watt, 1971], where actin is abundant. Much of the actin in nonmuscle cells exists in a stable filamentous form that requires high salt to dissociate monomeric actin [Cano et al., 1992]. The Tris-Triton X-100 extraction buffer also was useful for the preparation of erythrocyte actin because extraction of whole erythrocytes eliminated having to isolate erythrocyte membranes, a process that requires multiple, high-speed centrifugations with large volumes to isolate and adequately wash the erythrocyte membranes from 6 L of blood. This new method also differs from other protocols for purifying actin in that subjecting the actin to alternate cycles of polymerization and depolymerization is not required. This is advantageous for studies of post-translational modifications to actin, such as ADP-ribosylation [Just et al., 1994], that alter the ability of actin to polymerize.
The initial purification step using DE-53 cellulose anion-exchange chromatography [Gordon et al., 1976] removed considerable contaminating proteins, including most of the capping protein and hemoglobin, and enriches for the actin prior to running the DNase I affinity column. An added benefit of the DE-53 cellulose column is the ability to use the same column to isolate other proteins, such as capping protein [Schafer et al., 1996], that elute in adjacent fractions of the salt gradient. Attempts to omit the DE-53 column for preparations starting with large quantities of tissue were not successful because of extremely slow flow rates through the DNase I-agarose column. However, the DE-53 cellulose column could be omitted when isolating actin from small amounts of tissue or cells where the extract volume and starting protein concentrations are low. This approach was successful for isolating ~ 1 mg of chick embryo brain actin in about 1.5 days starting with 8 g of tissue.
To ensure that brief treatment of the actin with 50% formamide during elution from the DNase I affinity column does not result in denaturation of a portion of the actin, we measured the critical concentration for polymerization of 6 μM actin obtained from the DNase I column. The concentration of monomeric actin remaining in the supernatant at equilibrium was 0.35 μM, compared with a concentration of monomeric actin of 0.23 μM in the supernatant from the same actin preparation after one cycle of polymerization. Thus, a small portion of actin obtained from the DNase I column may be denatured or modified and does not polymerize.
References
- Allen PG, Shuster CB, Kas J, Chaponnier C, Janmey PA, Herman IM. Phalloidin binding and rheological differences among actin isoforms. Biochemistry. 1996;35:14062–14069. doi: 10.1021/bi961326g. [DOI] [PubMed] [Google Scholar]
- Cano ML, Cassimeris L, Fechheimer M, Zigmond SH. Mechanisms responsible for F-actin stabilization after lysis of polymorphonuclear leukocytes. J Cell Biol. 1992;116:1123–1134. doi: 10.1083/jcb.116.5.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casella JF, Barron-Casella EA, Torres MA. Quantitation of Cap Z in conventional actin preparations and methods for further purification of actin. Cell Motil Cytoskeleton. 1995;30:164–170. doi: 10.1002/cm.970300208. [DOI] [PubMed] [Google Scholar]
- Gordon DJ, Eisenberg E, Korn ED. Characterization of cytoplasmic actin isolated from Acanthamoeba castellanii by a new method. J Biol Chem. 1976;251:4778–4786. [PubMed] [Google Scholar]
- Gordon DJ, Boyer JL, Korn ED. Comparative biochemistry of non-muscle actins. J Biol Chem. 1977;252:8300–8309. [PubMed] [Google Scholar]
- Gunning P, Weinberger R, Jeffrey P. Actin and tropomyosin isoforms in morphogenesis. Anat Embryol. 1997;195:311–315. doi: 10.1007/s004290050050. [DOI] [PubMed] [Google Scholar]
- Hart MC, Korshunova YO, Cooper JA. Vertebrates have conserved capping protein a isoforms with specific expression patterns. Cell Motil Cytoskeleton. 1997;38:120–132. doi: 10.1002/(SICI)1097-0169(1997)38:2<120::AID-CM2>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Herman IM. Actin isoforms. Curr Opin Cell Biol. 1993;5:48–55. doi: 10.1016/s0955-0674(05)80007-9. [DOI] [PubMed] [Google Scholar]
- Janmey PA, Chaponnier C. Medical aspects of the actin cytoskeleton. Curr Opin Cell Biol. 1995;7:111–117. doi: 10.1016/0955-0674(95)80052-2. [DOI] [PubMed] [Google Scholar]
- Just I, Wollenberg P, Moss J, Aktories K. Cysteine-specific ADP-ribosylation of actin. Eur J Biochem. 1994;221:1047–1054. doi: 10.1111/j.1432-1033.1994.tb18823.x. [DOI] [PubMed] [Google Scholar]
- Kron SJ, Drubin DG, Botstein D, Spudich JA. Yeast actin filaments display ATP-dependent sliding movement over surfaces coated with rabbit muscle myosin. Proc Natl Acad Sci USA. 1992;89:4466–4470. doi: 10.1073/pnas.89.10.4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Crawford K, Close L, Madison M, Lorenz J, Doetschman T, Pawlowski S, Duffy J, Neumann J, Robbins J, Boivin GP, O’Toole BA, Lessard JL. Rescue of cardiac alpha-actin-deficient mice by enteric smooth muscle gamma-actin. Proc Natl Acad Sci USA. 1997;94:4406–4411. doi: 10.1073/pnas.94.9.4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lessard JL. Two monoclonal antibodies to actin: One muscle selective and one generally reactive. Cell Motil Cytoskeleton. 1988;10:349–362. doi: 10.1002/cm.970100302. [DOI] [PubMed] [Google Scholar]
- Ohshima S, Abe H, Obinata T. Isolation of profilin from embryonic chicken skeletal muscle and evaluation of its interaction with different actin isoforms. J Biochem. 1989;105:855–857. doi: 10.1093/oxfordjournals.jbchem.a122765. [DOI] [PubMed] [Google Scholar]
- Pinder JC, Gratzer WB. Investigation of the actin deoxyribonuclease-I interaction using a pyrene-conjugated actin derivative. Biochemistry. 1982;21:4886–4890. doi: 10.1021/bi00263a009. [DOI] [PubMed] [Google Scholar]
- Pinder JC, Sleep JA, Bennett PM, Gratzer WB. Concentrated Tris solutions for the preparation, depolymerization, and assay of actin: Application to erythroid actin. Anal Biochem. 1995;225:291–295. doi: 10.1006/abio.1995.1157. [DOI] [PubMed] [Google Scholar]
- Prassler J, Stocker S, Marriott G, Heidecker M, Kellerman J, Gerisch G. Interaction of a Dictyostelium member of the plastin/fimbrin family with actin filaments and actin-myosin complexes. Mol Biol Cell. 1997;8:83–95. doi: 10.1091/mbc.8.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puszkin S, Maimon J, Puszkin E. Erythrocyte actin and spectrin. Interactions with muscle contractile and regulatory proteins. Biochim Biophys Acta. 1978;513:205–220. doi: 10.1016/0005-2736(78)90174-8. [DOI] [PubMed] [Google Scholar]
- Ronnov-Jessen L, Petersen OW. A function for filamentous alpha-smooth muscle actin: Retardation of motility in fibroblasts. J Cell Biol. 1996;134:67–80. doi: 10.1083/jcb.134.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein PA, Spudich JA. Actin microheterogeneity in chick embryo fibroblasts. Proc Natl Acad Sci USA. 1977;74:120–123. doi: 10.1073/pnas.74.1.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DA, Korshunova YO, Schroer TA, Cooper JA. Differential localization and sequence analysis of capping protein β-subunit isoforms of vertebrates. J Cell Biol. 1994;127:453–465. doi: 10.1083/jcb.127.2.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DA, Jennings PB, Cooper JA. Dynamics of capping protein and actin assembly in vitro: Uncapping barbed ends by polyphosphoinositides. J Cell Biol. 1996;135:169–179. doi: 10.1083/jcb.135.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaier SR. Purification and characterization of platelet actin, actin-binding protein, and α-actinin. Methods Enzymol. 1992;215:58–77. doi: 10.1016/0076-6879(92)15053-f. [DOI] [PubMed] [Google Scholar]
- Segura M, Lindberg U. Separation of non-muscle isoactins in the free form or as profilactin complexes. J Biol Chem. 1984;259:3949–3954. [PubMed] [Google Scholar]
- Shuster CB, Herman IM. Indirect association of ezrin with F-actin-isoform specificity and calcium sensitivity. J Cell Biol. 1995;128:837–848. doi: 10.1083/jcb.128.5.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spudich JA, Watt S. The regulation of rabbit skeletal muscle contraction. J Biol Chem. 1971;246:4866–4871. [PubMed] [Google Scholar]
- Towbin H, Staehlin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber A, Nachmias VT, Pennise C, Pring M, Safer D. Interaction of thymosin b4 with muscle and platelet actin: Implications for actin sequestration in resting platelets. Biochemistry. 1992;31:6179–6185. doi: 10.1021/bi00142a002. [DOI] [PubMed] [Google Scholar]
- Weir JP, Frederiksen DW. Preparation of cytoplasmic actin. Methods Enzymol. 1982;85:371–373. doi: 10.1016/0076-6879(82)85036-2. [DOI] [PubMed] [Google Scholar]