

Abstract
Idiopathic hypercalciuria (IH) is the commonest metabolic abnormality in patients with calcium kidney stones. It is characterized by normocalcemia, absence of diseases that cause increased urine calcium, and calcium excretion that is above 250 mg/day in women and 300 mg/day in men. Subjects with IH have a generalized increase in calcium turnover, which includes increased gut calcium absorption, decreased renal calcium reabsorption, and a tendency to lose calcium from bone. Despite the increase in intestinal calcium absorption, negative calcium balance is commonly seen in balance studies, especially on a low calcium diet. The mediator of decreased renal calcium reabsorption is not clear; it is not associated with either an increase in filtered load of calcium or altered PTH levels. There is an increased incidence of hypercalciuria in first-degree relatives of those with IH, but IH appears to be a complex polygenic trait with a large contribution from diet to expression of increased calcium excretion. Increased tissue vitamin D response may be responsible for the manifestations of IH in at least some patients.
Definition of Hypercalciuria
Calcium, the most abundant cation in human beings, is an important participant in many physiologic processes, including hormonal secretion, cardiac contraction, blood clotting, and neurotransmission. In addition, growth and repair of our skeleton demands an adequate supply of calcium, and careful maintenance of extracellular calcium concentrations. For this reason, prevailing levels of intracellular and extracellular calcium are tightly regulated, by a complex system of control mechanisms. These mechanisms control calcium absorption by gut and renal tubular epithelia, as well as fluxes of calcium on and off bone, during varying dietary calcium intakes. In healthy, non-pregnant adults, whose skeleton is no longer growing, net intestinal calcium absorption is matched by renal excretion, and provides sufficient calcium for skeletal maintenance; however, under pathologic conditions, some urine calcium may derive from bone loss.
Abnormalities of calcium homeostasis could result in a number of undesired consequences, including abnormally high or low serum calcium levels, or inadequate maintenance of bone mineral. Hypercalciuria might be defined as any level of urine calcium that exceeds net intestinal absorption, leading to net loss of calcium. However, in practice net intestinal absorption is seldom measured, and under the assumption that urine calcium is primarily derived from intestinal absorption, the working definition of hypercalciuria is urine calcium excretion that exceeds that found in the majority of healthy adults eating their usual diets. In practice, this is usually defined as a daily calcium excretion over 250 mg/day in women or 300 mg/day in men [1], or 4 mg/kg body weight in either sex [2;3]; these excretions are higher than those in 80% of normal individuals in our experience.
One of the sequelae of increased daily urine calcium excretion is the potential for formation of insoluble calcium salts, calcium oxalate or calcium phosphate (as brushite or apatite), in the urinary tract, resulting in nephrolithiasis or nephrocalcinosis. High urine calcium concentrations, which may be due to increased calcium excretion, low urine volume, or both, lead to elevated urine supersaturations with respect to these minerals in the distal nephron and urine, where crystals may form. Elevated urinary calcium excretion has been recognized as a risk factor for kidney stone formation since Flocks [3] reported that calcium stone formers had higher daily calcium excretion than non-stone formers, whether they ate diets containing 7.5 or 62.5 mmol calcium/day. In most cases, the elevated urine calcium excretion was accompanied by normal serum calcium, normal or slightly low serum phosphorus, and no overt evidence of bone disease; known causes of hypercalciuria such as primary hyperparathyroidism, sarcoidosis, Cushing’s syndrome, cancer, excess vitamin D intake, hyperthyroidism, glucocorticoid use, Paget’s disease, or renal tubular acidosis were absent. The term idiopathic hypercalciuria (IH) was coined to describe this constellation of findings [4]. IH is the most common metabolic abnormality found in adult calcium stone formers, present in 30–60% of such patients; it is common among pediatric calcium stone formers, as well [5].
Mechanisms of IH
IH is the result of the interplay between genetic background and environment. Studies have shown an increased incidence of nephrolithiasis and hypercalciuria among first degree relatives of stone formers. Among families of nine patients with kidney stones and IH, hypercalciuria was found in 19 of 44 first-degree relatives, often in multiple generations, indicating that IH has a genetic basis [6]. The precise gene or genes that contribute to IH are still unknown; the trait is most likely polygenic in origin, with contributions from several genes which may differ from one individual with IH to another. In addition, there is a large effect of diet on expression of hypercalciuria in individuals with IH.
Calcium homeostasis involves the coordinated control of mineral handling by the intestine, kidney and bone, under the influence of parathyroid hormone (PTH) and 1,25(OH)2D3. These hormones regulate the synthesis and activity of transporters responsible for calcium translocation in these sites. In addition, the calcium sensing receptor (CaSR) allows cells of the parathyroid, renal tubule, and potentially intestine and bone to monitor local calcium ion levels, and alter cell function in response to changes. Abnormalities of calcium regulation in these sites have been implicated in causing IH, however the underlying mechanisms are not completely understood and the relative contributions of these sites may vary from one individual to another.
Patients with IH have sometimes been categorized by the presumed site of the primary abnormality [7;8]. The major subtypes have included 1) ‘absorptive’ hypercalciuria in which a primary increase in intestinal calcium absorption may result in increased urine calcium; 2) ‘resorptive’ hypercalciuria, caused by an increase in bone turnover, leading to loss of bone calcium in the urine; and 3) ‘renal leak’ hypercalciuria, in which a primary defect in renal tubule calcium transport allows loss of calcium in the urine, with compensatory increase in calcium absorption from gut or mobilization from bone. Classification has rested on measurement of fasting and 24-hour urine calcium, and urinary calcium responses to low calcium diet (400 mg/day) and to a calcium load (1 gm). In particular, those patients with a low fasting calcium excretion, and normal or low PTH, have been categorized as ‘absorptive’; their urine calcium may or may not fall into the normal range on low calcium diet. In contrast, those patients with fasting hypercalciuria and a mildly elevated PTH (but normocalcemia) have been diagnosed with ‘renal leak’. Resorptive-type hypercalciuria would manifest fasting hypercalciuria with a suppressed PTH. However, in many cases, classification into one of these groups has proved difficult. Most individuals with IH appear to have a more generalized acceleration of calcium transport, which affects all these processes, and the clinical value of classification appears modest, at best.
Intestinal Absorption of Calcium in IH
Increased gut absorption of calcium is a feature noted in essentially all patients with IH. Studies using radio-labeled calcium that have compared intestinal calcium absorption in patients with IH to that in normal subjects find consistently increased calcium uptake in IH (Figure 1, panel a) [9;10]. Although not measured in these studies, other work has shown that gut calcium secretion is not altered in subjects with IH, meaning that net absorption is also increased [11]. Absorption is higher in IH at all levels of calcium intake [12], and reflects an increase in active calcium transport by the intestine. One potential mechanism for increased absorption may be the higher levels of serum calcitriol found in patients with IH compared to normal subjects in many studies (Figure 1, panel b) [9;10]. Calcitriol upregulates expression of calcium transport proteins in the intestine, including the apical calcium channel, TRPV6 [13], and calcium absorption is directly related to calcitriol levels [14]. In some hypercalciuric subjects, however, intestinal calcium absorption appears to be increased out of proportion to the calcitriol level [14]. Patients with IH maintain normal fasting serum calcium levels, and PTH levels have generally been reported as normal or slightly low [15].
Figure 1. Intestinal Calcium Absorption and Serum 1,25(OH)2D3 Levels in IH and Normals.

Fractional absorption of radio-labeled calcium by patients with IH (y-axis, left panel) exceeds that of normal subjects (x-axis, left panel) in 7 studies as shown by the displacements of points about the diagonal line of identity. Each point compares mean values for IH vs. normal within a single study. The same was true for serum 1,25(OH)2D3 levels: those of IH patients (y-axis, right panel) exceeded normals (x-axis, right panel) in 11 of 13 studies. (From reference [10]).
Urine calcium rises in normal subjects as net intestinal calcium absorption increases (Figure 2, solid lines). However, in patients with IH, urine calcium is higher than normal at any level of net calcium absorption (calculated as [diet calcium-fecal calcium]) (Figure 2) [9]. Thus, while a normal subject with net calcium absorption of 200 mg/day will excrete about 150 mg calcium in the urine, a patient with IH with the same absorption may have a calcium excretion of 250–400 mg/day, implicating abnormal renal calcium conservation in the production of hypercalciuria. The balance studies shown in Figure 2, using controlled diets, demonstrate that urine calcium is often higher than net absorption, meaning that some of the urine calcium must be derived from bone.
Figure 2. Balance Studies of IH.
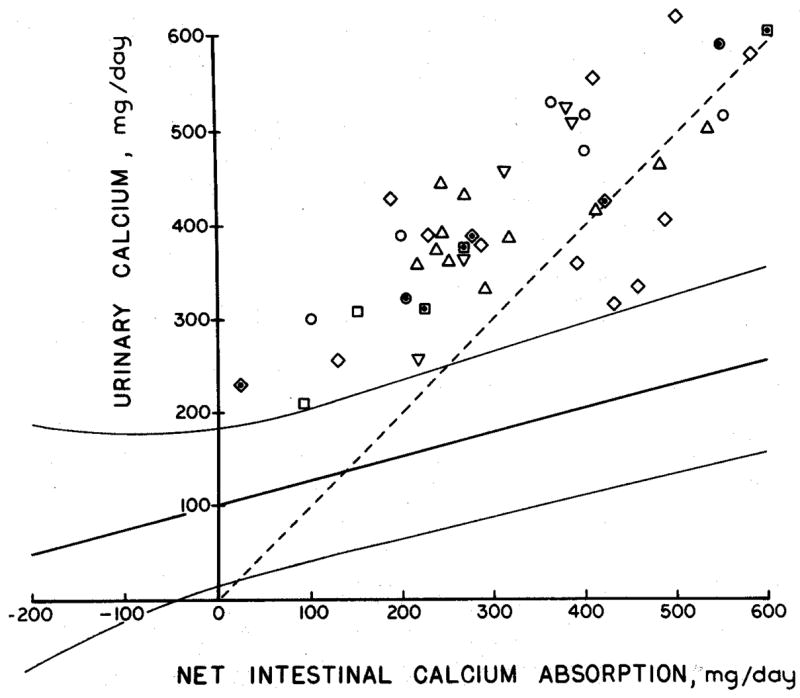
In each of 42 metabolic balance studies of subjects with IH (individual laboratories reporting data are denoted by differing symbols) IH subjects excreted more calcium (y-axis) for any given level of net calcium absorption (x-axis) than did 203 normal subjects represented by the curving 95% confidence limits. Moreover, in all but 7 subjects, calcium excretion exceeded or equaled net calcium absorption as indicated by points lying above the diagonal dashed line of identity. (From reference [9]).
A critical prediction that arises from Figure 2 is that IH patients will lose bone mineral more readily than normal when challenged by an extremely low calcium diet. This was demonstrated in a study in which 27 subjects with IH were compared to 9 normal subjects on days 7–9 of an experimental diet containing very little calcium (2 mg/kg body weight/day) but otherwise composed of normal foods [16]. Patients with IH frequently excreted more calcium in the urine than was ingested (Figure 3), whereas normal subjects did not. The loss of calcium in the urine above that in the diet is definitive proof of abnormal bone mineral wasting in IH, and again suggests that alterations in renal calcium handling that limit calcium conservation may be a common feature of IH. It is notable that the IH patients did not drop their serum calcium (unlike the normal subjects) or increase their serum PTH while on low calcium diet compared to levels seen on a free choice diet; serum PTH levels were lower in IH than in the normal subjects on both diets. Overall, it appears that renal mineral conservation must be abnormal in IH, and bone mineral loss somehow facilitated so that despite the absence of diet calcium to balance renal losses, serum calcium can remain normal and serum PTH suppressed. This is further suggested by studies of calcium kinetics using 47Ca which demonstrate a larger exchangeable calcium pool and higher calcium turnover, together with increased rates of both bone formation and resorption, in 9 subjects with IH compared to 3 normal subjects [11].
Figure 3. Response of IH and Normal Subjects to very Low Calcium Diet.
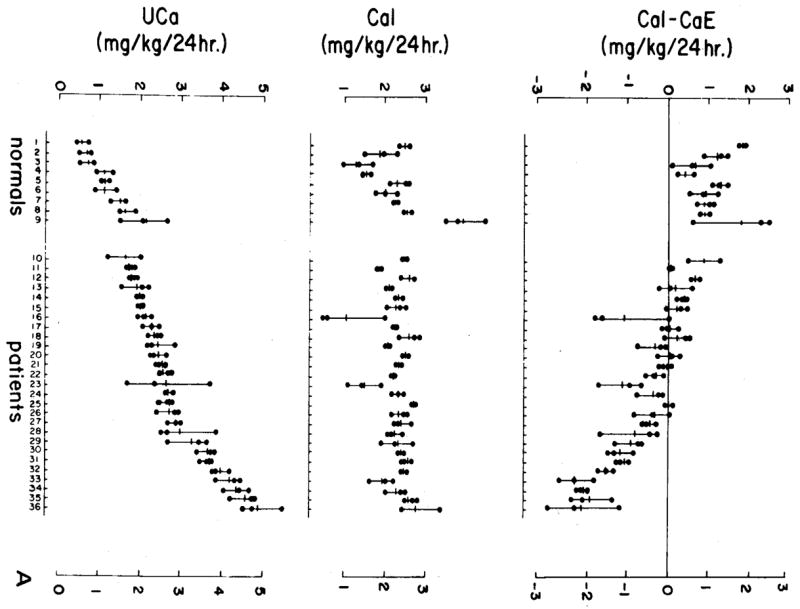
Nine normal people and 27 people with IH ate a diet containing 2 mg/kg/day of calcium (middle panel) for 9 days; the actual amounts of calcium ingested during days 7–9 (middle panel) by normal and IH overlapped. Urine calcium excretion of IH subjects (lower panel) matched or exceeded the highest values for the normals, as did the difference between ingested and excreted calcium (upper panel, CaI – CaE). Many IH subjects excreted more calcium than they ingested, whereas no normal did so; the loss of calcium above diet content can only mean loss of bone mineral into urine. (From reference [16])
A possible mechanism that could explain bone mineral lability and abnormal renal tubule conservation of calcium, as well as increased gut absorption of calcium, would be altered tissue vitamin D response. A direct test of this conjecture was carried out in normal men [17;18] who were fed diets containing varying amounts of calcium - normal calcium (22 mmol/day), low-normal (9.3 mmol/day) or very low calcium (4 mmol/day) - with or without calcitriol supplementation, comparing calcium balance after an equilibration period (Figure 4). On both the 22 and 9.3 mmol calcium diets, calcitriol increased both gut calcium absorption and urine calcium; calcium balance was not affected (Figure 4). However on the 4 mmol calcium diet, urine calcium rose far above calcium absorption, and calcium balance became decisively negative when subjects were given calcitriol. Subjects were not hypercalcemic while on calcitriol, suggesting that filtered loads of calcium did not differ between the two groups. These results support the idea that high tissue vitamin D response can produce all the manifestations of IH: increased gut calcium absorption, decreased renal reabsorption, and increased mobilization of calcium from bone. They leave uncertain what mechanisms account for increased tissue vitamin D response: serum calcitriol itself, the vitamin D receptor, or other factors.
Figure 4. Response of Normal Men Given 1,25(OH)2D3 Supplements.
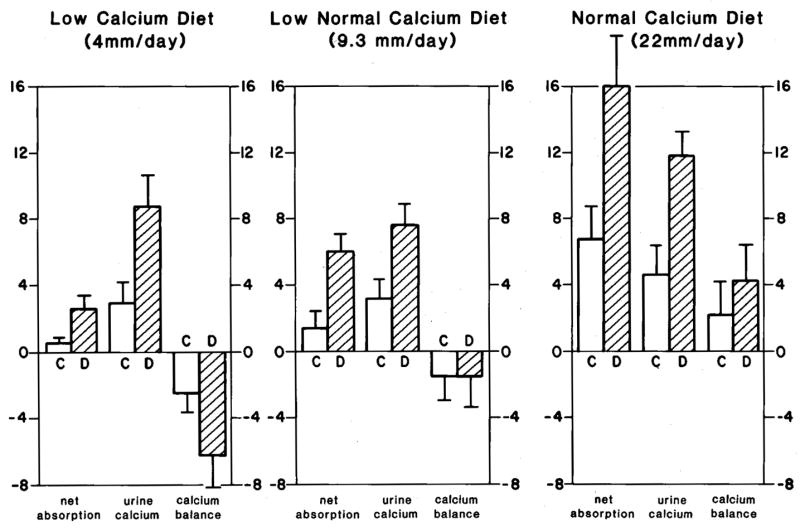
On a low calcium diet (left panel) vitamin D (hatched bars) increased net calcium absorption and urine calcium excretion compared to control periods (clear bars), but because diet calcium was limiting urine calcium rose more than calcium absorption could and bone mineral balance became more negative than during the control periods. With 9.3 and 22 mM/day calcium diets (middle and right panels) calcium balance became neutral (middle panel) and positive (right panels) respectively. (From Coe FL, Parks JH: Nephrolithiasis: Pathogenesis and Treatment, 2nd ed. Chicago, Year Book Medical Publishers, 1988, p. 113. By permission.)
The Genetic Hypercalciuric Rat
A rat model of hypercalciuria has been generated by breeding rats for increased urine calcium excretion, and has been called the genetic hypercalciuric stone-forming (GHS) rat [19]. These rats have now been bred for 70 generations, and consistently excrete 8–10 times as much calcium as control rats from the same background strain; they form kidney stones composed of calcium oxalate or calcium phosphate, depending on dietary components [20].
Like humans with IH, the GHS rats have increased gut absorption of calcium, decreased renal calcium reabsorption, and a tendency to bone demineralization; when placed on low-calcium diet, they excrete more calcium than is absorbed, indicating a loss of bone mineral [19]. Overall, they have a systemic abnormality in calcium homeostasis, which appears to be similar to that found in human stone formers with IH. Unlike many humans with IH, 1,25(OH)2D3 levels are normal in the GHS rat. However, significantly higher levels of vitamin D receptors (VDR) have been found in bone, kidney and intestine, compared to normal rats, which would explain the increased sensitivity to 1,25(OH)2D3 that was found in tissues taken from GHS rats [21]. In the gut, vitamin D-responsive genes are expressed at increased levels in response to small doses of 1,25(OH)2D3 [22], with consequent increased expression of proteins related to calcium homeostasis, such as epithelial calcium channels (TRPV6 in gut), and calbindins. The calcium sensing receptor (CaSR) is another 1,25(OH)2D3-responsive gene, and increased levels of CaSR protein and mRNA have also been found in the kidneys of GHS rats compared to control rats fed the same diet [23].
Altered 1,25(OH)2D3 Activity in IH
As shown previously, humans with IH frequently have high 1,25(OH)2D3 levels, and administration of calcitriol to normal men can replicate much of the picture seen in IH [17]. The importance of 1,25(OH)2D3 was further demonstrated by a study of 19 hypercalciuric subjects treated with the P450 inhibitor ketoconazole, which suppresses 1,25(OH)2D3 levels by 30–40% [24]. Twelve subjects had significant decreases in intestinal 47Ca absorption (76 ± 8 vs 62 ± 8%, p < 0.03) and urine calcium excretion (7.6 ± 1.4 vs 5.7 ± 1.1 mmol/day, p < 0.03) during the ketoconazole treatment, suggesting that these abnormalities were at least in part secondary to 1,25(OH)2D3; however, 9 subjects had minimal or no change in gut and renal calcium handling, despite similar drops in 1,25(OH)2D3 levels, suggesting that in their cases, these abnormalities were independent of 1,25(OH)2D3. In addition, of the 12 subjects who responded to ketoconazole, nine had 1,25(OH)2D3 levels that were not elevated, leading to the possibility that increased sensitivity to 1,25(OH)2D3 rather than frankly elevated vitamin D levels caused the alterations. This would be consistent with the abnormalities seen in the GHS rat.
Whether VDR levels are increased in some patients with IH is unsettled. One study has looked at VDR in peripheral blood monocytes from 10 male stone formers with IH, compared with age matched normal controls [25]. The VDR levels were twice as high in the patients with IH (49 ± 21 vs 20 ± 15 fmol/mg protein, p < 0.008), while 1,25(OH)2D3 levels did not differ. Increased levels of VDR could lead to an increased tissue effect of 1,25(OH)2D3, despite comparable serum levels of the hormone. However, whether monocyte VDR expression reflects tissue levels of VDR is not known.
Renal Mineral Handling in IH
It is evident that renal calcium handling in most patients with IH differs from normal. As shown in Figure 3, when patients with IH are placed on a very low calcium diet, most are unable to reabsorb calcium with the same efficiency as normal subjects. Even on diets that provide normal or increased amounts of calcium, many subjects excrete calcium in excess of absorption, as shown in Figure 2. Studies have shown that patients with IH have an increased fasting fractional excretion of calcium, and many have fasting hypercalciuria, suggesting a role for decreased tubule calcium reabsorption [26]. Alternatively, the increased absorption of calcium with meals in IH could lead to a post-absorptive rise in blood calcium that increases the filtered load; this has been seen in studies using single high calcium foods or calcium alone [27]. Speculatively, PTH could be suppressed as well, which would decrease tubule calcium reabsorption. In order to better understand the mechanism of increased urine calcium excretion in IH, we studied ten stone formers with IH (5 female) and seven normal subjects (4 female) during a 3-meal day, while on diets composed of whole foods containing equivalent amounts of daily calcium (1160 mg), phosphorus (1240 mg), sodium (2141 mg) and potassium (2427 mg) [28]. Urine was collected hourly, and blood samples taken every 30–60 minutes, during both fasting and fed periods; iothalamate was used as a filtration marker. As shown in Figure 5, neither ultrafiltrable calcium nor filtered load of calcium differed between the normal subjects and those with IH, either while fasting or after meals. However, urine calcium excretion was significantly higher after meals in subjects with IH, and fractional reabsorption was lower both during the fasting and the post-absorptive periods. Therefore, the increased calcium absorbed with meals in IH is removed into the urine mainly via decreased tubule reabsorption. This occurred despite the fact that PTH levels did not differ between normal subjects and those with IH, either fasting or with meals (Figure 6). Although PTH levels fell with eating in both groups, PTH levels overlapped in the two groups, and at any given level of PTH calcium reabsorption was significantly lower in subjects with IH. Urine sodium excretion did not differ between the two groups, as would be expected on this controlled diet.
Figure 5. Response of IH Patients (Black circles) and Normals (Grey circles) to a Three Meal Day.

Serum ultra-filtratable calcium rose with meals (upper left panel) but values did not differ between IH and normal, nor did filtered load of calcium (lower left panel). Urine calcium of IH rose far above normals, and fractional calcium reabsorption of IH was below normals. Therefore, response of IH to feeding was an abnormally marked fall in tubule calcium reabsorption that accounts for hypercalciuria. (From reference [28]).
Figure 6. Serum PTH in IH (Black circles) and Normal Subjects (Grey circles).

Fasting (upper right values for each group) and during meals, values of PTH did not differ in IH vs. normal. At all values of PTH, calcium reabsorption of IH was below normal (points are ± SEM). (From reference [28]).
It is not clear what tubule segment is responsible for abnormal calcium absorption. Prior work has shown that hypercalciuric subjects respond with abnormally large increments in urinary sodium, magnesium and calcium excretion to acute hydrochlorothiazide administration; conversely, acetazolamide raises urine sodium in hypercalciuric patients less than normal [29]. These observations point to an underlying defect in proximal tubule reabsorption of sodium and calcium. As acute thiazide administration acts on the distal tubule to increase both sodium and calcium excretion, the greater increment in calcium excretion in IH may stem from either increased distal delivery due to a decrease in proximal reabsorption or abnormal sensitivity to the drug. By the same token, a pre-existing proximal tubule defect in absorption would lead to a smaller increment in sodium excretion from the effect of acetazolamide, a proximal-acting diuretic, in IH.
Both our work [28] and that of others [7] provides evidence for a defect in renal phosphate reabsorption in some patients with IH. Despite levels of PTH that are not different from normal, fractional reabsorption of phosphate after eating was decreased compared to controls in our study [28]. The ability to conserve phosphorus on a low-phosphorus diet may be compromised [30], and IH patients have a trend toward negative phosphorus balance, even on normal intakes [12]. Many patients with IH have slightly decreased serum phosphate levels, and perhaps 15–20% are frankly low [7]. It has been suggested that some patients with IH have a defect in phosphate metabolism, which may lead to elevated 1,25(OH)2D3 levels, and increased calcium absorption. To pursue this question, Prie et.al. [31] studied 207 calcium stone formers with normal PTH levels and compared them to 105 normal subjects of similar age (although there was a higher percentage of females among the normals, 53% vs. 31% of the stone formers). The stone formers of both sexes had a significantly decreased renal phosphate threshold (TmPi) compared to normal subjects; values were normally distributed, but shifted to lower values among the stone formers. Among the stone formers, 19% had a TmPi below 0.63 mmol/liter, while only 5% of normals did so. The stone formers with low TmPi had lower serum phosphate levels and higher urine calcium excretion than stone formers with TmPi > 0.63 mmol/liter, but their 1,25(OH)2D3 levels did not differ, although 1,25(OH)2D3 levels of both groups exceeded that in the normal subjects. It may be that abnormal phosphate excretion is part of a more general defect in proximal tubule function, but this defect may not explain raised 1,25(OH)2D3 levels in many patients with IH.
Effect of Dietary Components on Calcium Excretion
Diet is known to have an effect on urine calcium excretion, in both normal subjects and those with IH, and the increasing incidence of stones that has been noted in the Western world over the past several decades surely has more to do with diet than heredity. Several common components of the Western diet can increase urine calcium excretion.
Sodium
Increasing sodium intake is accompanied by increased urine calcium excretion; in healthy subjects the urine calcium increases about 0.6 mmol/day for each 100 mmol/day increment in sodium excretion [30]. Patients with IH have higher calcium excretion at any given sodium excretion, compared to normals [32]; lowering salt intake will decrease calcium excretion, but high sodium intake is seldom the sole cause of IH.
Protein
Increased dietary protein intake will also raise urine calcium excretion, by about 0.04 mmol/gram protein intake in healthy men and women [30]. Metabolism of ingested protein creates an acid load, due to the oxidation of organic sulfur in amino acids to sulfate, leading to increased excretion of net acid and calcium. Administration of alkali reduces both net acid and calcium excretion [12]. The increase in net acid production inhibits renal tubular reabsorption of calcium. Studies in mice have shown that induction of acidosis leads to decreased renal tubular expression of the distal tubular apical calcium channel, TRPV5; mice in which the TRPV5 gene was inactivated did not have an increase in calcium excretion with acidosis [33]. Decreasing protein intake (together with a lower salt intake) has been shown to decrease urine calcium excretion and recurrence of calcium stones [34]. However, urine calcium is higher in patients with IH than among normals at comparable levels of acid excretion suggesting that high protein intake worsens but does not usually cause IH.
Glucose, sucrose, and ethanol
Several rapidly metabolized nutrients cause increased excretion of urine calcium due to diminished tubular calcium and magnesium reabsorption [35], and patients with IH appear to have an exaggerated calciuric response to a carbohydrate load [36]. This effect is not accompanied by augmented sodium excretion; in fact, urine sodium excretion falls significantly with carbohydrate load, in most studies. Urine phosphate excretion may be increased at the same time [37]. The increase in calcium excretion seen with glucose ingestion is additive to that caused by acidosis, suggesting a different site of action [38]. Since the studies looking at this phenomenon used glucose ingestion in the absence of calcium intake, increased gut calcium absorption is not the source of the elevated urine calcium. Insulin may mediate changes in renal tubular calcium reabsorption [39], and some studies suggest an exaggerated insulin response to carbohydrates in patients with IH [40]. Both calcium stone formers and their relatives have an exaggerated calciuric response to a glucose load compared to normal subjects, suggesting a disturbance in renal tubular function that may be genetic in origin [36].
Bone Mineral in IH
When urine calcium exceeds gut absorption, some of the calcium in urine must be derived from bone. Recent studies have confirmed that bone mineral density is often mildly decreased in patients with IH [41]. Bone seems to participate in the accelerated calcium turnover in IH, but the mediators of this tendency to lose mineral are still unclear. A chapter in this seminar details bone mineral regulation in IH in more detail. (see article by J. Zerwekh).
Genetic Associations of IH and Hypercalciuria
A number of rare monogenic disorders are associated with hypercalciuria, with or without renal calcification, as part of their phenotype [42], however, most of these are unlikely to be confused with IH. Perhaps the best-studied of these disorders is Dent’s disease, which is caused by a mutation in a voltage-gated chloride channel, leading to defects in reabsorption in the proximal tubule. The disease is characterized by kidney stones, renal failure, and low molecular weight proteinuria; screening of a group of male patients with typical IH suggests that mutations of this gene are rarely the underlying cause [43].
IH is a complex trait, and is likely to be the result of contributions of several genes, which may differ from individual to individual. Studies searching for such genes have utilized various approaches. A genome-wide linkage study of three kindreds with IH and low bone density identified a locus that encodes a gene for the human soluble adenylyl cyclase [44]. Several sequence variations in the gene tracked with IH in an unrelated group of 80 hypercalciuric patients with kidney stones, compared with a group of non-stone formers [45]. The protein is found in kidney and in bone, and may function as a bicarbonate sensor [46]. Its link to hypercalciuria is still unclear.
Several candidate genes have been screened for possible association with IH; some examples include:
Vitamin D receptor
Polymorphisms of the Vitamin D receptor gene that segregate with hypercalciuria have been detected in some [47], but not all, studies [48]. A study of a cohort of 54 French Canadian sibships found evidence for linkage to nephrolithiasis of a gene in the region of the VDR [49].
TRPV5
The gene for the renal epithelial calcium channel was screened for mutations in 9 families with IH, but none were found [50].
Calcium sensing receptor
A polymorphism has been identified in the gene encoding the calcium sensing receptor which produces a gain-of-function of the receptor when transfected into HEK-293 cells [51]. In a study from Italy, the relative risk of hypercalciuria was increased in individuals carrying this haplotype, both among stone formers [52] and non-stone formers [51], but it explained only 4.1% of the total variance in urinary calcium excretion, and was not felt to be a major gene determining IH.
Sodium-phosphate co-transporter (NPT2a)
A study of 20 subjects with osteoporosis or stones found 2 with NPT2a variants, associated with renal phosphate leak [53]. However, a later study of a cohort of 98 French-Canadian families of hypercalciuric stone formers did not find an association between NPT2a gene variations and phosphate leak associated with hypercalciuria and stones [54].
Recently Described Regulators of Urine Calcium and Phosphate Handling
The last few years have given us a clearer picture of renal and intestinal calcium and phosphorus transporters. Regulated renal calcium transport involves a calcium channel, TRPV5, found in the distal nephron; it is closely related to the analogous channel, TRPV6, found in the gut [13]. Proximal tubule phosphate transport is primarily conducted by a sodium-phosphate co-transporter, NPT-2a, a member of the type 2 family of phosphate transport proteins [55]. Both of these transporters are controlled by the PTH - vitamin D axis, but in the last few years, new transport regulators have been identified.
Klotho, a gene product that co-localizes in distal convoluted tubule with TRPV5, appears critically important to calcium reabsorption; klotho-deficient mice exhibit reduced TRPV5 expression and renal calcium reabsorption [56]. The effect of klotho appears to involve its ability to cleave a carbohydrate residue from the calcium channel, which increases TRPV5 activity by trapping it in the plasma membrane [57]. Knockout mice over-express the 25(OH)D-1α-hydroxylase, and have high serum 1,25(OH)2D3 levels with consequent hypercalcemia and hypercalciuria. Subtle variations in klotho could possibly cause a syndrome like IH if 1,25(OH)2D3 increase were not so high as to cause hypercalcemia.
FGF-23, a bone derived hormone, reduces tubule phosphate reabsorption and 1,25(OH)2D3 synthesis. Klotho is required for FGF-23 to mediate its effects via its receptor [58]. Variations in the klotho-FGF23 axis could mediate alterations in calcium and phosphate handling by the kidney, and play a role in IH.
Leptin, an important regulator of nutrient uptake, suppresses formation of 1,25(OH)2D3 [59]. Mice lacking leptin or the leptin receptor manifest high serum 1,25(OH)2D3 levels, hypercalcemia and hypercalciuria. Therefore, variations of leptin expression or receptor could also lead to a syndrome resembling IH.
Data is lacking at present about the role that these interconnected mediators play in IH, if any, but future research may shed some light on this question.
Summary
Perhaps the most important thing to keep in mind about IH is that it is not so much a disease as it is the physiology of people who occupy one extreme of a continuous spectrum of urine calcium excretion rates. Apart from stones and possibly bone disease, IH is simply a benign trait. When we speak of the pathogenesis of IH, we are in all likelihood describing the normal regulation of systemic calcium traffic as viewed at one of its extreme end-points, and comparing that view, usually, to the same regulation at a middle zone of human behavior. Because it is not a disease, we are likely at all times to encounter the extreme complexity of a normal homeostatic regulation. Studies of genes will almost certainly reveal subtle allelic variations as opposed to mutations. The IH rat is instructive in this regard, because it represents simply the in-breeding for a single trait. Understanding the physiology of IH improves our understanding of mineral homeostasis overall, and allows us to provide optimal treatment for our patients who manifest kidney stones.
Acknowledgments
Supported in part by NIH NIDDK PO1 56788.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Hodkinson A, Pyrah LN. The urinary excretion of calcium and inorganic phosphate in 344 patients with calcium stone of renal origin. Br J Surg. 1958;48:10–18. doi: 10.1002/bjs.18004619504. [DOI] [PubMed] [Google Scholar]
- 2.Coe FL. Treated and untreated recurrent calcium nephrolithiasis in patients with idiopathic hypercalciuria, hyperuricosuria, or no metabolic disorder. Ann Intern Med. 1977;87:404–410. doi: 10.7326/0003-4819-87-4-404. [DOI] [PubMed] [Google Scholar]
- 3.Flocks RH. Calcium and phosphorus excretion in the urine of patients with renal or ureteral calculi. JAMA. 1939;113:1466. [Google Scholar]
- 4.Henneman PH, Benedict PH, Forbes AP, Dudley HR. Idiopathic Hypercalciuria. N Engl J Med. 1958;259:802–807. doi: 10.1056/NEJM195810232591702. [DOI] [PubMed] [Google Scholar]
- 5.van’t Hoff WG. Aetiological factors in paediatric urolithiasis. Nephron Clin Pract. 2004;98:45–48. doi: 10.1159/000080251. [DOI] [PubMed] [Google Scholar]
- 6.Coe FL, Parks JH, Moore ES. Familial idiopathic hypercalciuria. N Engl J Med. 1979;300:337–340. doi: 10.1056/NEJM197902153000703. [DOI] [PubMed] [Google Scholar]
- 7.Levy FL, Adams-Huet B, Pak CYC. Ambulatory evaluation of nephrolithiasis: An update of a 1980 protocol. American Journal of Medicine. 1995;98:50–59. doi: 10.1016/S0002-9343(99)80080-1. [DOI] [PubMed] [Google Scholar]
- 8.Pak CYC, Ohata M, Lawrence EC, Snyder W. The hypercalciurias: Causes, parathyroid functions, and diagnostic criteria. J Clin Invest. 1974;54:387–400. doi: 10.1172/JCI107774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coe FL, Favus MJ, Asplin JR. Nephrolithiasis. In: Brenner BM, Rector FC Jr, editors. The Kidney. 7. chap. 39. Philadelphia: Elsevier; 2004. pp. 1819–1866. [Google Scholar]
- 10.Coe FL, Parks JH, Evan AP, Worcester E. Pathogenesis and Treatment of Nephrolithiasis. In: Alpern R, Hebert S, editors. Seldin and Giebisch’s The Kidney. chap. 68. Elsevier, Inc; 2007. pp. 1945–1977. [Google Scholar]
- 11.Liberman JA, Sperling O, Atsmon A, et al. Metabolic and calcium kinetic studies in hypercalciuria. J Clin Invest. 1968;47:2580–2590. doi: 10.1172/JCI105940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lemann J., Jr . Idiopathic hypercalciuria. In: Coe FL, Favus M, editors. Disorders of Bone and Mineral Metabolism. 2. chap. 30. Lippincott: 2002. pp. 673–697. [Google Scholar]
- 13.Hoenderop JGJ, Nilius B, Bindels RJM. Calcium absorption across epithelia. Physiol Rev. 2005;85:373–422. doi: 10.1152/physrev.00003.2004. [DOI] [PubMed] [Google Scholar]
- 14.Kaplan RA, Haussler MR, Deftos LJ, et al. The role of 1 alpha 25 dihydroxyvitamin D in the mediation of intestinal hyperabsorption of calcium in primary hyperparathyroidism and absorptive hypercalciuria. J Clin Invest. 1977;59:756–760. doi: 10.1172/JCI108696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Monk RD, Bushinsky DA. Pathogenesis of idiopathic hypercalciuria. In: Coe FL, Favus MJ, Pak CYC, et al., editors. Kidney Stones: Medical and Surgical Management. chap. 32. Lippincott-Raven; 1996. pp. 759–772. [Google Scholar]
- 16.Coe FL, Favus MJ, Crockett T, et al. Effects of low-calcium diet on urine calcium excretion, parathyroid function and serum 1,25(OH)2D3 levels in patients with idiopathic hypercalciuria and in normal subjects. Am J Med. 1982;72:25–32. doi: 10.1016/0002-9343(82)90567-8. [DOI] [PubMed] [Google Scholar]
- 17.Maierhofer WJ, Gray RW, Adams ND. Bone resorption stimulated by elevated serum 1,25-(OH)2-vitamin D concentrations in healthy men. Kidney Int. 1983;24:555–560. doi: 10.1038/ki.1983.193. [DOI] [PubMed] [Google Scholar]
- 18.Adams ND, Gray RW, Lemann J, Jr, Cheung HS. Effects of calcitriol administration on calcium metabolism in healthy men. Kidney Int. 1982;21:90–97. doi: 10.1038/ki.1982.13. [DOI] [PubMed] [Google Scholar]
- 19.Bushinsky DA, Frick KK, Nehrke K. Genetic hypercalciuric stone-forming rats. Curr Opin Nephrol Hypertens. 2006;15:403–418. doi: 10.1097/01.mnh.0000232881.35469.a9. [DOI] [PubMed] [Google Scholar]
- 20.Bushinsky DA, Asplin JR, Grynpas MD, et al. Calcium oxalate stone formation in genetic hypercalciuric stone-forming rats. Kidney Int. 2002;61:975–987. doi: 10.1046/j.1523-1755.2002.00190.x. [DOI] [PubMed] [Google Scholar]
- 21.Li XQ, Tembe V, Horwitz GM, et al. Increased intestinal vitamin D receptor in genetic hypercalciuric rats. A cause of intestinal calcium hyperabsorption. Journal of Clinical Investigation. 1993;91:661–667. doi: 10.1172/JCI116246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yao J, Kathpalia P, Bushinsky DA, Favus MJ. Hyperresponsiveness of Vitamin D Receptor Gene Expression to 1,25-Dihydroxyvitamin D3. A New Characteristic of Genetic Hypercalciuric Stone-forming Rats. Journal of Clinical Investigation. 1998;101:2223–2232. doi: 10.1172/JCI1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yao J, Karnauskas AJ, Bushinsky DA, Favus M. Regulation of renal calcium-sensing receptor gene expression in response to 1,25(OH)2D3 in genetic hypercalciuric stone forming rats. J Am Soc Nephrol. 2005;16:1300–1308. doi: 10.1681/ASN.2004110991. [DOI] [PubMed] [Google Scholar]
- 24.Breslau NA, Preminger GM, Adams BV, et al. Use of ketoconazole to probe the pathogenetic importance of 1,25-dihydroxyvitamin D in absorptive hypercalciuria. J Clin Endocrinol Metab. 1992;75:1446–1452. doi: 10.1210/jcem.75.6.1464646. [DOI] [PubMed] [Google Scholar]
- 25.Favus MJ, Karnauskas AJ, Parks JH, Coe FL. Peripheral blood monocyte vitamin D receptor levels are elevated in patients with idiopathic hypercalciuria. J Clin Endocrinol Metab. 2004;89:4937–4943. doi: 10.1210/jc.2004-0412. [DOI] [PubMed] [Google Scholar]
- 26.Peacock M, Nordin BEC. Tubular reabsorption of calcium in normal and hypercalciuric subjects. J Clin Pathol. 1968;21:353–358. doi: 10.1136/jcp.21.3.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwille PO, Rumenapf G, Schmidtler JKR. Fasting and post calcium load serum calcium, parathyroid hormone, calcitonin, in male idiopathic calcium urolithiasis. Exp Clin Endocrinol. 1987;90:71–75. doi: 10.1055/s-0029-1210674. [DOI] [PubMed] [Google Scholar]
- 28.Worcester EM, Gillen DL, Evan AP, et al. Evidence that postprandial reduction of renal calcium reabsorption mediates hypercalciuria of patients with calcium nephrolithiasis. Am J Physiol Renal Physiol. 2007;292:F66–F75. doi: 10.1152/ajprenal.00115.2006. [DOI] [PubMed] [Google Scholar]
- 29.Sutton RAL, Walker VR. Responses to Hydrochlorothiazide and Acetazolamide in Patients with Calcium Stones. N Engl J Med. 1980;13:709–713. doi: 10.1056/NEJM198003273021302. [DOI] [PubMed] [Google Scholar]
- 30.Lemann J., Jr . Pathogenesis of Idiopathic Hypercalciuria and Nephrolithiasis. In: Coe FL, Favus MJ, editors. Disorders of Bone and Mineral Metabolism. 1. chap. 32. New York: Raven Press; 1992. pp. 685–706. [Google Scholar]
- 31.Prie D, Ravery V, Boccon-Gibod L, Friedlander G. Frequency of renal phosphate leak among patients with calcium nephrolithiasis. Kidney Int. 2001;60:272–276. doi: 10.1046/j.1523-1755.2001.00796.x. [DOI] [PubMed] [Google Scholar]
- 32.Lemann J, Jr, Worcester EM, Gray RW. Hypercalciuria and stones. [Review] American Journal of Kidney Diseases. 1991;17:386–391. doi: 10.1016/s0272-6386(12)80628-7. [DOI] [PubMed] [Google Scholar]
- 33.Nijenhuis T, Renkema KY, Hoenderop JGJ, Bindels RJM. Acid-base status determines the renal expression of Ca2+ and Mg2+ transport proteins. J Am Soc Nephrol. 2006;17:617–626. doi: 10.1681/ASN.2005070732. [DOI] [PubMed] [Google Scholar]
- 34.Borghi L, Schianchi T, Meschi T, et al. Comparison of two diets for the prevention of recurrent stones in idiopathic hypercalciuria. N Engl J Med. 2002;346:77–84. doi: 10.1056/NEJMoa010369. [DOI] [PubMed] [Google Scholar]
- 35.Lemann J, Jr, Lennon EJ, Piering WF, et al. Evidence that glucose ingestion inhibits net renal tubular reabsorption of calcium and magesium in man. J Lab Clin Med. 1970;75:578–585. [PubMed] [Google Scholar]
- 36.Lemann J, Jr, Piering WF, Lennon EJ. Possible role of carbohydrate-induced calciuria in calcium oxalate kidney-stone formation. N Engl J Med. 1969;280:232–237. doi: 10.1056/NEJM196901302800502. [DOI] [PubMed] [Google Scholar]
- 37.Lennon EJ, Lemann J, Jr, Piering WF, Larson LS. The effect of glucose on urinary cation excretion during chroinic extracellular volume expansion in normal man. J Clin Invest. 1974;53:1424–1433. doi: 10.1172/JCI107690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lennon EJ, Piering WF. A comparison of the effects of glucose ingestion and NH4Cl acidosis on urinary calcium and magnesium excretion in man. J Clin Invest. 1970;49:1458–1465. doi: 10.1172/JCI106363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.DeFronzo RA, Goldberg M, Agus ZS. The effects of glucose and insulin on renal electrolyte transport. J Clin Invest. 1976;58:83–90. doi: 10.1172/JCI108463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwille PO, Rumenapf G, Kohler R. Blood levels of glucometabolic hormones and urinary saturation with stone forming phases after an oral test meal in male patients with recurrent idiopathic calcium urolithiasis and in healthy controls. Journal of the American College of Nutrition. 1989;8:557–566. doi: 10.1080/07315724.1989.10720327. [DOI] [PubMed] [Google Scholar]
- 41.Bataille P, Achard JM, Fournier A, et al. Diet, vitamin D and vertebral mineral density in hypercalciuric calcium stone formers. Kidney Int. 1991;39:1193–1205. doi: 10.1038/ki.1991.151. [DOI] [PubMed] [Google Scholar]
- 42.Bushinsky DA, Coe FL, Moe OW. Nephrolithiasis. In: Brenner BM, editor. Brenner and Rector’s The Kidney. 8. chap. 37. Philadelphia: Saunders; 2007. pp. 1299–1349. [Google Scholar]
- 43.Scheinman SJ, Cox JP, Lloyd SE, et al. Isolated hypercalciuria with mutation in CLCN5: relevance to idiopathic hypercalciuria. Kidney Int. 2000;57:232–239. doi: 10.1046/j.1523-1755.2000.00774.x. [DOI] [PubMed] [Google Scholar]
- 44.Reed BY, Heller HJ, Gitomer WL, Pak CY. Mapping a gene defect in absorptive hypercalciuria to chromosome 1q23.3-q24. J Clin Endocrinol Metab. 1999;84:3907–3913. doi: 10.1210/jcem.84.11.6155. [DOI] [PubMed] [Google Scholar]
- 45.Reed BY, Gitomer WL, Heller HJ, et al. Identification and characterization of a gene with base substitutions associated with the absorptive hypercalciuria phenotype and low spinal bone density. J Clin Endocrinol Metab. 2002;87:1476–1485. doi: 10.1210/jcem.87.4.8300. [DOI] [PubMed] [Google Scholar]
- 46.Geng W, Wang Z, Zhang J, et al. Cloning and characterization of the human soluble adenylyl cyclase. Am J Physiol Cell Physiol. 2005;288:1305–1316. doi: 10.1152/ajpcell.00584.2004. [DOI] [PubMed] [Google Scholar]
- 47.Rendina D, Mossetti G, Viceconti R, et al. Association between vitamin D receptor gene polymorphisms and fasting idiopathic hypercalciuria in recurrent stone-forming patients. Urol. 2004;64:833–838. doi: 10.1016/j.urology.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 48.Zerwekh JE, Hughes MR, Reed BY, et al. Evidence for Normal Vitamin D Receptor Messenger Ribonucleic Acid and Genotype in Absorptive Hypercalciuria. J Clin Med Metab. 1995;80:2960–2965. doi: 10.1210/jcem.80.10.7559881. [DOI] [PubMed] [Google Scholar]
- 49.Scott P, Ouimet D, Valiquette L, et al. Suggestive evidence for a susceptibility gene near the vitamin D receptor locus in idiopathic calcium stone formation. J Am Soc Nephrol. 1999;10:1007–1013. doi: 10.1681/ASN.V1051007. [DOI] [PubMed] [Google Scholar]
- 50.Muller D, Hoenderop JGJ, Vennekens R, et al. Epithelial Ca2+ channel (ECaC) in autosomal dominant idiopathic hypercalciuria. Nephrol Dial Transplant. 2002;17:1614–1620. doi: 10.1093/ndt/17.9.1614. [DOI] [PubMed] [Google Scholar]
- 51.Vezzoli G, Terranegra A, Arcidiacono T, et al. R990G polymorphism of calcium-sensing receptor does produce a gain-of-function and predispose to primary hypercalciuria. Kidney Int. 2007;71:1155–1162. doi: 10.1038/sj.ki.5002156. [DOI] [PubMed] [Google Scholar]
- 52.Vezzoli G, Tanini A, Ferrucci L, et al. Influence of calcium-sensing receptor gene on urinary calcium excretion in stone-forming patients. J Am Soc Nephrol. 2002;13:2517–2523. doi: 10.1097/01.asn.0000030077.72157.d2. [DOI] [PubMed] [Google Scholar]
- 53.Prie D, Huart V, Bakouh N, et al. Nephrolithiasis and osteoporosis associated with hypophosphatemia caused by mutations in the type 2a sodium-phosphate cotransporter. N Engl J Med. 2002;347:983–991. doi: 10.1056/NEJMoa020028. [DOI] [PubMed] [Google Scholar]
- 54.Lapointe J-Y, Tessier J, Paquette Y, et al. NPT2a gene variation in calcium nephrolithiasis with renal phosphate leak. Kidney Int. 2006;69:2261–2267. doi: 10.1038/sj.ki.5000437. [DOI] [PubMed] [Google Scholar]
- 55.Murer H, Hernanado N, Forster I, Biber J. Proximal tubular phosphate reabsorption: molecular mechanisms. Physiol Rev. 2000;80:1373–1409. doi: 10.1152/physrev.2000.80.4.1373. [DOI] [PubMed] [Google Scholar]
- 56.Torres PU, Prie D, Molina-Bletry V, et al. Klotho: an antiaging protein involved in mineral and vitamin D metabolism. Kidney Int. 2007;71:730–737. doi: 10.1038/sj.ki.5002163. [DOI] [PubMed] [Google Scholar]
- 57.Chang Q, Hoefs S, Van der Kemp AW, et al. The beta-glucuronidase klotho hydrolyzes and activates the TRPV5 channel. Science. 2005;310:490–493. doi: 10.1126/science.1114245. [DOI] [PubMed] [Google Scholar]
- 58.Liu S, Quarles LD. How fibroblast growth factor 23 works. J Am Soc Nephrol. 2007;18:1637–1647. doi: 10.1681/ASN.2007010068. [DOI] [PubMed] [Google Scholar]
- 59.Matsunuma A, Horiuchi N. Leptin attenuates gene expression for renal 25-hydroxyvitamin D3-1α– hydroxylase in mice via the long form of the leptin receptor. Arch Biochem Biophys. 2007;463:118–127. doi: 10.1016/j.abb.2007.02.031. [DOI] [PubMed] [Google Scholar]