

Abstract
Previous studies have indicated that volume regulated anion channels (VRACs) may be involved in the pathology of the ischemic brain cortical penumbra due to activation of VRAC-mediated excitatory amino-acid (EAA) release. To assess this we had studied neuroprotection and EAA release inhibition by a potent VRAC inhibitor, tamoxifen. However, tamoxifen inhibits several other neuro-damaging processes. In the present study we use an ethacrynic acid derivative, 4-(2-butyl-6, 7-dichloro-2-cyclopentyl-indan-1-on-5-yl) oxobutyric acid (DCPIB), that has recently been shown to be a specific antagonist of volume regulated anion channels (VRAC), to measure the extent of neuroprotection provided and thus to better assess the role of VRAC-mediated release of excitatory amino acids in an intraluminal suture, reversible middle cerebral artery occlusion (rMCAO) model in adult rats. Rats given DCPIB intracisternally had significantly better neurobehavioral scores after 24 hours and showed significantly reduced infarct volumes. Mean infarct volumes were 208.0 (SD = 38.3) mm3 for the vehicle groups, compared with 68.5 (SD = 22.7) mm3 for intracisternally DCPIB-treated groups (p=0.02, Mann-Whitney test), a reduction of around 75%. However, a 500-fold higher dose of DCPIB given intravenously did not reduce infarct volume or improve behavior. The microdialysis study demonstrated statistically significant reduced brain extracellular fluid glutamate when DCPIB was present in the probe. Thus DCPIB, a specific inhibitor of VRACs, given i.c. provides strong neuroprotection in brain ischemia, but it appears to not cross the blood brain barrier as it is not effective when given i.v. These experiments support the hypothesis that EAA released via VRACs contributes to later ischemic-induced damage.
Keywords: Cerebral ischemia; Neuroprotection; Rat; Reversible focal middle cerebral artery occlusion; Volume regulated anion channels (VRACs); microdialysis; excitotoxicity; behavior; infarct size; 4-(2-butyl-6, 7-dichloro-2-cyclopentyl-indan-1-on-5-yl) oxobutyric acid (DCPIB)
Introduction
Astrocytes swell during ischemia and as a result can release excitatory amino acids (EAAs) via activation of Volume Regulated Anion Channels (VRACs). VRACs may be a major contributor to neuronal damage in ischemia (Kimelberg, 2000;Kimelberg, 2005;Phillis et al., 1997;Ochoa De La Paz LD et al., 2002;Law, 1991). Excess EAAs, primarily extracellular glutamate, initiate a cascade of events progressing over hours to many days (Lipton, 1999). The increase in extracellular EAAs may occur through a number of routes, including exacerbation by decreased reuptake into neurons and astrocytes, exocytotic release from neurons, reversal of the high affinity uptake system and swelling-induced opening of anion channels, especially in astrocytes (Nicholls and Attwell, 1990;Kimelberg and Mongin, 1998;Mongin and Kimelberg, 2004;Law, 1994;Quesada et al., 1998;Pasantes-Morales et al., 1990;Phillis et al., 1997;Phillis et al., 2000;Strange et al., 1994;Liu et al., 2006). Prevention or inhibition of ischemia-induced EAA release, either acute or prolonged, may protect neurons in ‘at risk’ locations. Swelling-activated VRACs play a major role in excitatory amino acids (EAAs) release in ischemic injury (reviewed in Kimelberg, 2005), especially in the ischemic cortical penumbra (Feustel et al., 2004). The inhibition of the VRACs seems a suitable target because VRACs are normally closed and their blockade presumably would not affect normal brain function.
Some years ago our laboratory in collaboration with E. Cragoe of Merck & Co., developed a series of compounds based on ethacrynic acid which were able confer protection in a closed head injury model. Synthesis of these compounds was guided by the principle of increasing their efficacy in inhibiting brain edema and reducing their renal saludiuretic effects (Nelson et al., 1979;Kimelberg et al., 1987;Kimelberg et al., 1990;Bourke et al., 1979;Cragoe et al., 1982;Cragoe, Jr. et al., 1986;Cragoe, 1987). Since that time one of these compounds 4-(2-butyl-6,7-dichloro-2-cyclopentyl-indan-1-on-5-yl) oxobutyric acid and therefore given the acronym DCPIB (Cragoe et al., 1982;Bourke et al., 1981;Nelson et al., 1982); and see Figure 1a), has been shown to specifically inhibit VRAC activity, but not a number of expressed chloride channels in Xenopus oocytes (Decher et al., 2001). VRACs are an important class of anion channels that are widely distributed, but whose molecular identity has so far eluded investigators’ best efforts (Nilius and Droogmans, 2003;Strange, 1998;Okada, 1997). DCPIB has recently been shown by us to completely inhibit cell swelling induced EAA release and Cl− currents in primary astrocyte cultures (Abdullaev et al., 2006). In the current study we have examined the effects of DCPIB in a rat model of reversible middle cerebral artery occlusion (rMCAo) and show that it results in robust reduction of infarct volume when given intracisternally (i.c.), but no effect when given intravenously (i.v.). Modest correlative improvements in behavior scores were also found. We also show that local administration of DCPIB via a microdialysis probe results in lower glutamate levels during ischemia.
Figure 1.
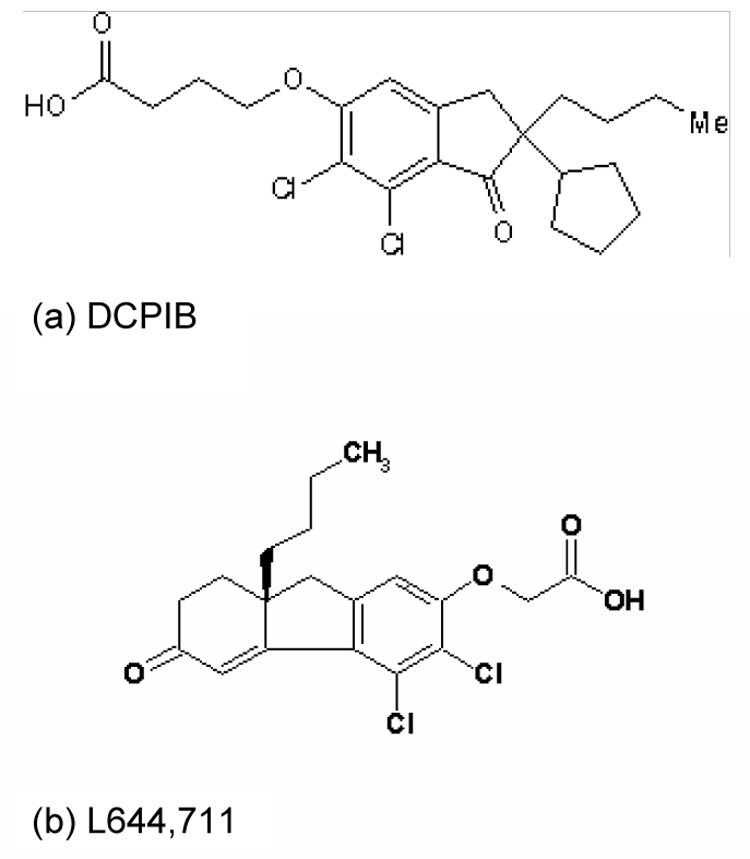
Structure of DCPIB (a) and L644, 711 (b). See text for details.
Materials and Methods
Animal preparation
All animal procedures were in accordance with the Guidelines for Care and Use of Laboratory Animals and were approved by the institutional animal care and use committee. Male Sprague–Dawley rats (300 to 350 g, Taconic,Germantown, NY) were anesthetized with isoflurane in a bell jar after 50 mg/kg atropine sulfate (Sigma, St.Louis, MO) had been given intramuscularly. Each animal was artificially ventilated using a respirator with 2.0% Isoflurane in a mixture of 30% O2 and 70% N2 after endotracheal intubation. Blood gas analysis was used to verify adequate ventilation. Rectal temperature was maintained at 37.0–37.5°C and temporalis muscle temperature was used to reflect brain temperature being maintained between 36.0-C and 37.0°C with a water-circulating heating pad placed under the rat during surgery and microdialysate sampling.
Middle Cerebral Artery Occlusion
Reversible middle cerebral artery occlusion was performed as described by Longa et al. (Longa et al., 1989), as used previously in our laboratory (Kimelberg et al, 2000; Zhang et al, 2005; 2007). The suture was left in place for 2 hours and then slowly withdrawn. In the protection experiments the animal was allowed to completely recover from anesthesia and returned to the cage.
Intravenous or Intracisternal Injection of DCPIB
For intracisternal (i.c.) injection, anesthetized rats were placed within the ear bars of a stereotaxic frame and the occipital membrane was punctured with the needle of a Hamilton syringe as described by Martinez et al (2000). Correct positioning of the needle into the cisterna magna was determined by the reflux of clear cerebrospinal fluid (CSF) into the syringe. Fifteen minutes before the onset of ischemia, 20 µg/kg DCPIB (Tocris, Ellisville, MO) in 10 µl of 3% dimethyl sulfoxide (DMSO), or DMSO alone, was administered manually over 20 seconds. For intravenous (i.v.) administrations, the right jugular vein was exposed for a catheter to be inserted. DCPIB (10 mg/kg) in 0.1 ml of DMSO or vehicle was slowly administered at the initiation of ischemia over a period of 15 minutes at a rate of 0.01 ml/minute controlled by a syringe pump (PHD 2000 Infusion, Harvard Apparatus). After 24 hours neurological status was scored, the animals were then sacrificed and the brains examined for infarct volume.
Measurement of Neurobehavioral Deficit Scoring and Infarct Volume
Neurobehavioral deficit scoring was based on the 18 point scale described by Garcia et al (Garcia et al., 1995), as used previously in our laboratory (Zhang et al., 2005). Neurological status was scored at 24 hours after ischemia initiation. The individual evaluating neurobehavioral deficits was blinded as to whether vehicle or drugs was administered. At twenty four hours after ischemia, rats were injected with 120 mg of pentobarbital; the brain was removed and cut into 2 mm sections. Infarct volume was assessed using 2,3,5-triphenyltetrazolium chloride (TTC) (Sigma, St. Louis, MO) staining, as described previously (Zhang et al., 2005,2007; Kimelberg et al., 2000).
Microdialysis Procedures
Two microdialysis probes (4-mm tip; CMA-12, Carnegie Med/BAS) were lowered slowly into the lateral cortex bilaterally through burr holes (from bregma, −1 mm anterior; 6 mm lateral; 5 mm down from the dura). This region experiences reduced but not zero blood flow during the occlusion period (Feustel et al., 2004), and is classified as penumbra. Artificial cerebrospinal fluid (aCSF) (Seki et al., 1999) was pumped through the dialysis probe by a syringe pump (Pump 22, Harvard Instrument Co) at a rate of 2 µl/min. After a one hour stabilization period, samples were collected in tubes cooled on ice and frozen at −80° until analysis. Two 15-minute samples were collected prior to introducing DCPIB into the dialysate. A liquid switch was used to switch dialysate to aCSF containing either 100umol/L DCPIB or vehicle and these dialysates were continued for the duration of the experiment. Three 20-minute samples were collected during drug administration to determine the effect of drug on nonischemic EAA levels. Reversible middle cerebral artery occlusion (rMCAo) was then performed as previously described. Eight 15-minute duration dialysate samples were collected during the 2 hours of ischemia and then the suture was withdrawn. Five 20-minute samples were taken after the reperfusion. Measurements of glutamate and aspartate concentrations in the dialysates were performed by reverse-phase high-performance liquid chromatography, with the use of precolumn derivatization and fluorescence detection, essentially as previously described (Seki et al., 1999;Feustel et al., 2004).
Statistical Analysis
Statistical assessment of infarct volume was by the Mann-Whitney non-parametric test, as infarct volumes have not been normally distributed. Behavioral scores were treated similarly. Microdialysis results were analyzed by two-way repeated measures analysis of variance with main effects of drug or vehicle presence, time, and their interaction. Planned comparisons were made at individual times if a significant main effect of drug or interaction were found. Differences were considered statistically significant at p < 0.05.
Results
Intracisternal (i.c.) treatment with DCPIB
The mean infarct volume was 208.0 ± 38.3 mm3 (n = 4) in vehicle - treated animals, which was significantly reduced to 68.5 ± 22.7 mm3 (n = 5) in DCPIB (i.c.) treated animals (Figure 2a, Figure 4). Behavioral scores were better in the DCPIB (i.c.) treated groups (16.8 ± 0.4) compared to the corresponding vehicle treated groups (15.3 ± 0.447, Mann-Whitney non-parametric test; p =0.012; Figure 2b).
Figure 2.
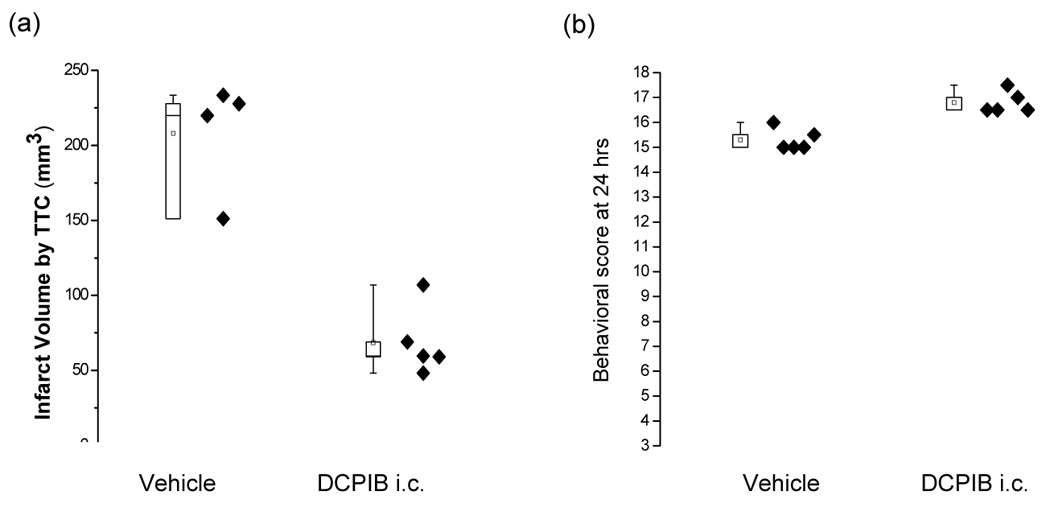
Box plots of the effects of treatment with intracisternal (i.c.) treatment of DCPIB on brain infarct size (a) and neurobehavioral scores (b). DCPIB (i.c., 20 µg/kg in 10 µl DMSO) given 15 minutes before the initiation of ischemia both reduced infarct volume significantly and improved neurobehavioral scores at 24 hours (p < 0.05; Mann-Whitney tests). Eighteen is the score exhibited by normal animals. Each data point represents one animal.
Figure 4.
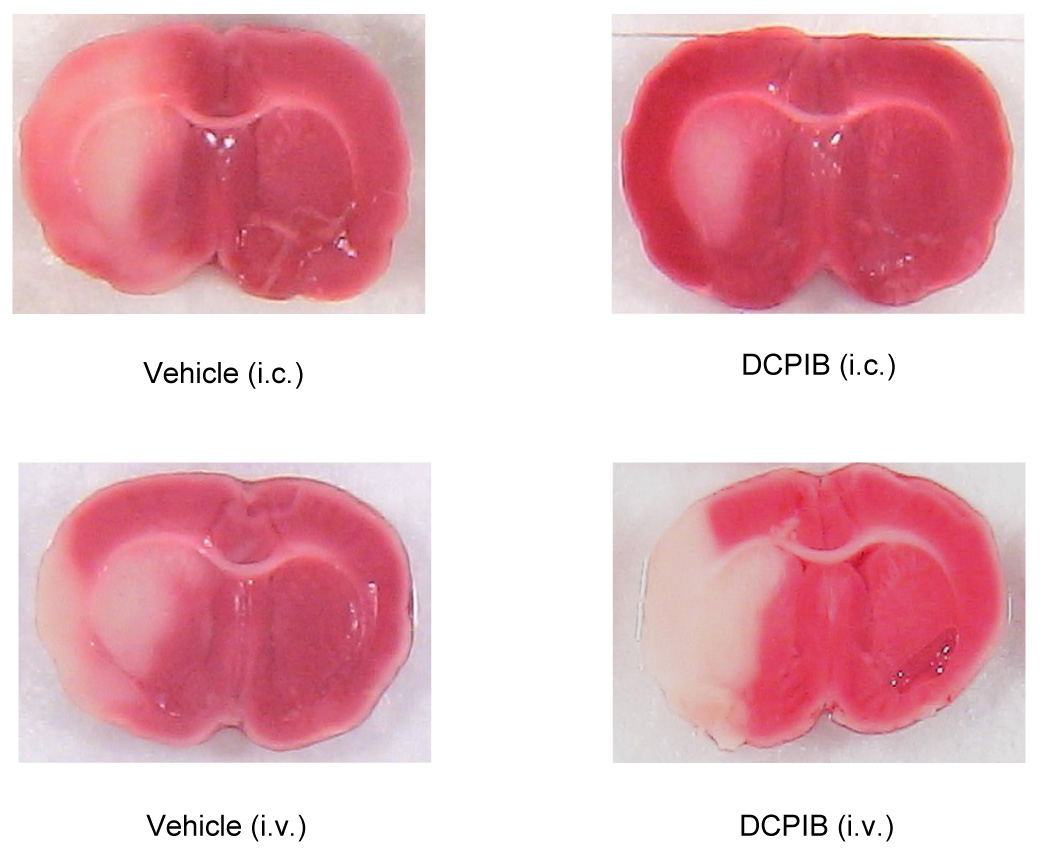
Representative sections from Vehicle - treated (i.c.) animal; DCPIB (i.c., 20 µg/kg) treated animal; Vehicle - treated (i.v.) animal; and DCPIB (i.v., 10 mg/kg) treated animals.
Intravenous treatment (i.v.) with DCPIB
Thus rats given DCPIB intracisternally had significantly improved neurobehavioral deficit scores after 24 hours and showed significantly reduced infarct volumes, giving a reduction of ~ 67%. In contrast to the effects with i.c. injection, i.v. injection of DCPIB led to no significant difference in infarct volume. These were 191.5 ± 86.8 mm3 (n = 14) in vehicle treated animals, which was not significantly different from 178.6 ± 98.2 mm3 (n = 12) in the animals given 10 mg/kg DCPIB (i.v.) (p = 0.73, Figure 3a, 4). Similarly, no differences were found in neurobehavioral score between vehicle and DCPIB (i.v.) treated groups (p =0.26, Figure 3b). The rat brain is ~1.5 g and we use 300g rats. Since DCPIB is likely restricted to the extracellular space (ECS) and while an ~ 20% ECS is found for normal brain and ~10% in ischemic brain due to cell swelling, we therefore increased the dose 500-fold.
Figure 3.
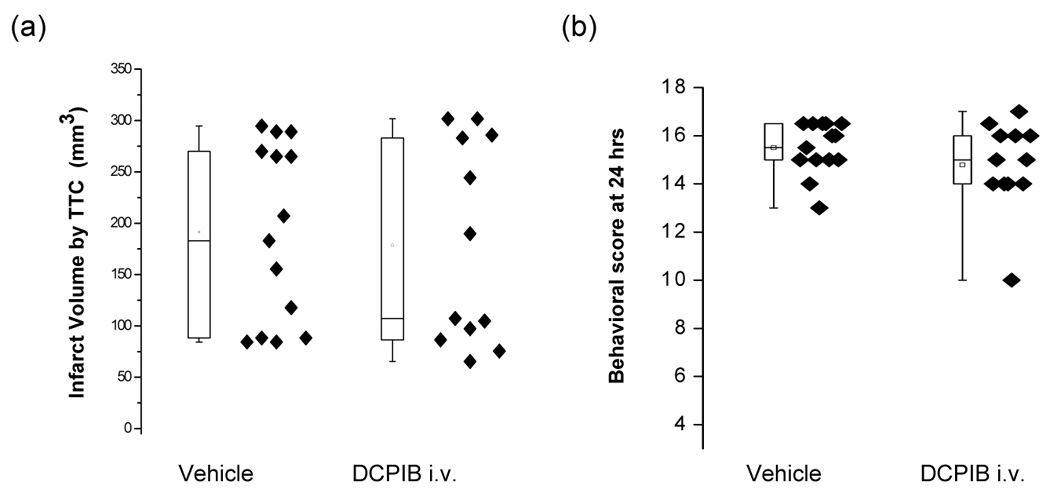
Box plots of the effects of treatment with intravenous (i.v.) treatment of DCPIB on brain infarct size (a) and neurobehavioral scores (b). DCPIB (i.v., 10 mg/kg in 0.1 ml DMSO) was given 15 minutes before the initiation of ischemia. Neither the behavioral scores nor the infarct volumes are significantly different (Mann-Whitney tests; p>0.05). Eighteen is the score exhibited by normal animals. Each data point represents one animal.
Effect of DCPIB on the extracellular concentration of aspartate and glutamate
During ischemia, microdialysate levels of both glutamate and aspartate rose less steeply in the presence of DCPIB (Figure 5a, b). DCPIB had no effect on contralateral glutamate or aspartate concentrations. The concentration peak of glutamate in the microdialysate on the rMCAO side not exposed to DCPIB was 8.1 ± 3.9 micromolar compared to 4.1 ± 1.3 in DCPIB treated cortex, and this effect was significant (p=0.007, see Fig.5 legend for more details). Upon reperfusion the concentrations of both glutamate and aspartate returned toward baseline levels.
Figure 5.
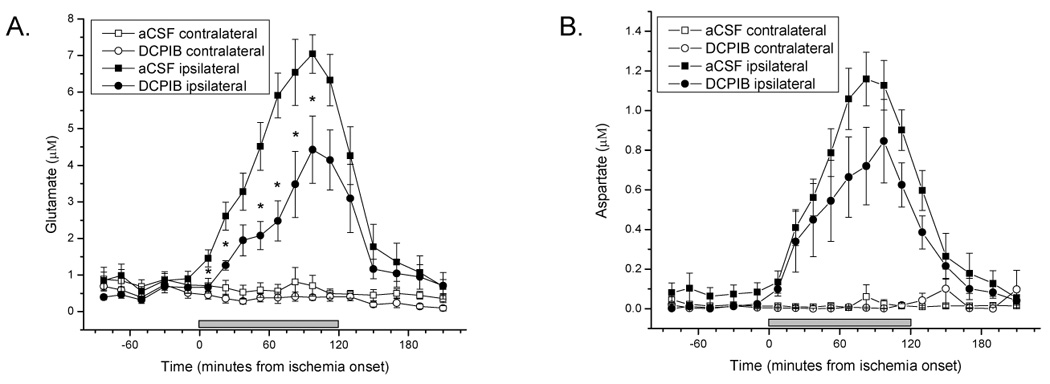
Mean and standard errors of EAA levels in microdialysate samples. Ischemia began at time zero and lasted 2 hours (gray bar). Statistical analysis was by two-way repeated measures analysis of variance with one within effect of time, one between effect of aCSF vs DCPIB, and their interactions. For glutamate (panel A) during ischemia there was a statistically significant effect of time (p<0.001), a significant effect of DCPIB (p=0.007) and a significant interaction (p=0.02) indicating both that the glutamate levels were overall lower and rose less steeply during the ischemia. Planned comparisons between the ipsilateral vehicle and DCPIB probes were done at each ischemia time point and significant differences (p<0.05) indicated with asterisks. For aspartate (panel B) during ischemia there was a statistically significant effect of time (p<0.001), and a significant interaction (p=0.04) indicating that aspartate levels rose less steeply during the ischemia although none of the planned comparisons at individual times achieved statistical significance.
Discussion
In this study we tested whether DCPIB can produce neuroprotective effects in a rat model of reversible middle cerebral artery occlusion (rMCAo). We first injected the compound intracisternally because we assumed that as a fully charged anion at physiological pH it will not effectively cross the BBB. We had previously seen this for a related compound (L-644,711), where the dose required to effectively reduce the mortality from closed head injury in cats was about 100 fold less than that needed if the drug was given intravenously (Kimelberg et al., 1987). The simplest interpretation of the data was that the drug did not cross the BBB, or at least in sufficient amounts to be effective in the TBI model. Similarly, in the present study, we also tested DCPIB via intravenous injection and found it did not achieve the protective effects as when given intracisternally in the same rMCAo model.
The aryloxalkanoic acid derivative, DCPIB, is a potent, selective blocker of VRACs in rat pancreatic β-cells (IC50 ~ 2 µM) and ICl,swell in various cardiovascular tissues and cultured cells (IC50 = 4.1 µM in CPAE cells). This blockade is voltage-independent. DCPIB displays minimal inhibition of other Cl− and K+ currents (< 10% inhibition at 10 µM), and inhibits glucose-stimulated insulin secretion in intact β-cells via VRAC inhibition and indirect KATP channel activation. It reverses cell swelling-induced action potential duration shortening in atrial myocytes and inhibits astroglial swelling in vitro (Abdullaev et al., 2006;Bourke et al., 1981;Best et al., 2004). DCPIB has been shown to be a specific antagonist of volume regulated anion channels (VRAC) using cell expression systems where it was shown not to have any effect on a variety of expressed Cl− channels , but to be a very effective inhibitor of VRAC activity in swollen cells (Decher et al., 2001). This does not exclude other possible effects that could lead to the observed reduction in infarct volumes and the modestly improved behavioral scores, but there are currently no reports of any other effects of DCPIB expected to give such results. Further we have shown previously (Seki et al., 1999) that presumed blockade of VRACs by the broad spectrum anion channel inhibitor DNDS, caused a ~50% reduction in ischemia-induced EAA release, and thus wanted to supplement these data with more specific channel inhibitor. During a number of pathological states such as ischemia or trauma, astrocytic swelling is an early event and leads to opening of VRACs (Kimelberg, 2000;Kimelberg, 2005). The number of different VRACs that are present, which ones open under particular types and degrees of swelling, and which compounds permeate these are all topics of current research in this area (Mongin and Kimelberg, 2004;Haskew-Layton et al., 2005).
Another reason for using DCPIB is that the inhibitors of VRACs used in previous studies, such as tamoxifen and DNDS, are known to have other effects in addition to blocking VRACs. Tamoxifen has been shown to inhibit nNOS (Osuka et al., 2001) and to also scavenge oxygen free radical (Zhang et al., 2007), both potential mechanisms for protection. Thus TAM generally shows more protection than DCPIB for both behavior and infarct volume (Kimelberg et al., 2000). This complicates the determination of the mechanisms by which it is protective. The present study is the first to report the neuroprotective effects produced by DCPIB, a more specific inhibitor of VRACs than others that have heretofore been studied in any cerebral ischemia model. Thus we may parsimoniously ascribe the strong protection afforded by intracisternal injection of DCPIB given at the time of ischemia, as measured by both neurobehavior and histology, to inhibition of VRACs. How long this conclusion will remain valid, as always in the empirical sciences, depends upon what new observations may be made in the future.
We did not explore the therapeutic window for DCPIB, yet further studies would be useful to establish the actual therapeutic window to show this correlates only with inhibition of the early transient increased EAAs, or a broader therapeutic window is found consistent with later smaller secondary increases in EAAs seen after reperfusion (Matsumoto et al., 1996).
We have previously showed that there was much greater protection in TBI when the fluoren derivative related to DCPIB, L-644,711 (Fig 1), was injected i.c. instead of intravenously, although, unlike the indane derivative DCPIB, L-644, 711 did have an equivalent effectiveness when injected i.v. at a 200 fold greater amount than when given i.c. (Kimelberg et al., 1987). Possibly a higher dose of DCPIB would be effective when injected intravenously. Tamoxifen like DCPIB inhibits VRAC channels (Abdullaev et al., 2006) but also readily penetrates the CNS (Biegon et al., 1996). It also inhibits other known neurodamaging effects that likely make it a very effective neuroprotectant after stroke in animal models when given i.v. (Kimelberg, 2005) Therefore the present work is not aimed at advocating the use of a pure blocker of VRACs as a therapeutic strategy, even if it were CNS accessible, but to show proof of the principle that blockade of VRACs is a likely component of the action of a better therapeutic such as tamoxifen, which additionally has other neuroprotective actions and penetrates the BBB because of its high lipid solubility.
Acknowledgments
This work was supported by NIH NS35205 (H.K.K) and Charitable Leadership Foundation, Latham NY. The authors gratefully acknowledge Yiqiang Jin and Layli Nazirova at the Nerve Cell Rescue Laboratory of Ordway Research Institute for their technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdullaev IF, Rudkouskaya A, Schools GP, Kimelberg HK, Mongin AA. Pharmacological comparison of swelling-activated excitatory amino acid release and Cl- currents in rat cultured astrocytes. J Physiol. 2006;572.3:677–689. doi: 10.1113/jphysiol.2005.103820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best L, Yates AP, Decher N, Steinmeyer K, Nilius B. Inhibition of glucose-induced electrical activity in rat pancreatic beta-cells by DCPIB, a selective inhibitor of volume-sensitive anion currents. Eur J Pharmacol. 2004;489:13–19. doi: 10.1016/j.ejphar.2004.02.030. [DOI] [PubMed] [Google Scholar]
- Biegon A, Brewster M, Degani H, Pop E, Somjen D, Kaye AM. A permanently charged tamoxifen derivative displays anticancer activity and improved tissue selectivity in rodents. Cancer Res. 1996;56:4328–4331. [PubMed] [Google Scholar]
- Bourke RS, Kimelberg HK, Daze MA, Popp AJ. Studies on the Formation of Astroglial Swelling and Its Inhibition by Clinically Useful Agents. In: Popp AJ, Nelson LR, Bourke RS, Kimelberg HK, editors. Neural Trauma. New York: Raven Press; 1979. pp. 95–113. [Google Scholar]
- Bourke RS, Waldman JB, Kimelberg HK, Barron KD, San Filippo BD, Popp AJ, Nelson LR. Adenosine-stimulated astroglial swelling in cat cerebral cortex in vivo with total inhibition by a non-diuretic acylaryloxyacid derivative. J Neurosurgery. 1981;55:364–370. doi: 10.3171/jns.1981.55.3.0364. [DOI] [PubMed] [Google Scholar]
- Cragoe EJ, Gould NP, Woltersdorf OW, Ziegler C, Bourke RS. Agents for treatment of brain injury I. (Aryloxy) Alkanoic Acids. J Med Chem. 1982;25:567–579. doi: 10.1021/jm00347a017. [DOI] [PubMed] [Google Scholar]
- Cragoe EJ, Woltersdorf OW, Jr, Gould NP, Pietruszkiewicz AM, Ziegler C, Sakurai Y, Stokker GE, Anderson PS, Bourke RS, Kimelberg HK, Nelson LR, Barron KD, Rose JE, Szarowski D, Popp AJ, Waldman JB. Agents for the treatment of brain edema. 2. [2,3,9,9a-Tetrahydro-3-oxy-substituted-1 H-fluoren-7-yl) oxyalkanoic acids and some of their analogues. J Med Chem. 1986;29:825–841. doi: 10.1021/jm00155a038. [DOI] [PubMed] [Google Scholar]
- Cragoe JE. Drugs for the treatment of traumatic brain injury. Medicinal Res. 1987;7:271–305. doi: 10.1002/med.2610070302. [DOI] [PubMed] [Google Scholar]
- Decher N, Lang HJ, Nilius B, Bruggemann A, Busch AE, Steinmeyer K. DCPIB is a novel selective blocker of I(Cl,swell) and prevents swelling- induced shortening of guinea-pig atrial action potential duration. Br J Pharmacol. 2001;134:1467–1479. doi: 10.1038/sj.bjp.0704413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feustel PJ, Jin Y, Kimelberg HK. Volume regulated anion channels are the predominant contributors to release of excitatory amino acids in the ischemic cortical penumbra. Stroke. 2004;35:1164–1168. doi: 10.1161/01.STR.0000124127.57946.a1. [DOI] [PubMed] [Google Scholar]
- Garcia JH, Wagner S, Liu K-F, Hu Xj. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Stroke. 1995;26:627–635. doi: 10.1161/01.str.26.4.627. [DOI] [PubMed] [Google Scholar]
- Haskew-Layton RE, Mongin AA, Kimelberg HK. Hydrogen peroxide potentiates volume-sensitive excitatory amino acid release via a mechanism involving Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 2005;280:3548–3554. doi: 10.1074/jbc.M409803200. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK. Cell volume in the CNS: Regulation and implications for nervous system function and pathology. The neuroscientist. 2000;6:13–24. [Google Scholar]
- Kimelberg HK. Astrocytic swelling in cerebral ischemia as a possible cause of injury and target for therapy. Glia. 2005;50:389–397. doi: 10.1002/glia.20174. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK, Barron KD, Bourke RS, Nelson LR, Cragoe EJ. Brain Anti-Cytoxic Edema Agents. New York: Wiley-Liss; 1990. [PubMed] [Google Scholar]
- Kimelberg HK, Cragoe EJ, Jr, Nelson LR, Popp AJ, Szarowski D, Rose JW, Woltersdorf OW, Jr, Pietruszkiewicz AM. Improved recovery from a traumatic-hypoxic brain injury in cats by intracisternal injection of an anion transport inhibitor. Cent Nerv Syst Trauma. 1987;4:3–14. doi: 10.1089/cns.1987.4.3. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK, Feustel PJ, Jin Y, Paquette J, Boulos A, Keller RW, Tranmer BI. Acute treatment with tamoxifen reduces ischemic damage following middle cerebral artery occlusion. NeuroReport. 2000;11:2675–2679. doi: 10.1097/00001756-200008210-00014. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK, Mongin AA. Swelling-activated release of excitatory amino acids in the brain: Relevance for pathophysiology. In: Lang F, editor. Cell Volume Regulation. Basel: Karger; 1998. pp. 240–257. [DOI] [PubMed] [Google Scholar]
- Law RO. Amino acids as volume-regulatory osmolytes in mammalian cells. Comp Biochem Physiol. 1991;99A:263–277. doi: 10.1016/0300-9629(91)90001-s. [DOI] [PubMed] [Google Scholar]
- Law RO. Regulation of mammalian brain cell volume. J Experimental Zoology. 1994;268:90–96. doi: 10.1002/jez.1402680204. [DOI] [PubMed] [Google Scholar]
- Lipton P. Ischemic cell death in brain neurons. Physiol Reveiws. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- Liu HT, Tashmukhamedov BA, Inoue H, Okada Y, Sabirov RZ. Roles of two types of anion channels in glutamate release from mouse astrocytes under ischemic or osmotic stress. Glia. 2006 doi: 10.1002/glia.20400. [DOI] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Lo EH, Pierce AR, Halpern EF, Newcomb R. Secondary elevation of extracellular neurotransmitter amino acids in the reperfusion phase following focal cerebral ischemia. Journal of Cerebral Blood Flow and Metabolism. 1996;16(1):114–124. doi: 10.1097/00004647-199601000-00014. [DOI] [PubMed] [Google Scholar]
- Mongin AA, Kimelberg HK. Astrocytic Swelling in Neuropathology. In: Kettenmann HO, Ransom BR, editors. Neuroglia. New York, NY: Oxford University Press; 2004. pp. 550–562. [Google Scholar]
- Nelson LR, Auen EL, Bourke RS, Barron KD, Malik AB, Cragoe J, Popp AJ, Waldman JB, Kimelberg HK, Foster VV, Creel W, Schuster L. Comparison of animal head injury models developed for treatment modality evaluation. In: Grossman RG, Gildenberg PL, editors. Head Injury: Basic and Clinical Aspects. New York: Raven Press; 1982. pp. 117–127. [Google Scholar]
- Nelson LR, Bourke RS, Popp AJ, Cragoe J, Signorelli A, Foster VV, Creel W. Evaluation of treatment modalities in severe head injuries using an animal model. In: Popp AJ, Bourke RS, Nelson LR, Kimelberg HK, editors. Neural Trauma. New York: Raven Press; 1979. pp. 297–313. [Google Scholar]
- Nicholls D, Attwell D. Release and uptake of excitatory amino acids. TiPs. 1990;11:462–468. doi: 10.1016/0165-6147(90)90129-v. [DOI] [PubMed] [Google Scholar]
- Nilius B, Droogmans G. Amazing chloride channels: an overview. Acta Physiol Scand. 2003;177:119–147. doi: 10.1046/j.1365-201X.2003.01060.x. [DOI] [PubMed] [Google Scholar]
- Ochoa, De. La., Paz LD, Lezama R, Torres-Marquez ME, Pasantes-Morales H. Tyrosine kinases and amino acid efflux under hyposmotic and ischaemic conditions in the chicken retina. Pflugers Arch. 2002;445:87–96. doi: 10.1007/s00424-002-0883-0. [DOI] [PubMed] [Google Scholar]
- Okada Y. Volume expansion-sensing outward-rectifier Cl− channel: Fresh start to the molecular identity and volume sensor. Am J Physiol :Cell Physiol. 1997;273:C755–C789. doi: 10.1152/ajpcell.1997.273.3.C755. [DOI] [PubMed] [Google Scholar]
- Osuka K, Feustel PJ, Mongin AA, Tranmer BI, Kimelberg HK. Tamoxifen inhibits nitrotyrosine formation after reversible middle cerebral artery occlusion in the rat. J Neurochem. 2001;76:1842–1850. doi: 10.1046/j.1471-4159.2001.00198.x. [DOI] [PubMed] [Google Scholar]
- Pasantes-Morales H, Moran J, Schousboe A. Volume-sensitive release of taurine from cultured astrocytes: Properties and mechanism. Glia. 1990;3:427–432. doi: 10.1002/glia.440030514. [DOI] [PubMed] [Google Scholar]
- Phillis JW, Ren J, O'Regan MH. Transporter reversal as a mechanism of glutamate release from the ischemic rat cerebral cortex: studies with DL-threo-β-benzyloxyaspartate. Brain Res. 2000;868:105–112. doi: 10.1016/s0006-8993(00)02303-9. [DOI] [PubMed] [Google Scholar]
- Phillis JW, Song D, O'Regan MH. Inhibition by anion channel blockers of ischemia-evoked release of excitotoxic and other amino acids from rat cerebral cortex. Brain Res. 1997;758:9–16. doi: 10.1016/s0006-8993(97)00155-8. [DOI] [PubMed] [Google Scholar]
- Quesada O, González E, Morales-Mulia S, Bargas J, Pasantes-Morales H. Effects of NaCl removal on osmolyte fluxes and regulatory volume decrease in cultured astrocytes. J Neurosci Res. 1998;53:195–202. doi: 10.1002/(SICI)1097-4547(19980715)53:2<195::AID-JNR8>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Seki Y, Feustel PJ, Keller RW, Tranmer BI, Kimelberg HK. Inhibition of ischemia-induced glutamate release in rat striatum by dihydrokainate and an anion channel blocker. Stroke. 1999;30:433–440. doi: 10.1161/01.str.30.2.433. [DOI] [PubMed] [Google Scholar]
- Strange K, Emma F, Paredes A, Morrison R. Osmoregulatory changes in myo-inositol content and Na+/myo- inositol cotransport in rat cortical astrocytes. Glia. 1994;12:35–43. doi: 10.1002/glia.440120105. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Jin Y, Behr MJ, Feustel PJ, Morrison JP, Kimelberg HK. Behavioral and histological neuroprotection by tamoxifen after reversible focal cerebral ischemia. Exp Neurol. 2005;196:41–46. doi: 10.1016/j.expneurol.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Milatovic D, Aschner M, Feustel PJ, Kimelberg HK. Neuroprotection by tamoxifen in focal cerebral ischemia is not mediated by an agonist action at estrogen receptors but is associated with antioxidant activity. Exp Neurol. 2007;204:819–827. doi: 10.1016/j.expneurol.2007.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]